

Abstract
We present an analysis of trajectories from Brownian dynamics simulations of diffusional protein-protein encounter for the well-studied system of barnase and barstar. This analysis reveals details about the optimal association pathways, the regions of the encounter complex, possible differences of the pathways for dissociation and association, the coupling of translational and rotation motion, and the effect of mutations on the trajectories. We found that a small free-energy barrier divides the energetically most favorable region into a region of the encounter complex above the barnase binding interface and a region around a second energy minimum near the RNA binding loop. When entering the region of the encounter complex from the region near the RNA binding loop, barstar has to change its orientation to increase the electrostatic attraction between the proteins. By concentrating the analysis on the successful binding trajectories, we found that the region of the second minimum is not essential for the binding of barstar to barnase. Nevertheless, this region may be helpful to steer barstar into the region of the encounter complex. When applying the same analysis to several barnase mutants, we found that single mutations may drastically change the free-energy landscape and may significantly alter the population of the two minima. Therefore, certain protein-protein pairs may require careful adaptation of the positions of encounter and transition states when interpreting mutation effects on kinetic rates of association and/or dissociation.
INTRODUCTION
Association of two or more proteins is one of the most important biochemical processes in biological cells. The association process is typically divided into a process of diffusional encounter, which may be accelerated by long-range electrostatic interactions, and the final step of association, where short-range interactions play the most prominent role. For electrostatically attracting proteins, the first process includes the region of free diffusion, when the proteins are far apart, the region of electrostatic steering, and the encounter complex, which can be defined as the minimum of the free-energy landscape in this diffusional regime (for other suitable definitions and for a summary of several structural approaches, see Gabdoulline and Wade (1)). In this encounter complex, the proteins can reorient their interaction patches, which is required for formation of the bound complex. This diffusive process is followed by the second step, which includes the formation of the transition state, a possible second intermediate, and the bound complex (2). To get from the encounter complex to the transition state involves a loss of translational and orientational entropy and the main part of the unfavorable desolvation. This is later compensated by the energy gained due to the hydrophobic and van der Waals interactions and by the formation of hydrogen bonds and salt bridges. The diffusive protein motion in the first process can be well modeled by Brownian dynamics (BD) simulations, whereas the modeling of the second step needs more detailed simulation techniques such as molecular dynamics simulations.
Over the past years, BD simulations have emerged as a powerful method for the computational analysis of the kinetics of protein-protein interactions. In particular, the calculated association rates are in good agreement with experimental findings (3). Given this good agreement it should be possible to deduce further information from the simulation data about the association behavior of the proteins: What are the individual association pathways and the regions of the encounter complex? Is there a distinction between association and dissociation pathways? How strong is the coupling of the translational and rotational motions? And how do mutations affect these features? These issues will be addressed in this article.
For this work, we chose as a model system the well-studied protein-protein pair of barnase and barstar (see Fig. 1). Barnase is an extracellular ribonuclease and barstar is its intracellular inhibitor. They are rather small proteins with diameters of ∼30 Å. The interaction between barstar and barnase is among the strongest known interactions between proteins, with a very fast association rate of 108–109 M−1 s−1 at 50 mM ionic strength (4). The binding interface, characterized by mutagenic experiments (4,5) and by a crystal structure of the barnase-barstar complex (6), mainly consists of polar and charged residues, and shows a high electrostatic complementarity. Therefore, this protein pair provides a well characterized model system of electrostatically steered association between proteins. Their interaction has been extensively studied both experimentally and theoretically: First, binding constants and kinetic association rates for wild-type and mutant proteins were extensively measured by the Fersht group (4,5). Schreiber and Fersht also aimed at predicting the rate enhancement for various mutants and for varying ionic strengths using a transition-state-like theory and atomic modeling (7,8). Gabdoulline and Wade then calculated the association rates of the wild types and a set of mutants from BD simulations (3,9) and obtained good agreement with the experimental association rates. They also analyzed energetically favorable regions by a Boltzmann factor analysis, and recently studied the binding energetics of these proteins (10). Complementary to the BD simulations, Camacho et al. systematically sampled the translational and rotational space to compute the free-energy landscape, and compared the contributions of the electrostatic and desolvation energies of encounter complexes (11). Janin and co-workers performed an extensive analysis of protein-protein recognition sites for barnase and barstar and a large series of protein pairs (12). The electrostatic contribution to the binding stability of barnase and barstar was analyzed by Zhou and colleagues (13).
FIGURE 1.

(A) Barnase and (B) barstar, colored according to their electrostatic potentials ranging from −7 kT/e (red) to +7 kT/e (blue). The residues of the interaction patches are drawn as white sticks. The complex structure can be obtained by placing barstar in panel B rotated by 180° around the vertical axis on top of barnase in panel A. In panel A, the mutated residues are highlighted in yellow. R59 and E60 are located in the RNA binding loop (residues 57–60). Residues D54 and K66 have no direct binding partners on the barstar interface, although D54 is located in the binding region. The 3D images in this article were generated using the visualization molecular dynamics program (VMD) (33).
The barnase mutations considered in this article are located in different regions on the protein surface and therefore they each have a different impact on the association and binding behavior of barnase and barstar. The positively charged residues at the inner region of the binding interface (K27, R59, R83, and R87) serve for binding and stabilization of the negatively charged RNA substrate (6). The negatively charged residues D54 and E60 are located in the periphery of the interface. Their function is probably to stabilize the positive charges around the active site of barnase (14). Residues 57–60 form the guanine binding loop. It was shown that replacement of the positively charged residues of the barstar interface reduces the association rate to barstar, whereas mutations D54A and E60A increase these rates (4,5,9). An exception to this rule is K66. A replacement of this positively charged residue by alanine reduces the association rate only slightly. It is located outside of the interface and was suggested to be responsible for incorrect steering of barstar toward barnase (3).
The studies in this article are based on previous work by Gabdoulline and Wade (3,9), in which they characterize the association kinetics of barnase and barstar from BD simulations, and on a recent study (15) in which Spaar and Helms developed a method for uncovering the underlying entropy and free-energy landscapes from a large set of BD simulations. Here, we focus on the analysis of the free-energy landscape to analyze the encounter complex regions and the optimal association/dissociation pathways. It has been pointed out several times that protein-protein association events are steered by a funnel-shaped free-energy landscape (11,16,17) such as protein folding. Therefore one may assume identical pathways for association and dissociation as long as the free-energy surfaces are the same. This results from the principle of detailed balance. Things may look different if one takes into account induced-fit effects that may alter the free-energy surface for dissociation. In further computations, we studied the mean orientation of barstar and the coupling of the translational and rotational motions. Identifying the actual pathways of the proteins on the six-dimensional configuration space is one of the features that can only be obtained from dynamic simulations and not by computing energies on a grid. To analyze effects of barnase mutations on the association behavior of the proteins, we studied the occupancy and free-energy landscape in the same way as for the barnase wild type. In this mutant study, we particularly focused on the position and shape of the encounter complex region and the optimal center-of-mass positions for increasing distance as derived from the occupancy and free-energy landscapes.
In a recent study, Miyashita et al. (18) characterized the conformations of the encounter and transition states for the system of cytochrome c2 and reaction center using Monte Carlo simulations. They assumed that the conformations of the encounter and transition states are nearly the same for all mutants and identified spatial regions where the computed free-energy differences between wild type and mutants matched the experimental values. In the last part of this article, the question is addressed whether this assumption holds true for the barnase-barstar system.
MATERIALS AND METHODS
Protein structures
We used the coordinates of barnase and barstar as prepared by Gabdoulline and Wade (9). The coordinates of the docked wild-type barnase-barstar complex, determined to 2.0 Å resolution by Buckle and co-workers (6), were taken from the Protein Data Bank (PDB code 1brs). Chain A was used for barnase and chain D for barstar. The two other pairs of chains in the crystal structure were used to model the missing side-chain atoms in the A and D chains. Crystallographic water molecules were removed. Mutants were modeled by replacing the mutated side chain. The barnase mutants considered here are K27A, D54A, R59A, E60A, D54A/E60A, K66A, K66A/D54A, R83Q, and R87A. Polar hydrogen atoms were added and their positions optimized by energy minimization with the CHARMM program (19) using the QUANTA molecular graphics package (Molecular Simulations, San Diego, 1992). Side-chain conformations were kept the same as in the barnase-barstar complex.
Computation of forces
The results of our work, the analysis of the association pathways and the encounter-complex regions, are complementary to the results of recent studies of association rates on barnase and barstar by Gabdoulline and Wade (3). To allow a comparison with their results we used the same parameters as in their work for the computation of forces and the simulations.
For the modeling of the long-range electrostatic interaction of the proteins, solutions of the linear Poisson-Boltzmann equation were computed for each protein using the University of Houston Brownian Dynamics program (UHBD) (21). Partial charges and atomic radii were assigned from the OPLS data set (22). The protonation states of titrable residues were assigned according to their standard protonation states at the experimental pH of 8.0 (5). Grids with dimensions of 150 × 150 × 150 nodes and a 1.0-Å spacing centered on each of the proteins were used. The ionic strength was set to 50 mM and the temperature was set to 300 K. The solvent dielectric was assigned a value of 78.0.
The effective charge method (ECM) (23) was used to derive charges that represent the external electrostatic potential in a uniform dielectric medium. The effective charges were fitted to reproduce the electrostatic potential in a 3-Å-thick layer starting at the accessible surface defined by a probe of radius 4 Å and extending outward of the protein. To compute forces and torques acting on protein 2(1), the array of effective charges for protein 2(1) is placed on the electrostatic potential grid of protein 1(2).
Short-range repulsive forces are treated by an exclusion volume prohibiting van der Waals overlap of the proteins. The exclusion volume is precalculated on a grid with 0.5-Å grid spacing. If a move during the BD simulation would result in van der Waals overlap, the BD step is repeated with different random numbers until no overlap occurs. The surface-exposed atoms of the smaller protein are listed and steric overlap is defined as occurring when one of the surface-exposed atoms is projected on a grid point that represents the interior of the larger protein (24).
Charge desolvation penalties are computed in an approximate fashion that treats the solvation of each charge independently (25) The charge desolvation penalty of one protein is taken as the sum of desolvation penalties of each charge of that protein. The desolvation penalty of each charge is the sum of desolvation penalties due to the low dielectric cavity of each atom of the other protein. The desolvation energy of protein 1 due to the presence of protein 2 is approximated as:
 |
where κ is the Debye-Hückel parameter, ɛs and ɛp are the dielectric constants of the solvent and the protein, respectively, qi is the effective charge on the ith atom of protein 1, aj is the radius of the jth atom of protein 2, and rij is the distance between the two atoms. The summation is carried out over all possible pairs of effective charges on protein 1 and atoms on protein 2. The scaling factor α for the weighting between electrostatic interaction and desolvation terms was set to 1.67. The validity of this choice may be assessed from the electrostatic interaction free-energy calculations shown in Figs. 1 and 2 of Gabdoulline and Wade (3).
FIGURE 2.

Maps of the (A) occupancy and (B) free-energy landscapes for the association of barstar to barnase. The maps are colored from blue (high occupancy/low free energy) to red (low occupancy/high free energy). (C) Energy profiles along the reaction pathway: the electrostatic energy ΔEel, the desolvation energy ΔEds, the entropy loss –TΔS, and the free energy ΔG (from left to right).
The atom-atom contacts of the reaction patches are assigned in a fully automated way independent of which residue each atom is in (9). Possible contacts are those pairs between hydrogen-bond donor and acceptor atoms having a separation distance of <5.0 Å in the x-ray structure of the complex.
Brownian dynamics simulations
For the BD simulations we used the software package SDA (26) which was modified to allow for a detailed analysis of the trajectories. Here, the proteins are modeled as rigid bodies, and short-range interactions such as van der Waals forces and the formations of hydrogen bonds and salt bridges are not modeled. However, these simplifications become important only at small protein-protein separations.
To get a high statistics for the occupancy maps, 200,000 trajectories were simulated for each protein pair. The trajectories start with the two proteins at a center-to-center distance b with randomly chosen orientations, and finish when the proteins reach a center-to-center distance c > b. In a previous work (15), b was chosen to be 100 Å and c 500 Å, which are the same values used by Gabdoulline and Wade (3). These values correspond to 7.4 and 36.8 Debye lengths at 300 K and 50 mM. In the simulations presented here we decreased c to 110 Å since we focused on the analysis of the trajectories and aimed to get a high statistics instead of improving the association rates. By that we gained a factor of 10 in computation time, compared to the simulations with c = 500 Å.
The diffusion equation is solved by the Ermak-McCammon algorithm (27). The translational Brownian motion of two interacting proteins is simulated as the displacement Δr of the relative separation vector r during a time step Δt according to the relation
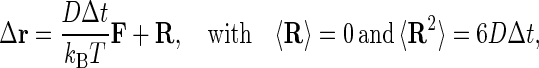 |
where F is the systematic interparticle force, kB is the Boltzmann constant, T is the temperature, and R is the stochastic displacement arising from collisions of the proteins with solvent molecules. Two analogous formulas are used to generate the rotational motions of the two proteins in terms of rotation angle wj = (w1j, w2j, w3j), torque Tij acting on protein i due to protein j, and rotational diffusion constant DiR of each protein i (i, j = 1, 2, i ≠ j):
 |
The diffusional properties of the molecules are assumed to be isotropic. In Gabdoulline and Wade (3), a relative translational diffusion constant of 0.030 Å2/ps was used, based on the individual diffusion constants of 0.015 Å2/ps assigned to both barnase and barstar in aqueous solution, and rotational diffusion constants of 4.0 × 10−5 and 4.5 × 10−5 rad2/ps for barnase and barstar, respectively.
The time step was set to 1.0 ps for center-to-center distances up to 75 Å and for larger distances it increased linearly with the intermolecular separation. This corresponds to an average random displacement of 0.4 Å at small and medium separations.
To reduce the computational cost of the simulations, no hydrodynamic interactions were considered. It was shown that the effect on the protein-protein association rates is rather small. (28) Also, in previous BD studies with simplified cytochrome c molecules it was found that hydrodynamics has only a small influence. (29) Hydrophobic forces are not included, since they are assumed to become important only at small contact distances.
The simulation software SDA also allows for the computation of association rates. This has been covered in detail in previous work by others (3,9,30). In our study, we concentrate on the analysis of the trajectories, largely omitting the computation of association rates.
Computation of the occupancy landscape
The analysis of the trajectories with the aim to compute the occupancy maps, the entropy and free-energy landscape is described in detail in (15). After each time step of the simulated trajectories the positional and orientational coordinates of protein 2 (relative to protein 1) are computed with respect to a reference coordinate system. During the simulations these coordinates are assigned to the nodes of a six-dimensional grid on which the occupancy, energy, and entropy maps are computed. The maps for the positional and orientational coordinates were computed separately to reduce the computational effort. At the same time the statistics in two three-dimensional grids is much higher compared to populating a six-dimensional grid.
The computation of the occupancy maps with respect to the positional coordinates can be understood as projecting the position of protein 2 onto a plane that is perpendicular to the center-to-center vector of the proteins in the bound state of the crystal structure. The origin of these occupancy maps is the position of protein 2 in the bound state. As a reference system, a spherical coordinate system was used, θ and φ denoting the corresponding polar and azimuthal angles between the center-to-center vector of the proteins at a trajectory position and the center-to-center vector of the proteins in the bound state. After each time step, the occupancy value of the grid element associated with the position of protein 2 is increased by 1. Note that for positions of protein 2 above the denoted plane (for θ ≤ π) and below (for θ ≥ π), two distinct sets of matrices are needed to represent the upper and lower half-spheres. Since the matrices are computed with respect to a “semi-Cartesian” coordinate system, the axes are called φx and φy, which are the x and y components of the angle φ. Here, the x axis is defined by the vector from the center of the reaction patch of protein 1 to the first atom in the list of reaction atoms. In the case of barnase wild type this is SER38:OG. Thus, the negative x axis points approximately in the direction from the patch center toward the guanine binding loop.
The occupancy maps with respect to the orientational coordinates are computed similarly. The normal vector of protein 2 is projected onto a plane that is perpendicular to the normal vector of protein 1. Here, the origin is the center of the reaction patch of protein 1. The axes are φn,x and φn,y, according to the azimuthal angle φn between the normal vector of the interaction patch of protein 2 and that of protein 1. The index n refers to the normal vectors of the reaction patches. The xn axis is defined analogously to the x axis using the first reaction atom of protein 1. The normal vector of protein 1 is defined to point outward from the protein, whereas the normal vector of protein 2 is defined to point inward, so that in the bound state the two vectors are approximately parallel. Again, two distinct sets of matrices are needed to represent the upper and lower half-spheres. The third orientational coordinate, the angle ωn, is given by the rotation of protein 2 around the normal vector of the reaction patch, ωn = 0 denoting the orientation of protein 2 in the bound state. Fig. 2 A displays the resulting occupancy maps for the positional coordinates. A detailed description is given in the Results section. The electrostatic and desolvation energies, as well as the translational and rotational entropy losses (see below) are recorded in further sets of matrices (not shown). In the matrices assigned to the electrostatic and desolvation energies, the minimum values for the given position/orientation are stored, thus allowing us to finally identify the minimum free-energy paths.
Along the angles φx and φy, as well as φn,x and φn,y, we used a discretization with 2 × 101 nodes (for the upper and lower half-spheres), i.e., an average step size of 1.8°. For ωn we used a step size of 4°, i.e., 90 nodes. Along the distance axis a spacing of 2 Å was used over a distance of 80 Å. The initial value was set by the center-to-center distance of the proteins in the bound state. Finally, the occupancy values are normalized according to the volume of the corresponding element of the configuration space. In contrast to our previous work, where we compared several definitions for the so-called contact distance, the distance between the interaction patches, throughout this article we are using the center-to-center distance d1–2 as the reaction coordinate.
Calculation of the entropy landscape
By interpreting the computed occupancy maps as probability distributions, the contribution of the translational and rotational entropy to the free-energy landscape is computed by the restriction of the degrees of motional freedom and by applying a local entropy function. The total entropy loss of protein-protein encounter is calculated as the sum of the translational and rotational entropies ΔS = ΔStrans + ΔSrot. The splitting into the translational and rotational parts allows the separate computation of the entropy maps with respect to the positional and orientational coordinates.
Since the proteins are simulated as rigid bodies we do not take account of their internal entropy loss through changes in vibrational modes and side-chain conformations. However, it can be assumed that this entropy contribution only becomes important for protein-protein separations of a few Ångstroms, i.e., in a regime where the protein motions are not diffusion-limited anymore. Also, the entropy of the solvent is not considered here. Again, this contribution can be assumed to be limited to small protein-protein distances.
Here, the process of protein encounter is considered as a step-by-step process on a molecular level, meaning that a protein at a certain position and with a certain orientation cannot explore the full configuration space within a Brownian Dynamics time step. Therefore, we are interested in calculating the position- and orientation-dependent (local) entropy loss, rather than the total (global) entropy loss during the association process. For the computation of local entropy loss we only take into account the occupancy values of all those configurations (positions and orientations) that are reachable from the particular position and orientation within one Brownian dynamics time step, i.e., that are within its “accessible volume” of configuration space. The size of the accessible spatial and angular volumes V and Y should be in the range of the mean positional and orientational displacements during a Brownian dynamics time step. We chose V as a sphere around the position of the protein, and Y as a sphere around its orientation. The corresponding radii are Δρ and Δω, respectively. To compute the local entropy loss at the given position and orientation from the occupancy of the positions and orientations (the states) within the accessible volumes V and Y, the occupancy landscape is interpreted as a probability distribution, which allows the application of the basic entropy formula  where the Pn are the probabilities for each state n. This configuration-dependent entropy value is then compared to the entropy with a constant, isotropic probability distribution, which is the reference state if the proteins are far apart. Note that the entropy loss is computed separately for all grid nodes of the positional and orientational space, which together represent the entropy landscape. See Spaar and Helms (15) for details.
where the Pn are the probabilities for each state n. This configuration-dependent entropy value is then compared to the entropy with a constant, isotropic probability distribution, which is the reference state if the proteins are far apart. Note that the entropy loss is computed separately for all grid nodes of the positional and orientational space, which together represent the entropy landscape. See Spaar and Helms (15) for details.
The average translational and rotational displacements of barnase and barstar during a time step of Δt = 1.0 ps are ∼0.4 Å and 1.3°, respectively. These represent minimum values for the radii of the accessible volumes V and Y, in particular for a combined translational and rotational motion. In our previous work (15), we showed that the entropic contribution is only weakly dependent on the size of the volumes V and Y. In the simulations, we used Δρ = 3 Å and Δω = 3°.
Free-energy landscape
In the sections above we described the computation of the energy and entropy landscapes. The contributions of electrostatic and desolvation energies, as well as the translational and rotational entropy loss, are stored in matrices that represent the six-dimensional configuration space. These matrices have the same grid sizes as the occupancy maps and they are computed separately for the positional and orientational coordinates. With the energy and entropy contributions as functions of the translational and rotational coordinates, the free-energy landscape of the encounter process is given by the sum of the electrostatic energy, the desolvation energy, and the translational/rotational entropy:
 |
Fig. 2 B displays maps of the free-energy landscape corresponding to the occupancy maps in Fig. 2 A. Fig. 2 C shows the energy profiles along the reaction path, which is defined as the path along the minima of the free-energy landscape: the electrostatic interaction energy ΔEel, the desolvation energy ΔEds, the (negative) translational and rotational entropy loss –TΔS, and the free energy ΔG of the protein-protein association.
Calculation of the mean orientation
In each step during the simulation, the position of barstar is assigned to a three-dimensional grid. In every step, the value of the corresponding grid element was increased by 1 to count the occupancy. The orientation of barstar was recorded by storing the components of the normal vector in separate three-dimensional grids with the same spacing. During the simulation, the values of the grid nodes are summed; the mean orientation is computed after the simulation. The spacing of this grid was varied from 1 Å to 5 Å. The vectors are scaled according to occupancy of the corresponding grid element and point from the center-of-mass of barstar to the center of its binding interface.
RESULTS
Several methods were used for analyzing the trajectories of BD simulations of barnase and barstar to get detailed insight into the diffusional association behavior of these proteins and to find the most prominent effects of mutations on the association. Barnase and barstar are shown in Fig. 1, A and B, with their surfaces colored according to the electrostatic potential at the surface, ranging from −7 kT/e (red) to +7 kT/e (blue). The residues of the interaction patches, defined by a distance of <4.5 Å between the proteins in the complex structure, are highlighted in white. Fig. 1 A shows barnase, with those residues that have been computationally mutated in this study highlighted in yellow.
Computation of the occupancy and free-energy landscapes
The occupancy and free-energy landscapes were computed by a detailed analysis of the BD trajectories (15). First, the particle positions sampled during 200,000 trajectories were collected in occupancy landscapes for the positional and orientational space. Due to the high statistics, this occupancy landscape could then be interpreted as a probability distribution from which we computed the entropy landscape by applying a locally defined entropy function. The free-energy landscape was then calculated as the sum of the electrostatic and desolvation energies and the entropy contributions. Resulting maps of the occupancy landscape of the association of barstar to barnase (wild type) are shown in Fig. 2 A for protein-protein separations d1–2 ranging from 32 to 44 Å. The positional coordinates φx and φy are the x and y components of the angle φ between the vector from the center of protein 1 to that of protein 2 and the vector between the centers of the proteins in the bound state. In these maps, regions of high occupancy are colored in blue, and low-occupancy regions in red. Complementary to the occupancy maps, Fig. 2 B shows maps of the free-energy landscape. Here, the color scale ranges from −4.0 (blue) to 0.0 kcal/mol (red). The regions of low (high) free energy coincide very well with the corresponding regions of high (low) occupancy in Fig. 2 A, demonstrating that the computed free-energy landscape correctly describes the association behavior of barnase and barstar as inferred from the analysis of the simulated trajectories.
The high density at the lower left of the maps suggests that up to a distance of ∼40 Å barstar is encountering barnase toward the barnase RNA binding loop (see below). At 36 Å the occupancy cloud has moved to near the center of the map, with a loose connection to the encountering region on the left side of the maps at d1–2 ≥ 40 Å. The region of high occupancy at 32 Å corresponds to the encounter-complex region, i.e., the energetically most favorable region. At shorter distances, the conformation of barnase converges toward the bound conformation (the central position of each map lies perpendicular to the binding patch).
Fig. 2 C shows the energy profiles along the reaction path, defined as the path along the minima in the free-energy landscape: the electrostatic energy, the desolvation energy, the entropy loss (the sum of the translational and rotational entropy losses), and the free energy (from left to right). The minimum position in the free-energy profile at 32 Å was used as the definition of the encounter complex. In addition to this minimum, a shoulder in the free-energy profile can be observed at ∼36 Å. This shoulder reflects a small barrier, which is discussed in more detail below.
Encounter-complex regions
Fig. 2 shows the results of a simulation where the motion of barstar is computed relative to barnase. We added a second set of simulations using the same input parameters but with the proteins being exchanged to exclude possible artifacts arising from this setup. In this simulation the motion of barnase is modeled relative to barstar. The resulting energy profiles (not shown) agree very well with the results displayed in Fig. 2 C.
The “encounter-complex regions” shown in Fig. 3 are deduced from the computed occupancy and free-energy landscapes from the simulations of barstar to barnase and vice versa. The encounter-complex regions for barnase (green) and barstar (orange) in Fig. 3, A and B, represent all center-of-mass positions with a free energy less than a certain threshold. In these images, barnase and barstar are shown in the bound configuration of the crystal structure as blue and red ribbons, respectively. The representation and colors were chosen to allow a direct comparison with Fig. 5 a in Gabdoulline and Wade (3), where the authors computed the energetically favorable regions for each protein by a Boltzmann factor analysis. In Fig. 3 A, the free-energy threshold was set to −3.0 kcal/mol, just below the above-mentioned shoulder. The corresponding encounter-complex region of barstar (barnase) has a volume of 5,338 (6,239) Å3 and the average lifetime of the proteins in these regions is ∼20 (14) ps. The encounter-complex regions agree well with the energetically favorable regions as computed by Gabdoulline and Wade, only the front part of their distribution is missing. Fig. 3 B shows the same picture with the free-energy threshold at −2.5 kcal/mol. Now, the whole region is reproduced, including the part missing before. These extended encounter-complex regions confirm the results from the Boltzmann factor analysis. The volume and the average lifetime increased to 15,870 (20,280) Å3 and 24 (16) ps for barstar (barnase). Isosurfaces of the occupancy landscapes for barstar (light brown) and barnase (light gray) are displayed in Fig. 3 C. A comparison with Fig. 3 B shows that these largely overlap with the free-energy isosurfaces. To analyze the entropic influence on the free-energy landscape of barstar, we compared the isosurfaces of the free energy and the enthalpy, which is defined as the sum of the electrostatic and desolvation energies. According to the definition of the locally defined entropy, the entropy loss is large where the occupancy is strongly varying. Therefore, the neck of the association funnel at the position of the small free-energy barrier gets even more restricted by the effect of the entropy loss. As already mentioned, the proteins are oriented in Fig. 3 in such a way as to allow a direct comparison with Fig. 5 a in Gabdoulline and Wade (3). However, in the following figures, barnase and barstar are shown in an orientation optimized for the best visualization of our results. In these images the proteins are slightly tilted compared to Fig. 3.
FIGURE 3.

Encounter-complex regions of barstar and barnase. The proteins are shown in their bound conformations in the crystal structure as blue and red ribbons for barnase and barstar, respectively. The orange isosurface represents the positions of the center-of-mass of barstar in the encounter-complex region, defined by (A) ΔG < −3.0 kcal/mol and (B) ΔG < −2.5 kcal/mol. Correspondingly, the green isosurface represents the positions of the center-of-mass of barnase. (C) Occupancy isosurfaces for barnase and barstar with an isocontour level of 300 events/Å3.
FIGURE 5.

(A) Mean orientations of barstar are represented by white arrows with pink tips for each volume element. The arrows point from the center of barstar toward its interaction patch. Barnase and barstar are shown in the same representation as in Fig. 4. (B) Close-up view at a finer resolution of the arrows (1 Å instead of 3 Å).
Association and dissociation pathways
To identify the optimal association pathway of barstar toward barnase we selected the configurations of maximal occupancy and minimal free energy from the computed maps as shown in Fig. 2, A and B, for center-to-center distances from 50 to 28 Å. The corresponding center-of-mass positions are shown in Fig. 4 A as orange and blue spheres, respectively, together with the isosurface of the free energy at −2.5 kcal/mol. The positions of the blue and orange spheres suggest that barstar is encountering barnase from the right, i.e., toward the RNA binding loop into the region of the small free-energy minimum.
FIGURE 4.

(A) Center-of-mass positions of maximum occupancy (orange) and minimum free energy (blue) for barstar encountering barnase, together with the isosurface of the free energy at −2.5 kcal/mol. For clarity, the isosurface of the free energy is shown in a transparent mode. Barnase is shown in gray with the RNA binding loop highlighted in yellow. Barstar is shown as a white ribbon in its bound conformation and its center of mass is shown as a white sphere. (B) Comparison of the center-of-mass configurations of maximum occupancy computed from all trajectories as in A and from only the successful trajectories. The maximal-occupancy configurations of all trajectories are shown in orange; those of the successful trajectories are displayed in green and blue for the associating and dissociating parts, respectively.
However, this representation does not provide exact information about the path of barstar from this region toward the region of the encounter complex. Therefore, we separately analyzed the “successful” trajectories, i.e., those which actually lead to complex formation. The criterion for the formation of the complex was the formation of two contacts at a distance of 6.5 Å as was used by Gabdoulline and Wade for the computation of the association rates (3). Here, it additionally served to divide the trajectories into their associating and dissociating parts. Fig. 4 B shows the conformations of the maximal occupancy obtained from this analysis. The maximal-occupancy conformations of all trajectories are shown in orange; those of the successful trajectories are displayed in green and blue for the associating and dissociating parts, respectively. Surprisingly, the successful trajectories do not seem to follow the path toward the RNA binding loop as was suggested by Fig. 4 A. However, when analyzing all successful trajectories by visualizing the center-of-mass positions of barstar along the trajectories we found that in ∼50% of these trajectories barstar enters into the region of the second, small free-energy minimum before and/or after matching the contact criteria. On the other hand, the pathways of association and dissociation seem to be very similar, as evidenced by Fig. 4 B. Also, the corresponding free-energy profiles (not shown) agree very well. They differ from the free-energy profiles obtained from all trajectories by ∼0.3 kcal/mol. This is probably due to the limited statistics: only ∼100 successful trajectories were obtained from the 200,000 started.
Orientation of barstar
Fig. 5, A and B, shows the mean orientation of barstar in each volume element by a white arrow with a magenta tip. These arrows are defined as pointing from the center of barstar toward its interaction patch. Barnase and barstar are shown in the same representation as in Fig. 4. In the lower right part of Fig. 5 A, in the region of the small free-energy minimum near the RNA binding loop, the arrows point away from barnase, whereas in the region of the encounter complex they roughly point in the opposite direction with the arrows parallel to the barnase surface. This indicates that in both places, barstar is oriented quite differently with respect to the surface of barnase than in the complex. The region of the free-energy barrier seems to be an inflection point where barstar has to change its orientation to enter the encounter-complex region from the region near the RNA binding loop. Fig. 5 B displays a close-up view at a finer resolution of the orientation arrows (1 Å instead of 3 Å). For positions closer than the encounter complex (orange sphere), the arrows start pointing toward the binding interface, meaning that the orientation of barstar indeed converges toward the conformation of the bound structure.
Mutant effects
So far we have presented the analysis of the association behavior of wild-type barnase and barstar. In this section, we describe the results from mutant studies, where seven charged residues of the barnase interaction patch were replaced by neutral residues. As shown in Fig. 1, A and B, the interaction patch of barnase is mostly positively charged, whereas the interaction patch of barstar is very negatively charged. This complementary charge distribution is reflected by the high association rate. Fig. 1 A shows barnase with the mutated residues K27, D54, R59, E60, K66, R83, and R87 highlighted. E60 and D54 are negatively charged, whereas the others are positively charged. Upon mutation they are all replaced by alanine, except for R83, which is replaced by glutamine. One would expect that by removing the negative charges of residues D54 and E60 from the positive barnase binding patch, the association rate should increase due to the enhanced charge complementarity. In the same way, the other mutations should cause a decrease of the association rate. Indeed, this effect was observed experimentally (4,5) and in previous BD simulations (3).
To investigate these effects in more detail, we performed BD simulations with the same simulation parameters as used by Gabdoulline and Wade (3) and analyzed the trajectories as described above for barstar and the wild type of barnase. For a better overview we divided the mutant studies into two series: one with K27A, R59A, R83Q, R87A, and the other one with D54A, E60A, D54A/E60A, K66A, K66A/D54A. Fig. 6, A and B, displays the energy profiles along the minimum free-energy pathways for both series. Shown are the electrostatic and desolvation energies, the translational and rotational entropy losses, and the free energies resulting from the sum of the three terms. In Fig. 7, the encounter-complex regions are shown together with the maximum-occupancy positions for increasing distance in the same representation as in Fig. 4. Here, the encounter-complex regions are defined by a threshold of 1.5 kcal/mol above the minimum of the corresponding free-energy profile in Fig. 6, A and B. Fig. 7, B–E, displays the results obtained from the analysis of all simulated trajectories for the mutants in the first series; those of the second series are shown in Fig. 7, F–K. For comparison, the encounter-complex region and the optimal association pathway for the wild type are displayed in Fig. 7 A. The maximum-occupancy positions give an indication for the “optimal association pathway”, although real association pathways may look different (see above). Table 1 lists the center-to-center distances of the encounter complex, the values of the free energy in the encounter complex, i.e., at the minimal position of the free-energy profile, and the volumes of the encounter-complex regions in Fig. 7 for barnase wild-type and the mutants from both series. Please note that the free-energy values only give an indication about the interaction strength between the proteins and cannot be correlated with the free-energy values of the transition state or the association rate constants.
FIGURE 6.

Energy profiles for the wild type of barnase and its mutants, split into the first (A) and second (B) sets of mutants: The electrostatic energy ΔEel (upper left), the desolvation energy ΔEds (lower left), the translational/rotational entropy loss –TΔS (upper right), and, as the sum of all, the free energy ΔG (lower right).
FIGURE 7.

Regions of the encounter complex and the center-of-mass positions of maximum occupancy for increasing distance for the wild type of barnase (A) and for its mutants as obtained from the analysis of all simulated trajectories. (B–E) Results for the first set of mutants. (F–K) Results for the second set of mutants. The encounter-complex regions are defined by the center-of-mass positions of barstar with a free energy within 1.5 kcal/mol from the minimum free energy, i.e., those that fulfill ΔG ≤ ΔGmin + 1.5 kcal/mol.
TABLE 1.
Center-to-center distance d1–2,enc, value of the free energy ΔGenc, and volume of the encounter-complex region Venc for barnase wild type and mutants
| d1–2,enc (Å) | ΔGenc (kcal/mol) | Venc (Å3) | |
|---|---|---|---|
| WT | 31.6 | −3.96 | 19,670 |
| K27A | 40.1 | −2.80 | 40,810 |
| R59A | 40.2 | −2.69 | 41,600 |
| R83Q | 40.2 | −2.88 | 41,990 |
| R87A | 40.1 | −2.82 | 48,910 |
| D54A | 29.6 | −9.47 | 943 |
| E60A | 30.5 | −5.79 | 6086 |
| D54A/ E60A | 28.6 | −10.58 | 1569 |
| K66A | 32.3 | −3.77 | 10,940 |
| K66A/ D54A | 30.8 | −5.94 | 5909 |
The values given are based on the encounter-complex region shown in Fig. 7. The values of the center-to-center distances were obtained by a parabolic fit around the free-energy minimum.
As expected for the first series, the electrostatic energy profiles in Fig. 6 A clearly reflect the decreased interaction due to the decreased charge complementarity, whereas the profiles of the desolvation energies and of the entropies are very similar to those of the wild type. Thus, the volume directly above the interaction patch of barnase (the encounter-complex region of the wild type) is much less favorable for these mutants, as indicated by the resulting free-energy profiles. The positions of the corresponding encounter complexes are shifted to a center-to-center distance of ∼40 Å, to the region of the second, small free-energy minimum of the barnase wild-type. The corresponding free energies are ∼1.2 kcal/mol higher than that of the wild-type. Fig. 7, B–E, shows that the encounter-complex region of barnase wild type is energetically less favorable for all mutants in this series. Correspondingly, the larger parts of the encounter-complex regions are located in the vicinity of the barnase RNA binding loop. The resulting encounter-complex regions are very similar for these mutants. Only the encounter-complex region of the r59a mutant shows a constricted neck near the RNA binding loop. The optimal association pathways are close to that of the wild type.
The second mutant series shows a different picture. The corresponding energy profiles and encounter-complex regions are displayed in Figs. 6 B and 7, F–K, respectively. Due to the increased electrostatic interaction, the association funnels for the E60A and D54A mutants are much deeper compared to that of the wild type and the encounter-complex positions are shifted slightly closer to the bound state. These effects are even more pronounced for double mutants of both residues. For the D54A/E60A mutant the association funnel is almost three times deeper compared to the wild type, and the encounter complex is 3 Å closer to the position of the bound state. Due to the steepness of the energy funnel, the encounter-complex regions are much more compact. However, the maximum-occupancy positions indicate that the region of the second free-energy minimum of the wild type is still energetically favorable when either the E60 or the D54 residue is mutated. Only by mutating both residues does the interaction between barstar and barnase become strong enough to change the optimal association pathway.
We described above how the mutants of the first set largely abolish the favorable encounter-complex region of the wild type. Is it possible to remove the region of the second minimum as well? Fig. 7 I shows that it is the K66 residue that is responsible for the energetically favorable region near the RNA binding loop. For a K66A mutant, this region becomes unfavorable, an effect which is also reflected by the missing shoulder in the free-energy profile at ∼40 Å. The free-energy funnel is almost as deep as that of the wild-type. This is a surprising effect because the mutation of K66 decreases the electrostatic interaction between the proteins. The K66A/D54A double mutant, which has the same total charge as the wild type, shows a behavior similar to that of K66A, and the energy funnel is even deeper than that of the E60A mutant.
DISCUSSION
The diffusional association behavior of barstar and barnase was analyzed in detail. The methods of analysis involved the computation of the occupancy and the free-energy landscapes and were extended for the analysis of specific features such as the encounter-complex regions, the optimal pathways, the difference between the pathways of association and dissociation, and the coupling of the translational and rotational motions. In the second part, these methods were applied to analyzing the effects of various barnase mutations on the diffusional association behavior.
Diffusional association behavior of the wild types
The energy profiles resulting from the diffusional association of barstar with wild-type barnase were discussed extensively in a previous study (15), which also described in detail the method for computing the occupancy and free-energy landscapes. In this study, we focused attention on the association behavior of the proteins, in particular with respect to the small free-energy barrier in the free-energy profile (see Fig. 2 C). We found that this barrier divides the energetically favorable region into the region near the RNA binding loop and the region of the encounter complex, which is located above the interaction patch of barnase and from which barstar can continuously reorient to finally bind to barnase. (In this article, the encounter complex is defined as the state of minimal free energy. In addition to this definition in terms of energy, the encounter complex may be defined by structural criteria (1).)
The maps of the occupancy and free-energy landscapes (see Fig. 2) suggest that the association pathway of barstar proceeds toward the RNA binding loop of barnase as was proposed in previous studies (9,11). The region near the RNA binding loop is actually the region of the second, small free-energy minimum along the reaction path (see the graph on the right in Fig. 2 C). Visualizing the optimal center-of-mass position of barstar, derived from the maps of the occupancy and free-energy landscapes (see Fig. 4 A), indeed supports this hypothesis. However, a different picture was obtained from a detailed analysis of the successful trajectories, i.e., those trajectories that fulfill the contact criterion and most probably lead to the formation of the final complex. The optimal center-of-mass positions derived from those trajectories only show no overlap with the energetically favorable region near the RNA binding loop. These results suggest that this region is not essential for the binding of barstar to barnase, but is probably helpful in steering barstar into the region of the encounter complex. It is possible that the presence of barstar in this region may also decrease the probability of barnase binding to the RNA. In this way, the region of the second, small free-energy minimum could support the biological function of barnase as an inhibitor of barstar. Furthermore, we found no evidence for a difference between the association and dissociation pathways during this analysis. Within the limitations of this study, which only considered the relative motion of two rigid proteins, we therefore conclude that statistically the two pathways are very close, if not equal.
By analyzing the position-dependent (mean) orientation of barstar, we found that the orientational motion was more strongly coupled to the positional motion than we expected. This analysis revealed a surprising detail of the association behavior: When encountering barnase on the side of its RNA binding loop, the barstar interface is pointing “backward”; i.e., in this position barstar orients in such a way as to avoid contact between its negatively charged interaction patch and the likewise negatively charged binding-loop region of barnase. To cross the free-energy barrier, however, barstar has to reorient to increase the attraction between the oppositely charged interaction patches by moving in a “forward” direction.
As mentioned above, Brownian dynamics simulations of relative interprotein motion typically do not: 1), sample the internal degrees of freedom of the proteins; 2), account for the perturbative effect at close distances of the moving protein on the electrostatic field created by the other protein; 3), consider the molecular nature of the solvent; and 4), include short-range hydrophobic interactions. Moreover, this study did not consider hydrodynamic interactions of the proteins as these have been estimated to be of minor importance for the diffusional encounter of proteins (28,29). Recently, Garcia de la Torre and co-workers have developed methods for evaluating the translational and rotational diffusion coefficients of arbitrarily shaped particles (31) and for simulating Brownian trajectories of nonisotropic particles (32). Estimating such effects on the diffusive protein motion close to the encounter region is worth addressing in future work. One may further consider the possibility that residues at the binding interface undergo changes of their titration states upon binding. However, we believe that this point is of minor importance at the distances up to the encounter region that were sampled in this study.
The effect of mutations
From the mutant studies we gained detailed insight into modulations of the diffusional association behavior, which is finally reflected in the different association rates. At the beginning of this work, it was not completely clear to us whether the energetically favorable region near the RNA binding loop facilitates or hinders the association of barstar to barnase. Therefore, we studied two series of mutations. The first set removes the positively charged residues K27A, R59A, R83Q, and R87A from the binding interface. The second set consists of D54A, E60A, D54A/E60A, K66A, and K66A/D54A. D54 and E60 are negatively charged residues of the interface. K66 is a positively charged residue at the RNA binding loop (see Fig. 2 C) (3).
The energy profiles in Fig. 6 A show that all mutations of the first series lead to a decreased interaction between the proteins, in particular at smaller distances. The decreased charge complementarity at the binding interface leads to a flatter energy funnel, and therefore the encounter-complex regions in Fig. 7, B–E, are larger compared to that of the wild type (Fig. 7 A). Most prominently, the region above the interface of barnase, which is the encounter-complex region of the wild-type, becomes much less favorable, whereas the association behavior up to this region seems to be similar to that of the wild type. The region near the RNA binding loop is energetically very similar for the mutants in this series. Their encounter-complex positions are located in this region as well, ∼8 Å farther away from the barnase center than the encounter complex of the wild type. For the R87A mutant and for the wild type, a two-step encounter process could be proposed, since the corresponding free-energy landscapes have a minimum at this position and one above the center of the interaction patch of barnase.
The mutations D54A and E60A of the second mutation series show a behavior opposite to that of the mutants of the first series: due to the increased electrostatic interaction the energy funnel is much deeper and the small free-energy barrier disappears. However, a shoulder remains in the free-energy profile in the region of the binding loop. The maximum-occupancy configurations are still close to that of the wild type, i.e., the region near the RNA binding loop is also energetically favorable for both mutants. Only by mutating both residues does the electrostatic interaction become strong enough to alter the occupancy in this region. In all three cases the encounter complex moved closer to the bound structure compared to the encounter complex of the wild type. The mutation of K66A shows a different effect: the region near the RNA binding loop becomes much less favorable, and therefore the occupancy in this region is strongly reduced. This shows that the K66 residue is responsible for this region near the RNA binding loop and also for the second, small free-energy minimum for the wild type. Correspondingly, also, the free-energy profiles of this mutant and of K66A/D54A do not show a shoulder at ∼40 Å distance. The energy funnel for this mutant is even deeper than that of the E60A mutant, although its total charge is the same as for the wild type.
One of the most important results from these mutant studies is the observation that the free-energy landscape can drastically change by mutating a single residue. Addressing changes of the position of the transition state is beyond the capabilities of the simulation approach due to the simplifications at small contact distances. However, one can expect that the position of the transition state will not be unaffected if the encounter-complex position varies that much. This result is inconsistent with the assumption of Miyashita et al. (18) that positions of the encounter complexes (and the transition states) are identical for all mutants. Maybe this assumption holds for the system of the cytochrome c2-reaction center that they investigated, but it is clearly not true for the barnase-barstar system studied here.
CONCLUDING REMARKS
For the barnase-barstar model system, which involves a protein-protein pair with strong electrostatic complementarity, this study provides details of diffusive motion on the interaction free energy surface. Here, we summarize our main findings:
From Brownian dynamics simulations, we identified a favorable free-energy minimum for barstar at ∼10 Å distance from its position the bound complex, which is termed the encounter-complex region. A second free-energy minimum was found near the RNA binding loop of barnase.
The two favorable binding regions are divided by a small free-energy barrier. When entering the region of the encounter complex from the region near the RNA binding loop, barstar must undergo a significant rotation to increase the electrostatic attraction between the proteins.
The region of the second binding minimum was visited by about half of the successful binding trajectories. Therefore this region does not appear to be essential for the binding of barstar to barnase but may nonetheless be helpful for steering barstar into the region of the encounter complex.
Single point mutations in barnase drastically changed the free-energy landscape and significantly altered the population of the two minima. For example, the K66A mutant completely removed the second binding region.
Whereas the main results obtained from Brownian dynamics simulations are kinetic on-rates of protein-protein association, we demonstrated that trajectory analysis may reveal additional details of the association pathways that are of mechanistic relevance.
Acknowledgments
A.S. and V.H. acknowledge financial support from the Deutsche Forschungsgemeinschaft via the Center for Bioinformatics in Saarbrücken. R.R.G. and R.C.W. acknowledge financial support from the Klaus Tschira Foundation and the Center for Modelling and Simulation in the Biosciences (BIOMS), Heidelberg.
References
- 1.Gabdoulline, R. R., and R. C. Wade. 1999. On the protein-protein diffusional encounter complex. J. Mol. Recognit. 12:226–234. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber, G. 2002. Kinetic studies of protein-protein interactions. Curr. Opin. Struct. Biol. 12:41–47. [DOI] [PubMed] [Google Scholar]
- 3.Gabdoulline, R. R., and R. C. Wade. 2001. Protein-protein association: investigation of factors influencing association rates by Brownian Dynamics simulation. J. Mol. Biol. 306:1139–1155. [DOI] [PubMed] [Google Scholar]
- 4.Schreiber, G., and A. R. Fersht. 1996. Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 3:427–431. [DOI] [PubMed] [Google Scholar]
- 5.Schreiber, G., and A. R. Fersht. 1995. Energetics of protein-protein interactions: analysis of the barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 248:478–486. [DOI] [PubMed] [Google Scholar]
- 6.Buckle, A. M., G. Schreiber, and A. R. Fersht. 1994. Protein-protein recognition: crystal structural analysis of a barnase-barstar complex at 2.0-Å resolution. Biochemistry. 33:8878–8889. [DOI] [PubMed] [Google Scholar]
- 7.Selzer, T., and G. Schreiber. 1999. Predicting the rate enhancement of protein complex formation from the electrostatic energy of interaction. J. Mol. Biol. 287:409–419. [DOI] [PubMed] [Google Scholar]
- 8.Vijayakumar, M., K. Y. Wong, G. Schreiber, A. R. Fersht, A. Szabo, and H. Z. Zhou. 1998. Electrostatic enhancement of diffusion-controlled protein-protein association: comparison of theory and experiment on barnase and barstar. J. Mol. Biol. 278:1015–1024. [DOI] [PubMed] [Google Scholar]
- 9.Gabdoulline, R. R., and R. C. Wade. 1997. Simulation of the diffusional association of barnase and barstar. Biophys. J. 72:1917–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang, T., S. Tomic, R. R. Gabdoulline, and R. C. Wade. 2004. How optimal are the binding energetics of barnase and barstar. Biophys. J. 87:1618–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camacho, C. J., Z. Weng, S. Vajda, and C. DeLisi. 1999. Free energy landscapes of encounter complexes in protein-protein association. Biophys. J. 76:1166–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conte, L. L., C. Chothia, and J. Janin. 1999. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 285:2177–2198. [DOI] [PubMed] [Google Scholar]
- 13.Dong, F., M. Vaijayakumar, and H.-X. Zhou. 2003. Comparison of calculation and experiment implicates significant electrostatic contributions to the binding stability of barnase and barstar. Biophys. J. 85:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meiering, E. M., L. Serrano, and A. R. Fersht. 1992. Effect of active site residues in barnase on activity and stability. J. Mol. Biol. 225:585–589. [DOI] [PubMed] [Google Scholar]
- 15.Spaar, A., and V. Helms. 2005. Free energy landscape of protein-protein encounter resulting from Brownian Dynamics simulations of barnase:barstar. J. Chem. Theory Comput. 1:723–736. [DOI] [PubMed] [Google Scholar]
- 16.Papoian, G. A., and P. G. Wolynes. 2003. The physics and bioinformatics of binding and folding—an energy landscape perspective. Biopolymers. 68:333–349. [DOI] [PubMed] [Google Scholar]
- 17.Tsai, C.-J., S. Kumar, B. Ma, and R. Nussinov. 1999. Folding funnels, binding funnels, and protein function. Protein Sci. 8:1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyashita, O., J. N. Onuchic, and M. Y. Okamura. 2004. Transition state and encounter complex for fast association of cytochrome c2 with bacterial reaction center. Proc. Natl. Acad. Sci. USA. 101:16174–16179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 20.Reference deleted in proof.
- 21.Davis, M. E., J. D. Madura, B. A. Luty, and J. A. McCammon. 1991. Electrostatic and diffusion of molecules in solution: simulations with the University-of-Houston-Brownian Dynamics program. Comput. Phys. Commun. 62:187–197. [Google Scholar]
- 22.Jorgensen, W. L., and J. Tirado-Rives. 1988. The OPLS potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110:1657–1666. [DOI] [PubMed] [Google Scholar]
- 23.Gabdoulline, R. R., and R. C. Wade. 1996. Effective charges for macromolecules in solvent. J. Phys. Chem. 100:3868–3878. [Google Scholar]
- 24.Northrup, S. H., J. O. Boles, and J. C. L. Reynolds. 1987. Electrostatic effects in the Brownian dynamics of association and orientation of heme proteins. J. Phys. Chem. 91:5991–5998. [Google Scholar]
- 25.Elcock, A. H., R. R. Gabdoulline, R. C. Wade, and J. A. McCammon. 1999. Computer simulation of protein-protein association kinetics: acetylcholinesterase-fasciculin. J. Mol. Biol. 291:149–162. [DOI] [PubMed] [Google Scholar]
- 26.Gabdoulline, R. R., and R. C. Wade. 1998. Brownian dynamics simulation of protein-protein diffusional encounter. Methods. 14:329–341. [DOI] [PubMed] [Google Scholar]
- 27.Ermak, D. L., and J. A. McCammon. 1978. Brownian dynamics with hydrodynamic interactions. J. Chem. Phys. 69:1352–1360. [Google Scholar]
- 28.Antosiewicz, J., J. M. Briggs, and J. A. McCammon. 1996. Orientational steering in enzyme-substrate association: ionic strength dependence of hydrodynamic torque effects. Eur. Biophys. J. 24:137–141. [DOI] [PubMed] [Google Scholar]
- 29.Gorba, C., T. Geyer, and V. Helms. 2004. Brownian Dynamics simulations of simplified cytochrome c molecules in the presence of a charged surface. J. Chem. Phys. 121:457–464. [DOI] [PubMed] [Google Scholar]
- 30.Flöck, D., and V. Helms. 2004. A Brownian Dynamics study: the effect of a membrane environment on an electron transfer system. Biophys. J. 87:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de la Torre, J. G., M. L. Huertas, and B. Carrasco. 2000. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 78:719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandes, M. X., and J. G. de la Torre. 2002. Brownian Dynamics simulation of rigid particles of arbitrary shape in external fields. Biophys. J. 83:3039–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]