

Abstract
CaMBP, a peptide corresponding to the 3614–3643 calmodulin (CaM) binding region of the ryanodine receptor (RyR1), is known to activate RyR1 Ca2+ channel. To analyze the mechanism of channel regulation by the CaMBP-RyR1 interaction, we investigated a), CaMBP binding to RyR1, b), induced local conformational changes in the CaMBP binding region of RyR1 using the fluorescent conformational probe badan attached to CaMBP (CaMBP-badan), and c), effects of “a” and “b” on SR Ca2+ release. We also monitored the interaction of CaMBP-badan with CaM and a peptide corresponding to the Met3534–Ala4271 region of RyR1 (R3534–4271) as a control. At lower peptide concentrations (≤15 μM), CaMBP binding to RyR1 increased the intensity of badan fluorescence emission at a shorter wavelength (the state resembling CaMBP-badan/Ca-CaM) and induced Ca2+ release. Further increase in CaMBP concentration (up to ∼50 μM) produced more binding of CaMBP accompanied by further increase in the badan fluorescence emission but at a longer wavelength (the state resembling CaMBP-badan/apo-CaM) and inhibited Ca2+ release. Binding of CaMBP-badan to R3534–4271 increased the intensity of badan fluorescence, showing the similar concentration-dependent red-shift of the emission maximum. It is proposed that CaMBP interacts with two classes of binding sites located in the Met3534–Ala4271 region of RyR1, which activate and inhibit the Ca2+ channel, respectively.
INTRODUCTION
Calmodulin (CaM) regulates Ca2+ channels of the skeletal and cardiac isoforms of the ryanodine receptor (RyR1 and RyR2, respectively) in a Ca2+-dependent manner (also in a manner characteristic of each RyR isoform (1–3)) presumably by binding via its C-terminal lobe (4). In RyR1, CaM increases RyR channel activity at lower (nanomolar to submicromolar) Ca2+ concentrations whereas it inhibits channel activity at higher (micromolar to submillimolar) Ca2+ concentrations (5–9). In the case of RyR2, however, CaM inhibits channel activity at all Ca2+ concentrations (6,10,11). In both RyR1 and RyR2 isoforms, one CaM binds to one subunit of RyR (6,12). In the case of RyR1, CaM binding seems to take place within the region encompassing residues 3614–3643 (12–17). The region encompassing residues 3583–3603 of RyR2 is highly homologous to the residues 3614–3643 of RyR1; furthermore, mutation of several critical residues in this region abolished CaM binding to RyR2 as it does to RyR1 (18). Thus, it appears that RyR1 and RyR2 have at least one common CaM binding domain (CaMBD) in the corresponding 3614–3643 and 3583–3603 regions, respectively, and this CaM binding region seems to play a central role in the mechanism of Ca2+/CaM-dependent channel regulation.
The mode of CaM regulation at low Ca2+ concentrations is quite distinguishable between RyR1 and RyR2, as described above. According to a recent report by Meissner and his colleagues (19), the two regions outside of the 3614–3643 CaM binding region—the region located at the N-terminal side of the CaM binding region and the one at the C-terminal side—seem to be involved in the isoform-specific Ca2+/CaM-dependent regulation. It has also been suggested that there are secondary CaM binding sites in the 1975–1999 residue region of RyR1 (20) and in the C-terminal tail region of the α1 subunit of the dihydropyridine receptor (21). These pieces of information suggest that multiple regions of the RyR polypeptide chain may be involved in the mechanism of Ca2+/CaM-mediated regulation of RyR1 Ca2+ channel, although the stoichiometry of binding is one CaM per subunit of the RyR. Of particular interest in this context is a recent report by Zhu et al. (22) that CaMBP, a synthetic peptide corresponding to the 3614–3643 CaM binding region of RyR1, activates RyR1 channels. This suggests that not only the CaM binding region but also yet unidentified regulatory domains of RyR1, with which the CaMBP peptide (perhaps the in vivo CaM binding domain as well) interacts, are involved in Ca2+ channel regulation. According to a recent work of Hamilton and her colleagues (23), there is a segment (residues 4064–4210) in the RyR1 polypeptide chain that resembles the sequence structure of CaM (CaM-like domain within the RyR1), and the expressed peptide corresponding to this segment has several CaM-like properties; importantly, the expressed peptide binds with CaMBP. Thus, the pieces of new information accumulated to date suggest a hypothesis that not only the 3614–3643 region of CaMBD but also the 4064–4210 CaMBP binding/CaM-like domain (CaMLD) are involved in the Ca2+- and CaM-mediated channel regulation. Since CaMBP seems to retain several important properties of its corresponding in vivo domain (i.e., CaMBD) and it binds to the expressed peptide corresponding to the CaMLD, it is not unreasonable to assume that the interaction between the CaMBD and the CaMLD are involved in channel regulation (cf. 23).
In our recent study of the interaction between CaMBP and CaM (24), we used a highly environment-sensitive fluorescent probe, 6-bromoacetyl-2-dimethylaminonaphthalene (badan), and demonstrated that the badan probe attached either to CaM (CaM-badan) or CaMBP (CaMBP-badan) can serve as an excellent reporter of the changes in the local environment of the CaM/CaMBP interaction interface and of protein conformational changes in the CaM molecule induced by its binding with CaMBP. Thus, the fluorescence intensity of the CaMBP-badan increased considerably upon binding with CaM. Furthermore, the magnitude of the fluorescence increase of CaMBP-badan induced by the interaction with CaM increased further with Ca2+ in a physiological range (0.1–2 μM), although CaMBP-badan alone (without interaction with CaM) showed no such change. Importantly, the Ca2+-dependent fluorescence changes of the CaM-CaMBP complex were identical regardless of whether the badan probe was attached to CaM or CaMBP. These findings indicate that CaMBP-badan can serve as an excellent spectroscopic probe to monitor local conformational changes in the CaMBP-interacting domain of the RyR1.
The strategy we have taken in this study is to utilize the aforementioned advantage of CaMBP-badan to monitor the peptide-induced conformational changes in its target domain of the RyR1 and resultant Ca2+ release from the SR. In this study, we investigated concentration-dependent effects of CaMBP on Ca2+ release from the SR vesicles; in parallel to the Ca2+ release experiments, we monitored CaMBP binding to the SR and the CaMBP-induced local conformational changes within the CaMBP-RyR interaction interface. To facilitate the interpretation of the data of these experiments, we also monitored the interaction between CaMBP-badan and CaM and the interaction between CaMBP-badan and the expressed peptide R3534–4271. We report here that the CaMBP-induced protein conformational changes in the RyR and those in the R3534–4271 share common properties in the basic aspects, suggesting that the 3534–4271 region of RyR1 containing both the CaMBD and the CaMLD is involved in the mechanism of CaMBP-mediated channel regulation. Coordinated analysis of the three parameters (CaMBP binding, local conformational changes, and effects on SR Ca2+ release) led us to a new hypothesis that CaMBP (presumably the corresponding in vivo domain, CaMBD, as well) interacts with two distinguishable sites located in the 3534–4271 region of RyR1, producing activation and inhibition of the RyR1 Ca2+ channel. The spectroscopic properties of the RyR1-bound CaMBP-badan at the activating concentration and at the inhibitory concentration were similar to those of Ca-CaM-bound CaMBP-badan and apo-CaM-bound CaMBP-badan, respectively, suggesting that the main CaMBP binding sites are located in the CaMLD described by Xiong et al. (23).
EXPERIMENTAL PROCEDURES
Preparation of SR
Triad-enriched microsomal fraction (SR) was prepared from the rabbit back paraspinous and hind leg skeletal muscle by the method of differential centrifugation as described previously (25). The microsomes from the final centrifugation were homogenized in a sample solution containing 0.3 M sucrose, 0.15 M KCl, proteolytic enzyme inhibitors (0.1 mM phenylmethanesulfonyl fluoride, 1 μg/ml leupeptin, 2.0 μg/ml soybean trypsin inhibitor), 20 mM 2-(N-morpholino)ethanesulfonic acid, pH 6.8 to a final concentration of 20–30 mg/ml, frozen immediately in liquid nitrogen and stored at −80°C.
Peptide synthesis
3614KSKKAVWHKLLSKQRRRAVVACFRMTPLYN3643 ……… CaMBP
3614KSKKAVWHKLLSKQRRRAVV ……… F1
AVVACFRMTPLYN3643 ……… F4
CaMBP corresponding to RyR1 residues 3614–3643 was used as a peptide model of the CaM binding region of RyR1. Two other peptides used, F1 and F4, are derived from the peptide CaMBP. Peptides were synthesized on an Applied Biosystems (Foster City, CA) model 431 A synthesizer employing Fmoc (N-(9-fluorenyl)methoxycarbonyl) as α-amino-protecting group and cleaved and deprotected with 95% trifluoroacetic acid and purified by reverse-phase high performance liquid chromatography. The peak fraction was extensively dialyzed against distilled water, lyophilized, and stored at −80°C.
Preparation of CaM
We used recombinant proteins overexpressed in Escherichia coli. The cDNA of human liver CaM used in this study was a generous gift from Dr. Richard Perham (Cambridge, UK) (26). The coding sequence with inclusion of a short untranslated region was subcloned into pAED4—the T7 expression vector/Phagemid (a generous gift from Dr. Don S. Doering, Whitehead Institute, Cambridge, MA) as previously described (27).
Fluorescence labeling of CaMBP
For site-specific labeling of CaMBP with the environment-sensitive fluorescent probe, badan (Molecular Probes, Eugene, OR) (28–30), 150 μg CaMBP was reacted with 2 mM badan in a total volume of 1.0 ml solution of 0.15 M KCl, 2 mM tris(2-carboxyethyl)phosphine, 20 mM 3-(N-morpholino)propanesulfonic acid (MOPS) (pH 7.2). After incubation for 2 h at 25°C in the dark, the reaction was stopped with 30 mM β-mercaptoethanol. The reaction mixture was loaded onto a Sephadex G-15 column. The fraction containing CaMBP-badan was used for the spectroscopic experiments. The concentration of the purified CaMBP-badan was determined using the molar absorbance of badan of 20,100 cm−1M−1 at 386 nm.
Spectroscopic studies of CaMBP binding and conformational changes in the CaMBP binding region of RyR1
Stock solution of CaMBP-badan probe (90 μM) was made of 3 μM CaMBP-badan as described above and 87 μM unlabeled CaMBP, and various concentrations of CaMBP-badan probe indicated were made by diluting this stock solution with the reaction buffer (for convenience, in the following part this solution is called CaMBP-badan solution). The fluorescence emission spectra of CaMBP-badan in the presence or absence of SR or CaM were collected in a PerkinElmer (Wellesley, MA) spectrofluorometer (model LS 50B) by exciting at 387 nm and by recording the fluorescence emission in the range 400–600 nm. Kinetic measurements of the changes in badan fluorescence produced by CaMBP binding and local conformational changes were performed in a stopped-flow spectrofluorometer with excitation at 368 nm and emission at 455 nm using an interference filter with 70 nm bandwidth. The free Ca2+ concentration was calculated from the amounts of 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′,-tetraacetic acid (BAPTA) (1 mM), calcium (added CaCl2), and MgATP (1.0 mM) that were present in the reaction solution by solving standard equilibrium equations using the modified Catlig solution mixing computer program; and various free Ca2+ concentrations indicated were created by varying the amount of calcium to be added on the basis of computer calculation.
CaMBP-induced Ca2+ release
SR (0.2 mg/ml) in a solution of 20 mM MOPS, 0.15 M KCl, pH 7.2, 2 μM fluo-3 (Ca2+ indicator) was loaded with Ca2+ by 1 mM MgATP. Various concentrations of CaMBP were added to the Ca2+ loaded SR to induce Ca2+ release. The time course of CaMBP-induced Ca2+ release was followed by recording the changes in the fluorescence intensity of fluo-3 either in a Perkin Elmer spectrophotometer (excitation at 488 nm, emission at 525 nm) or in a stopped-flow apparatus (Bio-Logic (Molecular Kinetics, Pullman, WA) SFM-4) (excitation at 440 nm and emission at 510 nm using an interference filter with a 40 nm bandwidth).
Construction and expression of R3534–4271
The peptide corresponding to residue 3534–4271 of RyR1 (R3534–4271) was cloned in pET 100/D-TOPO vector using the Champion pET directional TOPO Expression kit (Invitrogen, Carlsbad, CA). For this, the DNA segment corresponding to R3534–4271 was amplified by polymerase chain reaction with Pfu DNA Polymerase (Stratagene, La Jolla, CA) using the plasmid containing a longer RyR1 fragment R2900–4272 as template (a generous gift from Dr. James D. Fessenden, Boston Biomedical Research Institute, Watertown, MA). The insertion and directionality were checked by DNA sequencing. R3534–4271 construct was transformed in BL21 Star (DE3) and expressed by inducing with isopropyl β-D-thiogalactoside at 37°C for 3 h. The cells were harvested by centrifugation at 6500 × g for 10 min, resuspended in buffer A (20 mM sodium phosphate, pH 7.0, 0.5 M NaCl), and disrupted by sonication, followed by centrifugation at 16,500 × g for 15 min. As R3534–4271 was expressed in inclusion bodies, the pellet was resuspended in buffer B (buffer A supplemented with 8 M urea) to solubilize the inclusion bodies and centrifuged at 16,500 × g for 15 min. The supernatant was loaded on the Talon metal affinity column (BD Biosciences, Franklin Lakes, NJ) preequilibrated with buffer B. The column was washed with 2 × 1 ml of buffer B and 2 × 1 ml of buffer B supplemented with 10 mM imidazole. Elution was done with buffer B plus 150 mM imidazole. The purified protein was refolded by dialysis against 20 mM MOPS, 0.15 M KCl, pH 7.2 using 12,000–14,000 molecular weight cutoff membrane.
RESULTS
CaMBP is an on/off regulator of the RyR1 Ca2+ channel
In recent reports, Zhu et al. (22) have shown that submicromolar concentrations of CaMBP enhanced ryanodine binding to RyR1 in a Ca2+-dependent manner, and Rodney et al. (31) have shown that CaMBP increased the frequency of Ca2+ sparks and the activation is caused by a direct interaction of CaMBP with RyR1, rather than by the peptide-mediated dissociation of RyR1-bound CaM (22,31). According to Zhu et al. (22) a shorter peptide corresponding to the N-terminal portion of CaMBP, F1 (according to their terminology, see sequence diagram in ‘Experimental Procedures’), retained the function of CaMBP to enhance ryanodine binding, whereas its C-terminal portion, F4, was inhibitory. As described in an earlier report by Rodney et al. (32) and in our recent study (24), CaM binds to CaMBP producing conformational changes in both CaM and CaMBP (24,31,32), and the F1 portion of CaMBP is sufficient for the binding with Ca-CaM, but both F1 and F4 portions are required for the binding with apo-CaM, suggesting a Ca2+-dependent shift of CaM binding site toward the N-terminal side of CaMBP (24 cf. 32). Thus, CaMBP seems to retain some structural and functional properties of the CaMBD of RyR1 and may serve as a useful probe for the studies of the postulated interaction between the CaMBD and the putative CaMBP binding domain of RyR1 (see Introduction).
In the experiment shown in Fig. 1, we induced Ca2+ release by adding various concentrations of CaMBP to the SR vesicles and followed the release time course using fluo-3 as a Ca2+ probe in a spectrophotometer (Fig. 1 A) or in a stopped-flow apparatus (Fig. 1 B). Our purified SR preparation contained only a residual amount of the RyR1-bound CaM (cf. 33), and the amount of the residual CaM did not change after treatment of the SR with 50 μM CaMBP as determined by Western blot analysis. Furthermore, if dissociation of the bound CaM were the cause of the observed effect, the effect of CaMBP would have been inhibition rather than activation, because Ca2+ release was triggered at submicromolar Ca2+. Therefore the CaMBP-induced Ca2+ release seems to be caused primarily by its interaction with the CaMBP binding domain of the RyR1. As shown, both the rate and the amplitude of Ca2+ release increased with an increase of the concentration of CaMBP up to ∼15 μM, but further increases of CaMBP concentration resulted in a progressive decrease in the rate and the amplitude of Ca2+ release and reached nearly zero at 50 μM. Fig. 2 shows the plot of the initial rate of Ca2+ release calculated from the fitting of the data shown in Fig. 1 A as a function of the concentration of CaMBP. This steep concentration-dependent activation and inhibition curve suggests that CaMBP binds to two classes of binding sites on the RyR1 in a sequential manner: binding of CaMBP to the first binding site activates Ca2+ release (with the extent of activation, a, and the affinity of binding, Ka), and further binding of CaMBP to the second site inhibits Ca2+ release (with the extent of inhibition, b, and the affinity of binding Ki). The data shown in Fig. 2 were fitted by an equation based upon the above model: 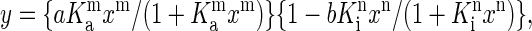 where y is the rate of Ca2+ release at x concentration of CaMBP, Ka and Ki are the association constants, and m and n are the Hill coefficients of peptide binding to the activation site(s) and the inhibition site(s), respectively. The calculated values of Ka, Ki, m, and n were 7.85 × 104M−1, 2.99 × 105M−1, 2.86, and 7.26, respectively. Interestingly, the estimated affinity of the CaMBP binding to the inhibitory site appears to be even higher than that to the activating site, even though the inhibition of Ca2+ release occurs after the activation of Ca2+ release with an increase of CaMBP concentration. This suggests that the first binding to the activating site prompts the second binding to the inhibitory site and the former is prerequisite for the latter.
where y is the rate of Ca2+ release at x concentration of CaMBP, Ka and Ki are the association constants, and m and n are the Hill coefficients of peptide binding to the activation site(s) and the inhibition site(s), respectively. The calculated values of Ka, Ki, m, and n were 7.85 × 104M−1, 2.99 × 105M−1, 2.86, and 7.26, respectively. Interestingly, the estimated affinity of the CaMBP binding to the inhibitory site appears to be even higher than that to the activating site, even though the inhibition of Ca2+ release occurs after the activation of Ca2+ release with an increase of CaMBP concentration. This suggests that the first binding to the activating site prompts the second binding to the inhibitory site and the former is prerequisite for the latter.
FIGURE 1.

Time courses of Ca2+ release induced by various concentrations of CaMBP. (A) Spectrophotometric traces of Ca2+ release in a longer timescale (60 s). (B) Stopped-flow traces of Ca2+ release in an earlier phase of the release time course (15 s).
FIGURE 2.

Initial rate of CaMBP-induced Ca2+ release shows a biphasic dependence on the concentration of CaMBP, showing bell-shaped curves. To calculate the initial rate of Ca2+ release, the time courses of Ca2+ release were fitted to the equation y = y0 + A1(1 − exp(−k1 × t)) + A2(1 − exp(−k2 × t)), where A is the amount of Ca2+ released, k is the rate constant, 1 and 2 are fast and slow phases, respectively, and (dy/dt)t→0 = A1 × k1 + A2 × k2 represents the initial rate.
In contrast to the biphasic activation/inhibition pattern of CaMBP-induced Ca2+ release, the rate and the amplitude of Ca2+ release induced by F1 peptide increased with an increase of the peptide concentration in monotonically up to as high as 50 μM without showing any sign of inhibition (Fig. 3). Fig. 3 also shows the time courses of Ca2+ release induced by 50 μM CaMBP and the release induced by a mixture of 50 μM F1 and 50 μM F4 peptides for comparison. As seen, both the rate and the magnitude of F1-induced Ca2+ release were considerably reduced by the equimolar concentration of F4 peptide mixed with F1 peptide, resulting in the release time course resembling the time course with 50 μM CaMBP. These results are consistent with the view deduced from the previous study of ryanodine binding (22) that the channel activating function of CaMBP is localized in its F1 region, whereas its inhibitory function is localized in the F4 region.
FIGURE 3.

Unlike the biphasic concentration dependence of CaMBP-induced Ca2+ release, F1 (activating segment of CaMBP)-induced Ca2+ release shows a monophasic concentration dependence of activation. Maximal activation of Ca2+ release by 50 μM F1 is considerably reduced if an equimolar concentration of F4 (inhibitory segment of CaMBP) is added together with F1.
CaMBP-badan serves as a useful spectroscopic probe to monitor the interaction of CaMBP with CaM
As shown in our recent study (24), the badan probe attached either to CaM (CaM-badan) or CaMBP (CaMBP-badan) can serve as an excellent reporter of the changes in the local environment of the CaM/CaMBP interaction interface and of protein conformational changes in the CaM molecule induced by its binding with CaMBP (Introduction). To facilitate the interpretation of the results of the fluorescent probe studies of the CaMBP-RyR1 interaction described below, we carried out the same type of studies using the combination of CaMBP-badan and CaM as a model. In these experiments, we collected emission spectra with increased concentrations of CaMBP-badan in the presence of 0.5 μM CaM or in its absence at 0.1 μM Ca2+ (apo-CaM model) or 200 μM Ca2+ (Ca-CaM model), and the spectra of CaMBP-badan alone were subtracted from the corresponding spectra of CaMBP-badan with CaM to see the net changes in the badan fluorescence due to the CaMBP-CaM interaction (Fig. 4 A). As shown, the fluorescence intensity increases with an increase of the concentration of CaMBP and plateaued with 1.0 μM CaMBP at 0.1 μM Ca2+ and with 0.7 μM CaMBP at 200 μM Ca2+. Interestingly, the wavelength at the emission maximum (λmax) remained constant at 500 nm at 0.1 μM Ca2+ and 475 nm at 200 μM Ca2+, regardless of the concentration of CaMBP added. This indicates that the CaMBP-CaM complex is in the two clearly distinguishable configurations depending on the Ca2+ concentration.
FIGURE 4.

Monitoring of interactions between CaMBP and CaM using big changes in the fluorescence properties of CaMBP-badan upon its binding to CaM as a measure. (A) Emission spectra with increasing concentrations of CaMBP-badan in the presence of 0.5 μM CaM at 0.1 μM and 200 μM Ca2+. The sensitivity of the fluorescence recording at 200 μM Ca2+ was reduced to ∼1/3 of that at 0.1 μM. (B) Emission spectra with 1 μM CaMBP-badan and 0.5 μM CaM at various Ca2+ concentrations. To calculate the CaM-bound CaMBP-badan fluorescence proper, the emission spectrum of CaMBP-badan alone was subtracted from the corresponding emission spectrum of CaMBP-badan in the presence of CaM.
An earlier study by Rodney et al. (32) and our recent study (24) suggest that at low Ca2+ (<0.1 μM) CaM binds to the central area (encompassing both F1 and F4 regions) of the CaMBP peptide, whereas at high Ca2+ (>1 μM) CaM binds to the F1 region of CaMBP. Based on this we propose that the fluorescence signal characterized by lower λmax represents a conformational state comparable to the state of CaM that is bound to the F1 region of CaMBP, whereas the signal characterized by higher λmax represents a state comparable with the CaM bound to the F1/F4 region. In the experiment of Fig. 4 B, we analyzed the emission spectra of the mixture of 1.0 μM CaMBP-badan and 0.5 μM CaM at various Ca2+ concentrations as indicated. The results indicate that there is a clear transition of the λmax from 500 nm to 475 nm with an increase of Ca2+ concentration from 0.1 μM to 0.3 μM. Importantly, this is the Ca2+ concentration range where the transition of the CaM function occurs from an activator to an inhibitor of the RyR1 Ca2+ channel. Thus, the distinct difference in the λmax value and in the shape of emission spectra characteristic of the CaMBP-CaM complex at low and high Ca2+ concentrations provides an important clue to interpret the data of the CaMBP-RyR1 (SR) interaction described below.
Spectroscopic monitoring of the interaction between CaMBP and the RyR1
CaMBP-badan is a powerful tool to study the CaMBP-CaM interaction, as shown in the experiments of Fig. 4 and in our recent study (24). Therefore, we exploited CaMBP-badan as a spectrometric probe for the study of the CaMBP-RyR1 interaction. We expected that CaMBP-badan would report two components: a very rapid (time-irresolvable) increase in the fluorescence intensity owing to the binding of CaMBP to its interacting domain of RyR1 and a time-resolvable portion of the fluorescence change owing to a local protein conformational change in the RyR induced by the binding of CaMBP. To monitor the fluorescence change corresponding to the peptide binding, we collected families of emission spectra at various concentrations of CaMBP-badan in the absence and in the presence of SR as shown in Fig. 5. We then calculated the fluorescence intensity of the protein-bound CaMBP-badan and the wavelength at the emission maximum by subtracting the emission spectrum of CaMBP-badan alone from the emission spectrum of CaMBP-badan obtained in the presence of SR, as we did in the experiment of Fig. 4. It should be noted that the experiments shown in Fig. 5, A and B, were carried out under the same conditions as those used for the Ca2+ release experiments shown in Figs. 1–3 (viz. in the presence of 1 mM MgATP) to correlate the peptide binding/conformation data with the pattern of channel activation and inhibition. Although we used unlabeled CaMBP in the Ca2+ release experiments, fluorescence labeling of CaMBP seemed to have virtually no effect on the activating function of CaMBP as tested with 30 μM CaMBP solution containing no CaMBP-badan used for stopped-flow experiments of Ca2+ release and 30 μM solution consisting of 1 μM CaMBP-badan and 29 μM CaMBP, the identical solution used for the spectroscopic measurements. Furthermore, 3 μM CaMBP-badan proper and 3 μM unlabeled CaMBP showed the identical Ca2+ release-inducing activity.
FIGURE 5.

(A) Changes in the emission spectra caused by the interaction of increasing concentrations of CaMBP-badan with SR in the presence of 1 mM MgATP (under the same conditions as those used for the Ca2+ release experiments). To calculate the RyR-bound CaMBP-badan fluorescence proper, the emission spectrum of CaMBP-badan alone was subtracted from the corresponding emission spectrum of CaMBP-badan in the presence of SR. B. The fluorescence intensity of the protein-bound CaMBP-badan (•) and changes in the wavelength of the emission maximum (○) zas a function of CaMBP-badan concentration.
As shown in Fig. 5, A and B, there are several important changes in the property of the emission spectrum upon binding of CaMBP-badan to the SR in the presence of 1 mM MgATP (the same conditions as Ca2+ release experiments). The intensity of badan fluorescence corresponding to the RyR1-bound CaMBP increases with an increase of CaMBP concentration and levels off at ∼20 μM, at which inhibition of Ca2+ release begins. Interestingly, there is a small but appreciable change in the λmax from 467 nm to 474 nm with an increase of CaMBP-badan concentration from 0.54 μM to 20 μM. In light of the findings with the CaMBP-badan/CaM model (Fig. 4), this suggests that low concentration of CaMBP produced first the Ca-CaM-like signal to activate Ca2+ channel and higher concentration of CaMBP produced the apo-CaM-like signal to inhibit the channel.
To monitor rapid local protein conformational changes within the RyR1 induced by CaMBP binding, we carried out stopped-flow measurements in parallel to the Ca2+ release experiments shown in Fig. 1 B. The data are shown in Fig. 6 (note that the initial jump in the fluorescence intensity ascribable to the CaMBP binding to the RyR1, i.e., time-unresolvable portion, is not shown in the figure). In the activating concentration range of CaMBP (1–15 μM), CaMBP binding produces a rapid and monophasic increase of badan fluorescence in a concentration-dependent manner, reflecting a local conformational change in the CaMBP binding domain of the RyR1. In the higher concentration range of CaMBP, coincident with the function of CaMBP becoming inhibitory, the initial rapid increase in the badan fluorescence is followed by a rapid spontaneous decay. This is due to the aforementioned red-shift of λmax caused by the conformational transition of the CaMBP/CaMLD complex from the Ca-CaM type to the apo-CaM type, because the stopped-flow fluorescence signal was recorded through an interference filter with a slit opening at 455 nm. Collectively, the results shown in Figs. 5 and 6 suggest that CaMBP-mediated channel activation is accompanied by the formation of the CaMBP-RyR1 complex characteristic of the CaMBP-CaM complex at higher Ca2+ (shorter λmax, Ca-CaM type), whereas CaMBP-mediated channel inhibition is accompanied by the formation of the complex characteristic of the CaMBP-CaM complex at lower Ca2+ (longer λmax, apo-CaM type).
FIGURE 6.

Time-resolvable portion of the stopped-flow traces of the changes in the badan fluorescence upon mixing various concentrations of CaMBP-badan with SR, showing the time course of CaMBP-induced local conformational changes. A large initial jump of the badan fluorescence due to CaMBP binding to the RyR1 (time-unresolvable portion) is not shown in the figure. The experiments were carried out in the same reaction solution as used for the Ca2+ release experiments, except that fluo-3 was excluded. Note that in the activating concentration range of CaMBP (≤10 μM), the peptide induces monophasic fluorescence increase, whereas in the inhibitory concentration range (≥15 μM) the time course of the badan fluorescence change becomes biphasic (an initial increase is followed by a rapid spontaneous decay).
The above results of the Ca2+ release and CaMBP-badan experiments together suggest that there are two classes of CaMBP binding sites in the CaMBP binding domain of the RyR1 and that CaMBP binding to these sites induced local conformational changes: the binding to the first site produces the badan fluorescence change characterized by a shorter λmax (465 nm) and produces channel activation, and the binding to the second site produces the badan fluorescence change characterized by a longer λmax (474 nm) and produces channel inhibition.
In the experiment shown in Fig. 7 A, we used R3534–4271, an expressed peptide corresponding to the Met3534–Ala4271 stretch (containing both CaMBD and CaMLD) of RyR1 to carry out the same type of CaMBP-badan experiment as done with SR and CaM. R3534–4271 was expressed in inclusion bodies, which could be refolded in a soluble form by dialysis. As shown, upon addition of increasing concentrations of CaMBP-badan to R3534–4271 (0.5 μM) at 0.1 μM Ca2+, the intensity of badan fluorescence increased. This indicates that the CaMBP binding domain is localized in the 3534–4271 region of RyR1. In support of the notion that the fluorescence increase represents CaMBP binding to the expressed peptide, the addition of unlabeled CaMBP (5 μM) reversed the fluorescence increase that had been produced by CaMBP-badan (Fig. 7 C). Interestingly, there is a small but appreciable change in the λmax from 460 nm to 470 nm with an increase of CaMBP-badan concentration from 10 nM to 100 nM. This suggests that as in the case of CaMBP-badan binding to the RyR1 moiety of the SR (Fig. 5, A and B), CaMBP binds to the two classes of binding sites in a sequential manner: the binding to the first site produces a conformational state resembling that of the CaMBP bound with Ca-CaM, and the binding to the second site produces the state similar to that of the CaMBP bound with apo-CaM. The first binding may correspond to the channel activation state, and the second binding to the channel inhibition state, although we can't monitor the activity of the channel in this case.
FIGURE 7.

(A) Changes in the emission spectra caused by the interaction of increasing concentrations of CaMBP-badan with R3534–4271. To calculate the fluorescence of the CaMBP-badan bound with R3534–4271, the emission spectrum of CaMBP-badan alone was subtracted from the corresponding emission spectrum of CaMBP-badan (•) in the presence of R3534–4271. (B) The fluorescence intensity of the protein-bound CaMBP-badan (•) and changes in the wavelength of the emission maximum (○) as a function of CaMBP-badan concentration. The fluorescence measurements were performed at 0.1 μM Ca2+ adjusted with a BAPTA/calcium buffer. (C) Addition of unlabeled CaMBP (5 μM) to the CaMBP-badan/R3534–4271 produced a considerable decrease in the fluorescence intensity that had been achieved by the binding of CaMBP-badan.
DISCUSSION
The region encompassing residues 3614–3643 of RyR1 has been identified as a major CaM binding region, as supported by various pieces of evidence as outlined in the Introduction. A considerable amount of information accumulated to date suggests that CaMBP, a synthetic peptide corresponding to residues 3614–3643, retains several functional properties of its in vivo counterpart (i.e., the CaMBD of RyR1). As shown in an earlier study by Hamilton and her colleagues (32) and our recent study (24), CaM binds to slightly different regions of the CaMBP depending upon the Ca2+ concentration; this is interesting in view of the report that there is a significant Ca2+-dependent shift in the location of the RyR1-bound CaM as shown in a cryo-EM single particle analysis (34,35). More precisely, both F1 and F4 portions of CaMBP are required for CaM binding at lower Ca2+ (submicromolar), but the F1 portion is sufficient for CaM binding at higher Ca2+ (larger than micromolar), suggesting that with an increase of Ca2+ from submicromolar to micromolar (or higher) ranges, the site of CaM binding shifts from the C-terminal side to the N-terminal side of CaMBP (cf. 32). According to a recent report by Zhu et al. (22), CaMBP activated the RyR1 Ca2+ channel, as evidenced by its strong enhancement of ryanodine binding activity and its ability to induce SR Ca2+ release in the whole cell system. A synthetic peptide corresponding to the F1 region (F1 peptide) activated ryanodine binding, whereas F4 peptide was inhibitory (22). A more recent study by Rodney et al. (31) has shown that CaMBP increases the frequency of Ca2+ sparks in the saponin-permeabilized skeletal muscle fiber in a highly cooperative concentration-dependent manner. These findings suggest that CaMBP binding to its target domain in the RyR1 activates—or under certain conditions inhibits—the Ca2+ channel and the CaMBP binding domain may represent an important, but not yet well explored, regulatory domain of RyR1. According to a recent report by Xiong et al. (23), the 4064–4210 residue segment of RyR1 shows a CaM-like sequence; the expressed peptide corresponding to this region in fact showed CaM-like properties in Ca2+ binding and Ca2+-induced conformational changes, and importantly it did bind with CaMBP. Since CaMBP corresponds to the CaM binding domain of RyR1, Xiong et al. (23,36) suggested that the interaction between the CaMLD, to which CaMBP binding takes place, with the CaMBD may serve as an intrinsic regulator of the RyR1 Ca2+ channel.
As shown in our recent study (24), the highly conformationally sensitive fluorescent probe badan that was attached to the Cys residue of CaMBP could report conformational changes within the CaM molecule induced by the interaction with CaMBP. In this study, we utilized this unique capability of CaMBP-badan to investigate the CaMBP-induced local conformational changes and resultant Ca2+ release from the SR. In this study, we correlated the three key functional parameters using CaMBP and SR as an experimental model: i), effects of CaMBP on channel regulation (Fig. 1 B) (the kinetics of CaMBP-induced Ca2+ release and its inhibition); ii), binding of CaMBP to the putative CaMLD of RyR1 (Fig. 5) (titration of the protein-bound CaMBP-badan fluorescence); and iii), rapid conformational changes in the CaMBP binding domain upon binding of CaMBP (Fig. 6) (rapid changes in the badan fluorescence upon CaMBP binding). Each of these three parameters was investigated in the presence of MgATP at near threshold concentrations of Ca2+. Under these conditions, CaMBP induced faster and larger Ca2+ release from the SR upon increasing the peptide concentration in a range of relatively low concentrations (<15 μM). Upon increasing the peptide concentration further, the Ca2+ release activity was suppressed and almost completely inhibited at ∼50 μM. In contrast to the biphasic activation/inhibition pattern, the binding of CaMBP proceeded in a monotonic fashion throughout the activating and inhibitory concentration ranges (as determined by the fluorescence titration assay) and it plateaus at the concentration, where the maximal inhibition of Ca2+ release takes place. The correlation between the Ca2+ release data and the fluorescence titration data, also a tight correlation between the transition in the conformational states of the CaMBP/RyR1 complex and that in the channel activation/inhibition pattern, suggests the mechanistic model shown in Scheme 1.
SCHEME 1.

This model assumes that there are two types of CaMBP binding sites in the CaMBP binding domain: an A site and an I site. Binding of CaMBP to the A site via the F1, the Ca-CaM binding region of CaMBP (red region), produces a conformational state resembling the CaMBP-badan that is bound with Ca-CaM (emission maximum at a lower wavelength), activating the channel and inducing Ca2+ release. Upon increasing the concentration of CaMBP, additional CaMBP binding takes place at the I site. This produces a new conformational state, resembling the state of apo-CaM (emission maximum at a longer wavelength) that is bound via the F1/F4 region of CaMBP (black region), resulting in the inhibition of Ca2+ channel and Ca2+ release.
Scheme 1 assumes that there are two classes of CaMBP binding sites in the CaMBP binding domain of the RyR1: an A site and an I site. Binding of CaMBP to the A site produces a conformational state of the binding domain characterized by a lower λmax. The spectroscopic property of this state resembles that of the CaMBP-badan that is bound with Ca-CaM (Fig. 4) and the CaMBP/Ca-CaM interaction is mediated by CaM binding to the F1 region of CaMBP (22,24). Therefore, we assume that the binding of CaMBP to the A site of the binding domain takes place via the F1 region of CaMBP. This state activates the Ca2+ channel and induces a rapid Ca2+ release. At higher concentrations of CaMBP, additional CaMBP binds to the I site. This produces the conformational state characterized by a higher λmax, the state that corresponds to the state of apo-CaM interacting with the F1/F4 region of CaMBP. Therefore, the binding of CaMBP to the I site seems to take place via the F1/F4 region of CaMBP, and this produces the inhibition of Ca2+ channel and Ca2+ release.
As shown in these stopped-flow traces of the badan fluorescence changes during the CaMBP-badan/SR interaction under the Ca2+ release conditions (in the presence of MgATP), there was a concentration-dependent transition from monophasic to biphasic time course of conformational change. This transition coincided with the concentration-dependent transition from activation to inhibition of Ca2+ release (Fig. 6, cf. Fig. 1 B). The monophasic time course in the activating concentration range reflects the formation of the A site-mediated Ca-CaM-like state with low λmax, whereas in the inhibitory concentration range the initial rapid fluorescence increase was followed by a spontaneous decay. The decay phase reflects the conformational transition from the A site-mediated state to the I site-mediated state because in the stopped-flow experiment, the change in the emission intensity was monitored using an interference filter with the window opening at 455 nm and the shift from shorter λmax to longer λmax accompanied by a decrease in the output signal.
The above model provides reasonable explanations for some unsolved questions in the recent literature. For instance, according to a recent report of Rodney et al. (31), CaMBP (0.2–0.7 μM) promoted the occurrence of Ca2+ sparks in a highly cooperative concentration-dependent manner. This apparent cooperativity can be explained by our finding that the CaMBP activation is immediately followed by inhibition with a rather small increase in the CaMBP concentration (cf. Fig. 2). According to our model (Scheme 1), this apparent cooperativity is caused by the second binding of CaMBP to the inhibition site prompted by the first binding to the activation site located in the CaMLD. The apparent cooperativity may have also been produced by the fact that the activation of Ca2+ release increases the local cytoplasmic Ca2+, prompting the formation of the A site-mediated Ca-CaM-like state, and spontaneous Ca2+ reuptake prompts the formation of the I site-mediated apo-CaM-like state. Rodney et al. (31) have shown that a single amino acid substitution L3624D resulted in a total loss of activating function of CaMBP without affecting its ability to bind with apo-CaM. This is consistent with our proposal that the interaction of CaMBP with the A site occurs via the N-terminal F1 region of CaMBP (Scheme 1), also consistent with previous reports of Hamilton and colleagues (32) and ours (24) that Ca-CaM binds to the F1 region, whereas apo-CaM binds to the C-terminal (or F1/F4) region of CaMBP.
This study of CaMBP-badan binding to the expressed peptide R3534–4271 corresponding to the Met3534–Ala4271 region containing both CaMBD and CaMLD provided us with some new insight into the location of the CaMBP binding domain. The binding of CaMBP-badan at a low peptide concentration produced the Ca-CaM-like low λmax state, and the binding at a higher peptide concentration produced a small but appreciable red-shift in the λmax. Interestingly, this peptide concentration-dependent shift in the λmax is identical to that seen in the interaction of CaMBP with the RyR1 moiety of SR. This suggests that both of the A and I sites of the CaMBP binding domain postulated here are located within the Met3534–Ala4271 region, most probably in the 4064–4210 CaMLD (cf. 23). However, there is an important difference between the R3534–4271 and CaM that upon binding of CaMBP-badan the R3534–4271 produces a Ca-CaM-like conformational state even at 0.1 μM Ca2+.
In conclusion, we used the interaction of CaMBP and its badan-labeled derivative with the RyR1 moiety of the SR membrane as a spectroscopic model for the study of the CaMBP-induced local conformational changes in the CaMBP binding domain of the RyR1 and induced Ca2+ release from the SR. Coordinated studies of Ca2+ release kinetics and spectrometric monitoring of the peptide binding and induced local conformational changes in the peptide binding region of the RyR1 suggest a new mechanism in which CaMBP binds to the two kinetically and spectroscopically distinguishable sites producing activating and inhibitory configurations of the CaMBP binding domain of RyR1 (Scheme 1). The studies of CaMBP binding to R3534–4271 peptide suggested that the CaMBP binding domain is located within the 3534–4271 region of RyR1, probably in the CaMLD, because spectroscopic studies of the CaMBP-CaM interaction suggested that the CaMBP binding domain retains CaM-like properties.
Acknowledgments
We thank Dr. Renne C. Lu, Dr. Paul Leavis, David Schrier, and Elizabeth Gowell for synthesis and purification of the peptides.
This work was supported by National Institutes of Health grants AR 16922 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and HL072841 from the National Heart, Lung, and Blood Institute.
References
- 1.Meissner, G. 2002. Regulation of mammalian ryanodine receptors. Front. Biosci. 7:d2072–d2080. [DOI] [PubMed] [Google Scholar]
- 2.Tang, W., S. Sencer, and S. L. Hamilton. 2002. Calmodulin modulation of proteins involved in excitation-contraction coupling. Front. Biosci. 7:d1583–d1589. [DOI] [PubMed] [Google Scholar]
- 3.Hamilton, S. L., I. Serysheva, and G. M. Strasburg. 2000. Calmodulin and excitation-contraction coupling. News Physiol. Sci. 15:281–284. [DOI] [PubMed] [Google Scholar]
- 4.Rodney, G. G., J. Krol, B. Williams, K. Beckingham, and S. L. Hamilton. 2001. The carboxyl-terminal calcium binding sites of calmodulin control calmodulin's switch from an activator to an inhibitor of RyR1. Biochemistry. 40:12430–12435. [DOI] [PubMed] [Google Scholar]
- 5.Tripathy, A., L. Xu, G. Mann, and G. Meissner. 1995. Calmodulin activation and inhibition of skeletal muscle Ca2+ release channel (ryanodine receptor). Biophys. J. 69:106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodney, G. G., B. Y. Williams, G. M. Strasburg, K. Beckingham, and S. L. Hamilton. 2000. Regulation of RyR1 activity by Ca2+ and calmodulin. Biochemistry. 39:7807–7812. [DOI] [PubMed] [Google Scholar]
- 7.Buratti, R., G. Prestipino, P. Menegazzi, S. Treves, and F. Zorzato. 1995. Calcium dependent activation of skeletal muscle Ca2+ release channel (ryanodine receptor) by calmodulin. Biochem. Biophys. Res. Commun. 213:1082–1090. [DOI] [PubMed] [Google Scholar]
- 8.Rodney, G. G., and M. F. Schneider. 2003. Calmodulin modulates initiation but not termination of spontaneous Ca2+ sparks in frog skeletal muscle. Biophys. J. 85:921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fruen, B. R., D. J. Black, R. A. Bloomquist, J. M. Bardy, J. D. Johnson, C. F. Louis, and E. M. Balog. 2003. Regulation of the RyR1 and RyR2 Ca2+ release channel isoforms by Ca2+-insensitive mutants of calmodulin. Biochemistry. 42:2740–2747. [DOI] [PubMed] [Google Scholar]
- 10.Meissner, G., and J. S. Henderson. 1987. Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. J. Biol. Chem. 262:3065–3073. [PubMed] [Google Scholar]
- 11.Fruen, B. R., J. M. Bardy, T. M. Byrem, G. M. Strasburg, and C. F. Louis. 2000. Differential Ca2+ sensitivity of skeletal and cardiac muscle ryanodine receptors in the presence of calmodulin. Am. J. Physiol. 279:C724–C733. [DOI] [PubMed] [Google Scholar]
- 12.Yamaguchi, N., C. Xin, and G. Meissner. 2001. Identification of apocalmodulin and Ca2+-calmodulin regulatory domain in skeletal muscle Ca2+ release channel, ryanodine receptor. J. Biol. Chem. 276:22579–22585. [DOI] [PubMed] [Google Scholar]
- 13.Moore, C. P., G. Rodney, J.-Z. Zhang, L. Santacruz-Toloza, G. M. Strasburg, and S. L. Hamilton. 1999. Apocalmodulin and Ca2+ calmodulin bind to the same region on the skeletal muscle Ca2+ release channel. Biochemistry. 38:8532–8537. [DOI] [PubMed] [Google Scholar]
- 14.Zang, J. Z., Y. Wu, B. Williams, G. Rodney, F. Mandel, G. M. Strasburg, and S. L. Hamilton. 1999. Oxidation of the skeletal muscle Ca2+ release channel alters calmodulin binding. Am. J. Physiol. 276:C46–C53. [DOI] [PubMed] [Google Scholar]
- 15.Moore, C. P., J. Z. Zhang, and S. L. Hamilton. 1999. A role for cysteine 3635 of RyR1 in redox modulation and calmodulin binding. J. Biol. Chem. 274:36831–36834. [DOI] [PubMed] [Google Scholar]
- 16.Xiong, L.-W., R. A. Newman, G. G. Rodney, O. Thomas, J.-Z. Zhang, A. Persechini, M. A. Shea, and S. L. Hamilton. 2002. Lobe-dependent regulation of ryanodine receptor type 1 by calmodulin. J. Biol. Chem. 277:40862–40870. [DOI] [PubMed] [Google Scholar]
- 17.Yoon, H. J., Z. Grabarek, S. L. Hamilton, and G. M. Strasburg. 2003. Conformational states of calmodulin bound to RyR1 peptide. Biophys. J. 84:107a. (Abstr.) [Google Scholar]
- 18.Yamaguchi, N., L. Xu, D. A. Pasek, K. E. Evans, and G. Meissner. 2003. Molecular basis of calmodulin binding to cardiac Ca2+ release channel (ryanodine receptor). J. Biol. Chem. 278:23480–23486. [DOI] [PubMed] [Google Scholar]
- 19.Yamaguchi, N., L. Xu, K. E. Evans, D. A. Pasek, and G. Meissner. 2004. Different regions in skeletal and cardiac muscle ryanodine receptors are involved in transducing the functional effects of calmodulin. J. Biol. Chem. 279:36433–36439. [DOI] [PubMed] [Google Scholar]
- 20.Zhang, H., J.-Z. Zhang, C. I. Danila, and S. L. Hamilton. 2003. A noncontiguous, intersubunit binding site for calmodulin on the skeletal muscle Ca2+ release channel. J. Biol. Chem. 278:8348–8355. [DOI] [PubMed] [Google Scholar]
- 21.Sencer, S., R. V. L. Papineni, D. B. Halling, P. Pate, J. Krol, J.-Z. Zhang, and S. L. Hamilton. 2001. Coupling of RyR1 and L-type calcium channel via calmodulin binding domains. J. Biol. Chem. 276:38237–38241. [DOI] [PubMed] [Google Scholar]
- 22.Zhu, X., J. Ghanta, J. W. Walker, P. D. Allen, and H. H. Valdivia. 2004. The calmodulin binding region of the skeletal ryanodine receptor acts as a self-modulatory domain. Cell Calcium. 35:165–177. [DOI] [PubMed] [Google Scholar]
- 23.Xiong, L., R. He, J.-Z. Zhang, S. Sencer, and S. L. Hamilton. 2006. A Ca2+ binding domain in RyR1 that interacts with the calmodulin binding site and modulates channel activity. Biophys. J. 90:173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gangopadhyay, J. P., Z. Grabarek, and N. Ikemoto. 2004. Fluorescence probe study of Ca2+-dependent interactions of calmodulin with calmodulin binding peptides of the ryanodine receptor. Biochim. Biophys. Res. Commun. 323:760–768. [DOI] [PubMed] [Google Scholar]
- 25.Ikemoto, N., D. H. Kim, and B. Antoniu. 1988. Measurement of calcium release in isolated membrane systems: coupling between the transverse tubule and sarcoplasmic reticulum. Methods Enzymol. 157:469–480. [DOI] [PubMed] [Google Scholar]
- 26.Wawrzynczak, E. J., and R. N. Perham. 1984. Isolation and nucleotide sequence of a cDNA encoding human calmodulin. Biochem. Int. 9:177–185. [PubMed] [Google Scholar]
- 27.Tan, R. Y., Y. Mabuchi, and Z. Grabarek. 1996. Blocking the Ca2+-induced conformational transitions in calmodulin with disulfide bonds. J. Biol. Chem. 271:7479–7483. [DOI] [PubMed] [Google Scholar]
- 28.Kipp, R. A., M. A. Case, A. D. Wist, C. M. Cresson, M. Carrell, E. Griner, A. Wiita, P. A. Albiniak, J. Chai, Y. Shi, M. F. Semmelhack, and G. L. McLendon. 2002. Molecular targeting of inhibitor of apoptosis proteins based on small molecule mimics of natural binding partners. Biochemistry. 41:7344–7349. [DOI] [PubMed] [Google Scholar]
- 29.Owenius, R., M. Osterlund, M. Lindgren, M. Svensson, O. H. Olsen, E. Persson, P.-O. Freskgard, and U. Calsson. 1999. Properties of spin and fluorescent labels at a receptor-ligand interface. Biophys. J. 77:2237–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hammaarstrom, P., R. Owenius, L.-G. Martensson, U. Calsson, and M. Lindgren. 2001. High-resolution probing of local conformational changes in proteins by the use of multiple labeling: unfolding and self-assembly of human carbonic anhydrase II monitored by spin, fluorescent and chemical reactivity probes. Biophys. J. 80:2867–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodney, G. G., G. M. Wilson, and M. F. Schneider. 2005. A calmodulin binding domain of RyR increases activation of spontaneous Ca2+ sparks in frog skeletal muscle. J. Biol. Chem. 280:11713–11722. [DOI] [PubMed] [Google Scholar]
- 32.Rodney, G. G., C. P. Moore, B. Y. Williams, J. Z. Zhang, J. Krol, S. E. Pedersen, and S. L. Hamilton. 2001. Calcium binding to calmodulin leads to an N-terminal shift in its binding site on the ryanodine receptor. J. Biol. Chem. 276:2069–2074. [DOI] [PubMed] [Google Scholar]
- 33.Moore, C. P., G. Rodney, J.-Z. Zhang, L. Santacruz-Toloza, G. Strasburg, and S. L. Hamilton. 1999. Apocalmodulin and Ca2+ calmodulin bind to the same region on the skeletal muscle Ca2+ release channel. Biochemistry. 38:8532–8537. [DOI] [PubMed] [Google Scholar]
- 34.Wagenknecht, T., and M. Samso. 2002. Three-dimensional reconstitution of ryanodine receptors. Front. Biosci. 7:d1464–d1474. [DOI] [PubMed] [Google Scholar]
- 35.Samso, M., and T. Wagenknecht. 2002. Apocalmodulin and Ca2+-calmodulin bind to neighboring locations on the ryanodine receptor. J. Biol. Chem. 277:1349–1353. [DOI] [PubMed] [Google Scholar]
- 36.Gangopadhyay, J. P., and N. Ikemoto. 2005. Peptide probe study of Ca2+ and calmodulin dependent regulation of the ryanodine receptor. Biophys. J. 88:484a. (Abstr.) [Google Scholar]