

Abstract
Purple membranes (PM) are two-dimensional crystals formed by bacteriorhodopsin and a variety of lipids. The lipid composition and density in the cytoplasmic (CP) leaflet differ from those of the extracellular (EC) leaflet. A new way of differentiating the two sides of such asymmetric membranes using the phase signal in alternate contact atomic force microscopy is presented. This method does not require molecular resolution and is applied to study the stiffness and intertrimer lipid mobility in both leaflets of the PM independently over a broad range of pH and salt concentrations. PM stiffens with increasing salt concentration according to two different regimes. At low salt concentration, the membrane Young's normal modulus grows quickly but differentially for the EC and CP leaflets. At higher salt concentration, both leaflets behave similarly and their stiffness converges toward the native environment value. Changes in pH do not affect PM stiffness; however, the crystal assembly is less pronounced at pH ≥ 10. Lipid mobility is high in the CP leaflet, especially at low salt concentration, but negligible in the EC leaflet regardless of pH or salt concentration. An independent lipid mobility study by solid-state NMR confirms and quantifies the atomic force microscopy qualitative observations.
INTRODUCTION
Most of the membranes of living organisms are composed of both lipids and proteins. Membrane proteins are essential for any communication, side-specific interactions, and molecular transport through the membrane (1). To fulfill these vectorial tasks, membrane proteins are extensively asymmetrically oriented (1,2). Lipids generally show less defined asymmetrical behavior between the two membrane leaflets and adapt their location around proteins (1,3). However, phospholipids tend to be located within the cytoplasmic (CP) leaflet of plasma membranes (4). Despite its ubiquity, it is experimentally difficult to observe membrane asymmetry and labeling techniques are generally required (5–7).
Purple membranes (PM) are two-dimensional crystals consisting of bacteriorhodopsin (bR) trimers assembled with unusual membrane lipids in a hexagonal array that is formed naturally in the CP membrane of Halobacterium salinarium (8). bR comprises seven transmembrane α-helices that enclose a retinal chromophore (9). bR acts as a light-driven proton pump, converting light energy into a proton gradient across the membrane (10). The important role of protein-lipid interactions in the assembly and activity of PM has been widely studied, mainly by diffraction spectroscopic techniques (8,11,12). In the bacteria, the lipid composition of PM is defined and differs from that of the surrounding membrane (13,14). The requirement of a fixed membrane composition indicates that selective interactions occur between bR and certain lipid molecules and that these interactions are essential for lattice assembly and bR function (8,11,12). A comprehensive understanding of the interactions within the membrane is hindered by the wide variety of lipids present (phospholipids, glycolipids, squalene, and traces of vitamin MK8 (13,15)). The densely charged phospholipids seem to interact with bR and each other mainly through nonspecific long range electrostatic forces (16,17). Diffraction experiments suggest that phospholipids are more mobile than glycolipids (18). Phospholipids are mainly located in the CP leaflet intertrimer space (6,16,19). The main phospholipid present in PM is phosphatidylglycerophosphate methylated (PGP-Me; see Fig. 4 D) (13) with a molar ratio of 2.4:1 to the retinal (15). The other archaeal phospholipids are phosphatidylglycerol (PG), phosphatidylglycerosulphate (PGS), and archaeal cardiolipin (BPG). The glycolipids are archaeal glycocardiolipin (GlyC) and triglycosyl lipid (S-TGA-1). Recently, the lipids molar ratio to the retinal has been precisely determined (15) giving a protein/lipid ratio of 10 lipid molecules per bR monomer. It has been shown that bR-DMPC reconstitution in the presence of >2 M NaCl exhibits the well-known hexagonal crystalline array formation of PM only when PGP or PGS are present (16,20). Reconstituted bR in model membranes without those natural lipids showed conformational changes (21), indicating that a specific interaction of bR with the charged phospholipids is important to maintain its three-dimensional structure. A phospholipid indeed mediates a specific interaction between bR monomers in the CP leaflet, critical for the assembly of the trimer (22). Glycolipids are specifically and tightly bound to bR (23,24) and show clear patterns in PM diffraction experiments (25). Neutron diffraction experiments confirmed the presence of two S-TGA-1 molecules per bR molecule, both located in the EC leaflet with one in the intertrimer and one in the intratrimer space (23). More generally, it has been proposed that glycolipids are located in the EC leaflet of the membrane (24).
FIGURE 4.

31P NMR spectra with proton decoupling of PM films obtained from water suspension (I), from 5 mM sodium citrate pH 6 suspension (II) at 100% (A) and 75% (B) relative humidity and dry (C), respectively. The spectra were measured at 20°C. The 31P NMR signals correspond mainly to PGP-Me (D) in PM.
Here atomic force microscopy (AFM) is used to study the overall cohesion of PM in buffer solution, particularly the influence of salt on PM stiffness and lipid mobility. AFM has recently provided molecular resolution images of PM (26,27). The two sides of PM have different surface charge and stiffness (5,6,28). We use these differences to distinguish the two sides of the membrane by alternate contact mode AFM (AC-AFM). Furthermore, probing with a nanometer size tip allows quantification of the interaction strength at the trimer level by pressing on the membrane (force curves), permitting investigation of intertrimer protein-lipid and lipid-lipid interactions in PM. Since the AFM tip charge depends on the solution pH, electrostatic changes in the membrane surface can be probed.
Additionally, we obtain complementary information about lipid mobility by solid-state NMR experiments. 31P solid-state NMR spectroscopy has been used to determine the morphology of phospholipid bilayers and to assess the degree of alignment of the lipids (29). Solid-state NMR can provide information about orientation and behavior of the phospholipid headgroups in PM (30). Oriented 31P NMR spectra (static) were used to characterize the mosaic spread of the oriented PM sample (31). Further information about the dynamics in the vicinity of the phosphate groups is obtained from the spin-lattice relaxation times in the laboratory frame (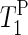 ), since it is sensitive to the motion at the frequency of the magnetic field (∼400 MHz = ∼2.5 × 10−9 s).
), since it is sensitive to the motion at the frequency of the magnetic field (∼400 MHz = ∼2.5 × 10−9 s).
MATERIALS AND METHODS
Purple membrane preparation
Wild-type PM from H. salinarium strain S9 and the M163C-bR mutant from strain L33 were grown by a standard method using a peptone media (L37, Oxoid, Basingstoke, UK) and isolated and purified using established protocols (32). The membranes were either frozen at −25°C for storage (4 months maximum) or kept at 4°C for immediate use.
AFM
Sample preparation
PM was diluted to ∼15 μg/ml in the imaging buffer. A drop (30 μl) of PM solution was adsorbed on freshly cleaved mica (9.9 mm mica discs, Agar Scientific, Essex, UK) for 5–10 min and then gently rinsed with imaging buffer (2 ml). For work in liquid with low salt concentration buffers (20–100 mM KCl), the discs were not rinsed because of the weak adsorption of PM on mica (33). For salt concentration <50 mM KCl, PM adsorption was carried out by putting a drop (50 μl) of PM in solution (50 mM KCl, 10 mM Tris-HCl) on freshly cleaved mica and by diluting with 10 mM Tris-HCl up to the required KCl concentration a few minutes later to prevent ill formation or no adsorption of the membrane. Imaging was performed after adding some more buffer (∼200 μl). For imaging in air, a drop (50 μl) of PM in solution (50 mM KCl, 10 mM Tris-HCl) was adsorbed for 5–10 min on mica, then gently rinsed with ultrapure water (2 ml of 18.2 MΩ, Maxima Ultrapure water system, ELGA, High Wycombe, Bucks, UK) and dried at 20°C in a closed petri dish. All the buffers were made with ultrapure water and renewed every 2 weeks. Buffer chemicals (KCl, HCl, Tris, and NaOH) were purchased from Sigma-Aldrich (Dorset, UK). AFM imaging and force curves were acquired at 20°C ± 1°C.
Imaging
High resolution images were recorded with a commercial Dimension 3100 AFM (Veeco, Santa Barbara, CA) equipped with a 90 μm scanner operated in open loop. An AC liquid-cell was used for imaging in buffer. Since AC-AFM is extremely sensitive to tip or sample contamination (26) the liquid cell was sonicated in ultrapure water, then cleaned with ethanol, and finally rinsed with ultrapure water before imaging in buffer. A new AFM tip was used for each sample. Imaging in liquid was carried out with Olympus TR800 tips (SiN, nominal spring constant kn = 0.57 N/m, Olympus, Tokyo, Japan). High resolution was achieved in 150 mM KCl 10mM Tris-HCl buffer at pH 8 (isoelectric point of the SiN (34)) according to Möller et al. (26). Imaging in air was performed with Olympus AC240 tips (SiN, kn = 2 N/m, Olympus). Before imaging, the system was left 2 or 3 h scanning a blank sample to reach equilibrium. For each image, height, amplitude, and phase information were acquired simultaneously. The scan speed was adjusted between 1 and 3 lines/s for low magnification frames (>400 nm) and between 4 and 9 lines/s for high resolution frames. Best results were obtained in ultrasoft AC, i.e., with a ratio of the free oscillation amplitude A0 over the set-point amplitude A as close as possible to 1 and at low oscillation amplitude (≤1 nm). The PM lattice was used for lateral calibration of the scanner and quantification of the drift. Acquired images were corrected for drift, but no averaging was done.
Force curves
Force curves were recorded in solution with a commercial MFP-3D AFM (Asylum Research, Santa Barbara, CA) with a close loop in the x, y, and z directions. Olympus TR400 tips (SiN, nominal spring constant kn = 0.08 N/m, Olympus) were used for increased sensitivity and to avoid sample damaging, especially in low salt buffer. AC images of the sample were acquired before and after force curves were measured to ensure reliability of the curves and that the sample had not been damaged. In each buffer condition, ∼300 curves were taken on both CP and EC sides of PM with the same cantilever and without reengaging the AFM. Force curves where also taken on mica before and after force curves acquisition on PM to calculate the inverse optical lever sensitivity (nm/V) and to ensure the stability of the system. The sampling rate was set to 300 nm/s. The z-piezo extension per curve was set to 100 or 150 nm to prevent excessive pressure on the membrane. The spring constant of the cantilever was calculated using the thermal noise method (35,36) and theoretical predictions (37). Thermal noise scans always indicated a similar value for the cantilevers used (k = 0.11 N/m within <10%), suggesting a good reproducibility of the cantilever stiffness, but theoretical calculations gave a spring constant of k = 0.085 N/m. Due to the discrepancy of the different force calibration methods, the nominal value of 0.08 N/m was used to calculate forces, including an error of 40%. To avoid systematic errors, each set of measurements carried out in a specific buffer condition was made in a random order.
Solid-state NMR
Sample preparation
For 31P NMR measurements, purified PM was suspended in buffer (5 mM sodium citrate; pH 6), with a bR concentration of ∼3 mg/ml (∼11 μM bR). Oriented PM films were prepared by slow evaporation of the aqueous PM suspension on thin glass slides (8 × 8 × 0.06–0.08 mm, Paul Marienfeld GmbH, Lauda-Königshofen, Germany). For “dry” PM samples, a sufficient number of these slides were stacked to fill a square NMR coil. Alternatively, the slides with dry PM films were kept at a controlled relative humidity of 75% or 100% by placing them in a sealed container at 37°C for 24 h with, respectively, a saturated NaCl solution or distilled water. The slides with (partially) hydrated PM were stacked and kept for another 24 h at controlled relative humidity. Subsequently, the stack was sealed with parafilm and placed in a polyethylene tube (RS Components, Northants, UK) to prevent dehydration during the NMR measurements. A total of 5 mg of PM was used for each sample.
NMR measurements
Solid-state 31P NMR measurements were performed on a CMX Infinity 400 spectrometer (Varian, Palo Alto, CA) with operating frequencies of 397.9 and 161.1 MHz for 1H and 31P, respectively. Static 31P NMR spectra of the hydrated PM samples were obtained with a Hahn-echo pulse sequence (90° − τ − 180° − τ1 − acquisition) and with a proton decoupling power of 40–45 kHz. Typical 90° and 180° pulse lengths for 1H and 31P were 5 and 10 μs, and an echo interval (τ) of 20 μs was used. For the dry PM sample, a cross-polarization pulse sequence was applied (1.0 ms contact time, 42 kHz proton decoupling). For all measurements, a recycling delay of 4 s was used, and 3000–12,000 scans were acquired. Before Fourier transformation and zero-filling to 2048 points, a Lorentzian line-broadening of 200 Hz was applied for the hydrated samples and of 500 Hz for the dry sample. The 31P chemical shift was referenced to external 85% H3PO4 (0 ppm). All spectra were recorded at 20°C.
The phosphorus spin-lattice relaxation times in the laboratory frame (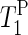 ) were determined by the inversion-recovery method using pulse durations from 0.001 to 6.0 s.
) were determined by the inversion-recovery method using pulse durations from 0.001 to 6.0 s. 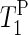 was evaluated using a nonlinear least-square fitting of the five data points of 31P NMR signal intensities.
was evaluated using a nonlinear least-square fitting of the five data points of 31P NMR signal intensities.
RESULTS
When PM solution is deposited on mica, adsorbed patches assemble into the hexagonal lattice (Fig. 1). A small band (typically 5–50 nm wide) of nonordered membrane can be observed around a developing patch. Patches are 5.3 ± 0.4 nm thick and 500 nm to 2 μm wide. They do not show any preferential orientation for the adsorption (EC or CP side facing mica).
FIGURE 1.

Distinction of PM EC and CP sides using AC-AFM (A–E). Topographic image (A). The phase image (B) reveals a contrast between EC patches (darker) and CP patches (lighter). A profile of (B) gives ∼7°CP-EC shift and ∼15°CP-mica shift (C). High resolution topographic images of PM CP (D) and EC (E) side obtained by zooming respectively on the lighter and darker patches in (B). The protrusions on the CP patches and a “smooth belt” around the EC patch are clearly visible in panels A and B (dashed line). Effect of salt removal on the PM imaged by AC-AFM in air (F–J). The membrane has been rinsed with ultrapure water and dried at room temperature. The EC (pitted) and CP (cracked) sides of PM after drying (F). Higher magnifications of the EC patch (G and H). Hexagonal depressions related to the CP leaflet and pits are visible. Higher magnification of the CP cracked patch (I and J). Arrows point to larger holes crossing EC patches in panels F and G and to trimers in panel J. The scale bar is 1 μm (A and B), 10° (C), 3 nm (D and E), 1 μm (F), 100 nm (G), 50 nm (H), 100 nm (I), and 50 nm (J). Panels F–I are topographic images, and J is a phase image.
AFM: distinction of EC and CP sides of PM
Phase and high resolution imaging
Due to the structural asymmetry of PM, molecular level studies on PM mechanical or electrostatic properties require leaflet-specific experiments. All AFM experiments showing side distinction reported to date used high resolution imaging (7,26). This method requires a careful balancing of the electrostatic double layer forces between the membrane and the AFM tip. The thickness of the double layer is characterized by the Debye length, 1/κ. In the case of KCl in aqueous solution (38)
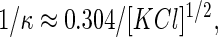 |
(1) |
where [KCl] is the concentration of KCl in mol per liter and 1/κ is in nanometers. Consequently, high resolution imaging imposes a specific salt concentration of the imaging buffer (39), which prevents PM side distinction away from the optimum conditions. We have overcome this problem by using the phase information provided by AC-AFM. Operated in very soft AC mode (A0/A close to 1, see Materials and Methods), the phase shift is very sensitive to changes in energy dissipation due to tip-sample interactions (40–42). The natural asymmetry between the EC and CP sides of PM (6,28) is sufficient to offer a clear phase contrast when scanned in any buffer studied. The EC patches appear darker than the CP patches in the phase image (Fig. 1, B and C). High resolution imaging under favorable buffer conditions confirmed the side assignment obtained through phase shift imaging (Fig. 1, D and E). Scanning in low salt buffer accentuates the asymmetry resolved by the AFM tip between the two sides of PM, and thus the phase shift. This new way of distinguishing the PM's two sides has been extensively used in this study, mainly for side-specific force spectroscopy.
Membrane protrusions on the CP side and pH effect
Additional features which help to distinguish the two sides of PM when adsorbed onto mica and present under any salt or pH conditions studied (20–300 mM KCl, pH 5–12) are the frequent and random protrusions exhibited by CP patches (patches having their CP side exposed for imaging and their EC side facing the substrate) in contrast to the smoothness of the EC patches (Fig. 1, A and B). These features, appearing only on CP patches, have been observed before (43) but never commented on or explained. High resolution images of CP patches showed that the PM lattice is still visible on top of the protrusions (data not shown). They are consequently due to material located underneath the membrane, between the EC leaflet and the substrate. EC patches do not exhibit such irregularities. Moreover they seem to prevent their formation, and CP patch regions adjacent to EC patches exhibit a smooth band without protrusions (Fig. 1 B), forming a “smooth belt” around the EC patches.
Finally, changing the pH of the buffer solution does not seem to affect the global cohesion of the membrane but more the tip-sample interaction by changing the surface charge of the AFM tip from negative (pH 10) to positive (pH 6). However, at pH ≥ 10, despite the good cohesion of the PM lattice, the assembly seems more difficult and large areas of a nonordered thinner membrane are visible (Fig. 2) around the well-assembled patches. The lattice then assembles from these nonordered regions (44). This misassembled membrane could be due to the lysine residues of bR loosing their positive charge at pH 10, thus weakening the bR interaction with highly negatively charged lipids and making the lattice assembly less favorable. Since almost all the lysine residues of bR are located in the CP leaflet, once the membrane is assembled, the cohesion is maintained by the EC leaflet-specific interactions. Consistently increasing the salt concentration of the buffer allowed a normal PM assembly. It should be noted that around the CP patches, the protrusions mentioned above are already present in this nonordered area (Fig. 2).
FIGURE 2.

PM assembly at pH 10. The arrows 1, 2, and 3 show CP patches, EC patches, and nonassembled membrane, respectively. The nonassembled membrane is ∼1 nm thinner than ordered lattice (profile line). Different lattice orientations are observable in assembled patches, and protrusions are visible in and around CP patches. The scale bar is 400 nm.
AFM: the asymmetric effect of salt on PM
Imaging with no salt in liquid and in air
Imaging PM in liquid under very low salt concentration (<20 mM KCl) is difficult due to a significant increase of the double layer thickness (Eq. 1). In water (ultrapure water), the AFM SiN tip feels a strong electrostatic interaction scanning over the negatively charged PM CP side, thus preventing any nondestructive imaging. However it is possible to obtain images of the less charged EC side, revealing a rough “pitted” membrane, with many circular depressions the diameter of few bR trimers and 1–2 nm deep (data not shown). To image the effect of salt removal on the CP side of the membrane, it is necessary to dry the sample for AC imaging in air. Two distinct sides are revealed: a pitted side similar to the membrane imaged in ultrapure water and a cracked side, with the cracks following a hexagonal lattice (Fig. 1 F). These pitted and cracked patches had previously been observed by cryoelectron microscopy (45) and assigned to the CP and EC membrane sides, respectively. However, contrary to this report, we attribute the cracks to the CP side and the pits to the EC one. This was confirmed using a mutant bR (M163C) that can covalently bind to gold via a cystein residue located on its CP side and prevents normal assembly of patches having their CP side in contact with gold. Patches with their EC side on gold were normally assembled and also exhibited hexagonal cracks, similar to those shown in Fig. 1 I (data not shown). Examination of the cracks in the CP leaflet reveals that they are following the bR hexagonal lattice (Fig. 1, I and J). The larger cracks traverse the membrane, but the smaller cracks are only ∼1 leaflet deep (2–3 nm) with occasional deeper holes corresponding to fully removed trimers. Phase imaging allows the identification of trimer rows (Fig. 1 J). Cracks propagate between these rows and are probably due to missing lipids, removed by capillary forces during the membrane drying, and condensation of the lipid chains (46). Some lipids are removed from the CP leaflet intertrimer space, weakening the membrane and occasionally allowing whole trimers to be extracted along with the lipids. The EC leaflet does not show clear cracks, but their presence is suggested in the opposite leaflet by depressions following the hexagonal lattice (Fig. 1 G). Small round pits similar to the holes observed in PM in ultrapure water (Fig. 1 H) and larger circular holes (50–150 nm in diameter) spanning the whole membrane width are visible.
Side-specific force spectroscopy
To quantify the differences reported above between the two PM leaflets, we have taken series of force curves on both sides separately, varying salt concentration and pH. All the curves are extension curves, showing the approach of the tip toward the sample. The corresponding retraction curves (not shown) are identical except for some possible adhesion of the tip to the sample.
Representative force curves are presented in Fig. 3; they are taken on both sides of PM and in a buffer containing 10 mM Tris and 20, 25, 30, 40, 150, and 300 mM KCl, respectively. The pH was set to 8.
FIGURE 3.

Force curves on the EC side (light shaded) and the CP side (dark shaded) of PM and on mica (solid) at different salt concentrations (20–300 mM KCl). The curves have been offset for better visibility. The maximum force applied by the tip is ∼2 nN (500 pN force scale) corresponding to the force beyond which all the curves look similar when superimposed, regardless of the sample.
At 20 mM and 25 mM KCl concentration, no obvious difference can be seen between the force curves on mica and on the CP side of PM. Both exhibit a double layer electrostatic repulsion zone (Eq. 1) followed by a linear deflection starting for forces <100 pN. This shows that the AFM tip feels almost no membrane repulsion pressing on the CP side. The tip penetrates very easily through the whole membrane to reach the mica underneath. However, imaging the sample after having taken force curves revealed no permanent damage of the membrane, suggesting an immediate healing after a hole was made by the tip. The CP leaflet is weakened by a lack of shielding between highly charged phospholipids and by the mobility of the intertrimer lipids. In contrast, the EC side of the membrane is much more resistant to the tip pressure and continuously deflects the cantilever.
At 30 mM KCl concentration in the buffer, force curves on the CP side of PM exhibit an intermediate behavior between the curves on mica and on the EC side due to partial shielding of electrostatic repulsion between the CP leaflet lipids. This could be explained by the tip perforating only the CP leaflets.
The critical salt concentration past which the electrostatic repulsion within the CP leaflet is fully shielded is 40 mM KCl. Curves on the EC and CP side are very similar. Both leaflets deform in the same way under the tip pressure. Increasing the salt concentration hardens the membrane symmetrically up to the point where force curves on the membrane and on mica show almost no difference (at 300 mM KCl).
Side-specific force curves have also been acquired at pH 6, 8, and 10 in a 150-mM KCl buffer. No obvious difference could be observed between the force curves taken on the CP and the EC side of PM (see Fig. 7 C).
FIGURE 7.

Influence of the pH on PM at 150 mM KCl. Evolution of PM Young's normal modulus with pH, measured from EC and CP side of PM (A). Curves are fit using Chadwick's model. Maximal tip indentation in PM, obtained from fit (B). Side-specific force curves on PM at different pH (C).
Solid-state NMR
To complement the AFM results, the effect of salt on PM was investigated by comparing 31P NMR spectra of PM aligned from a buffered suspension (5 mM sodium citrate, pH 6) and from a dispersion in distilled water. Phospholipids are predominantly located in the CP leaflet of PM, and thus NMR can probe this leaflet specifically (22). Fig. 4 shows 31P static NMR spectra of PM aligned from water and from the buffer suspension at different levels of hydration. At 100% relative humidity (Fig. 4 A), two signals were observed around 40 and 13 ppm. They can be assigned to the α-phosphate (phosphodiester) and the γ-phosphate (phosphomonoester) groups (Fig. 4 D) of the principal PM phospholipid, PGP-Me (47), respectively. For all samples, the line width of the γ-phosphate resonance was smaller than the line width of the α-phosphate resonance, indicating a higher mobility of the γ-phosphate moiety. This is due to the position of the γ-phosphate, which is located further away from the lipid backbone. At 75% relative humidity, the 31P line shape of the sample aligned from water became broad and the two signals were hardly separated (Fig. 4 B), whereas the two signals from the sample containing salt could still be clearly distinguished. For all the dry samples, the 31P NMR spectra showed extremely broad signals (Fig. 4 C), not only from 50 to 0 ppm but also from 0 to −50 ppm. However, resolved peaks could easily be obtained by rehydration of the dry samples, resulting in similar spectra as those shown in Fig. 4, A and B.
Table 1 summarizes the 31P spin-lattice relaxation times in the laboratory frame (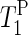 ) for the α- and γ-phosphate signals at 20°C.
) for the α- and γ-phosphate signals at 20°C. 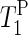 of the γ-phosphate groups is smaller than
of the γ-phosphate groups is smaller than 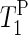 of the α-phosphate moieties. It can also be seen that
of the α-phosphate moieties. It can also be seen that 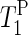 of PM aligned from water and from sodium citrate suspension decreases gradually as the hydration level increases from 75% to 100%. For the dry sample in sodium citrate, a
of PM aligned from water and from sodium citrate suspension decreases gradually as the hydration level increases from 75% to 100%. For the dry sample in sodium citrate, a 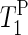 value ∼10 times larger than
value ∼10 times larger than 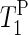 of the hydrated samples was obtained. Accordingly, the timescale of the anisotropic motion of the phosphate groups is on the “long correlation” side of the T1 minimum (ω0τc ∼ 1; τc ∼ 2.5 × 10−9 s).
of the hydrated samples was obtained. Accordingly, the timescale of the anisotropic motion of the phosphate groups is on the “long correlation” side of the T1 minimum (ω0τc ∼ 1; τc ∼ 2.5 × 10−9 s).
TABLE 1.
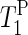 (s) of the α- and γ-phosphate groups of PGP in PM film at 20°C; α and γ are described in Fig. 4
(s) of the α- and γ-phosphate groups of PGP in PM film at 20°C; α and γ are described in Fig. 4
| Relative humidity
|
Water
|
5 mM Sodium citrate
|
||
|---|---|---|---|---|
| α | γ | α | γ | |
| 100% | 0.17 (±0.01) | 0.09 (±0.01) | 1.43 (±0.09) | 1.01 (±0.05) |
| 75% | 4.29 (±0.30) | 4.65 (±0.24) | 1.96 (±0.12) | 1.41 (±0.03) |
| Dry | – | 16.7 (±0.2) | ||
DISCUSSION
Lipid mobility and density
AFM
Despite the very high salt concentration of the H. salinarium native environment (∼4 M NaCl), PM still assembles on mica at very low salt concentration, and membrane patches extensively rinsed with ultrapure water exhibit the hexagonal lattice (Fig. 1, F and I) characteristic of native PM (48,49). However, the lack of salt affects the membrane in an asymmetric fashion. The CP leaflet becomes very soft with weakly attached intertrimer lipids that can be removed by capillary forces, whereas the EC leaflet remains almost unchanged. These observations indicate that highly branched and charged phospholipids located in the intertrimer space of the CP leaflet (PG, PGS, PGP-Me, and BPG) interact with bR and each other mainly through nonspecific long range electrostatic forces yet still possessing high mobility within the leaflet. This is consistent with other studies (6,47) in which the charge asymmetry of PM has been determined using fluorescence and NMR. The AFM images emphasize the importance of phospholipids for the overall membrane cohesion, since their removal induces the extraction of whole bR trimers by capillary forces (Fig. 1). However, the main intertrimer interactions stabilizing the membrane together appear to be specific and to take place in the EC leaflet, as previously reported for intratrimer interactions (25,50). These intertrimer interactions are sufficient to maintain the lattice structure of PM, even in very low salt conditions when the CP leaflet is dramatically affected or partially removed (Fig. 1). Sulfoglycolipids are known to be predominantly located in the EC leaflet (24) and in the intertrimer space (23) and to interact strongly and specifically with bR, mainly with its tryptophan residues. Tryptophan residues have been shown to be determinant in the anchoring of proteins in lipid membranes in general (51). In PM, the asymmetric interaction of sulfoglycolipids with bR is underscored by the position of the tryptophan residues in bR since almost all of them are located in the EC leaflet, some of them pointing toward the intertrimer space (9).
More evidence of the specificity of the lipids and the interactions in each leaflet can be found in Fig. 1, A and B, where the CP patches (EC side facing mica) exhibit many protrusions due to material located under the patch. Since these protrusions are already present in the membrane assembling edge (Fig. 2), they are probably lipid vesicles joining the membrane edge during the patch formation. These vesicles are indeed able to fuse with the CP leaflet of the membrane, allowing PM to be smooth if they are sandwiched between mica and the CP leaflet. This suggests that the vesicles are composed of phospholipids, which would also explain why they cannot cross the membrane to reach the CP leaflet when trapped between mica and the EC leaflet. The location of the vesicles exclusively under CP patches demonstrates the specificity of the interactions within the EC leaflet. Vesicles trapped close enough to an EC patch (CP side down) seem to reach it by lateral diffusion and fuse to it, thus creating this “smooth belt” around it. This phenomenon, however, raises an interesting question about lipid density within the CP leaflet; if the above deduction is correct, the CP leaflet can find room for more lipids than the number initially present just after the assembly, since vesicles are fusing to an already assembled patch. A possible explanation would be the high mobility of the phospholipids, allowing them to diffuse within the whole CP leaflet and leave it at its edges if the lipid density is too high and the headgroup repulsion excessive. The density of lipids has indeed been shown to be significantly higher in the CP leaflet (52).
NMR
31P NMR resonances shown in Fig. 4 indicate that the orientation of the phospholipids in fully hydrated samples does not have a clear dependence on the salt concentration. However reducing the hydration levels to 75% in the absence of salt produces misorientation of the lipids. Salt ions enable lipids to orient well even at low hydration levels. Reducing the salt concentration increases the interlipid headgroup repulsion, inducing disorientation. Complete removal of water produces a decrease in lipid long axis rotation, leading to condensation of their chains and a poorer membrane alignment (46,53). This is in good agreement with the AFM images of dried PM, where condensation and removal of the phospholipids are responsible for the cracks observed in Fig. 1, F–J.
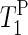 NMR relaxation times (Table 1) confirm the reduction of phospholipid mobility with decreasing humidity. The increase in
NMR relaxation times (Table 1) confirm the reduction of phospholipid mobility with decreasing humidity. The increase in 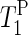 for the drier membranes indicates that the correlation time of the lipid motion lies to the right of the minimum (∼2.5 × 10−9 s), suggesting an increase in low frequency motion. Fully hydrated samples show increased lipid mobility in the absence of salt. The repulsion of the headgroups caused by reduced shielding of the charge seems to increase the phospholipid mobility. This result supports the interpretation of AFM force curves experiments. At low salt concentrations, the AFM tip easily perforates the softened CP patches, but they healed fast after having been perforated (Fig. 3). At lower hydration levels, the lack of charge shielding strongly decreases lipid mobility, producing disorder of the lipids and condensation of the chains.
for the drier membranes indicates that the correlation time of the lipid motion lies to the right of the minimum (∼2.5 × 10−9 s), suggesting an increase in low frequency motion. Fully hydrated samples show increased lipid mobility in the absence of salt. The repulsion of the headgroups caused by reduced shielding of the charge seems to increase the phospholipid mobility. This result supports the interpretation of AFM force curves experiments. At low salt concentrations, the AFM tip easily perforates the softened CP patches, but they healed fast after having been perforated (Fig. 3). At lower hydration levels, the lack of charge shielding strongly decreases lipid mobility, producing disorder of the lipids and condensation of the chains.
PM stiffness measured by side-specific force spectroscopy
We have estimated PM Young's normal bulk modulus by fitting the force curves with a theory recently developed by Chadwick (54). This theory assumes a sphere indenting a thin film located on a substrate composed of harder material. The modulus has been calculated independently for both sides of the membrane at each salt concentration. The AFM tip apex has a curvature radius of 10–20 nm, and thus presses on at least a whole trimer when indenting the membrane. As shown from imaging (Fig. 1), bR conserves its trimeric form both in the lattice and when extracted from the membrane. bR is indeed more stable in trimeric than in monomeric form (55,56); this implies that force curves are mainly representative of the intertrimer protein-lipid and lipid-lipid interactions within PM.
For a membrane not bound to its substrate, the indentation δ of a sphere of radius R pressing with a force F on the thin membrane is given by
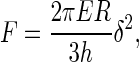 |
(2) |
where h is the membrane thickness and E the Young's modulus. The membrane is considered incompressible (Poisson's ratio of ½), which is a reasonable assumption for biological membranes. The major improvement of Chadwick theory over the traditional Sneddon's modification of the Hertzian approach (57,58) used in previous AFM experiments (59) is the assumption of a hard substrate under the membrane, which is relevant in our case. The Sneddon's approach assumes an indenting cone on a semiinfinite medium, also giving a quadratic dependence of the force in δ, but with different constant parameters:
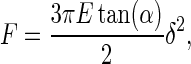 |
(3) |
where α is the half opening angle of the indenting cone and Poisson's ration is also ½. We have fitted our data with both theories for comparison. Fig. 5 shows the principle used to do it, taking the curve on mica as a reference. The indentation curve is superimposed on the reference curve (on mica) for measuring δ. Fδmax corresponds to the applied tip force beyond which the maximal possible indentation δmax on the membrane is reached and the curve does not then represent the indentation properties of the membrane, but more those of the substrate underneath (substrate region) (59). If F ≥ Fδmax, then δ = δmax and the force curve on the membrane is similar to that for the substrate underneath (substrate region). When the AFM tip approaches the membrane, electrostatic effects arise from the tip crossing the electrostatic double layer (electrostatic region). Membrane force curves were fitted in the region where the tip is in contact with the sample and the corresponding curve on mica is linear, between the electrostatic and the substrate regions. The tip radius R, the membrane thickness h, the cantilever spring constant k, and the half opening angle α of the tip cone were assumed to be identical for each curve analyzed. Many independent fitting trials suggested values of R = 20 nm and α = 30°. The membrane thickness h is 5.3 nm (Fig. 1 A), and k = 0.08 N/m.
FIGURE 5.

Principle of measurement of the effective indentation of the membrane by the AFM tip. The region taken for fitting the models, labeled “Indentation region”, corresponds to the region where the equivalent force curve on mica is linear and the applied force is smaller than Fδmax.
Fig. 6 A shows the evolution of the Young's modulus with increase of KCl concentration in the buffer (pH 8), calculated using the Chadwick theory. Young's modulus calculated from Sneddon's modification of the traditional Hertzian theory shows a similar evolution with values systematically 35% higher than for Chadwick treatment and are thus not shown for more clarity.
FIGURE 6.

Young's normal bulk modulus of PM evolution with KCl concentration at pH 8, measured from each side independently by fitting force curves with Chadwick's model (A). The evolution of the values obtained is fitted with a biexponential curve. Evolution of the maximum tip indentation δmax in the membrane, also obtained from force curves fitting (B).
Two different regimes are distinguishable, and the evolution of the Young's modulus has been fitted with a biexponential curve of type:
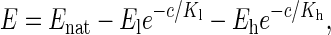 |
(4) |
where Enat is the Young's modulus of PM close to its natural medium salt concentration (4 M NaCl), c is the KCl concentration, and the two exponential terms represent the changes in E at low and high salt concentration, respectively. The salt concentration parameters Kl = 12 mM and Kh = 650 mM have been fixed as global variables for both sides of the membrane. This choice reflects the fact that when free, these parameters give similar final results within 20% and that their globalization allows an easier interpretation of the results.
Interestingly, PM stiffness increases with the salt concentration despite a better surface charge shielding. This is because at low salt concentration, the phospholipids are mobile within the CP leaflet and can accommodate the indentation without significantly increasing the electrostatic potential. The effective membrane stiffness is therefore decreased because the structure of the CP leaflet is temporarily modified.
At low salt concentration (between 20 mM KCl and 40 mM KCl), the measured Young's modulus differs depending on the side indented. The EC side gives a value of 14.4 MPa for 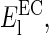 whereas
whereas 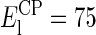 MPa for the CP side, giving a ratio for
MPa for the CP side, giving a ratio for 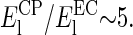 The stiffening is five times more important in the CP leaflet.
The stiffening is five times more important in the CP leaflet.
At higher salt concentration, the parameter Eh gives comparable values for both sides of the membrane: 17 MPa for the EC side and 16.4 MPa for the CP side. Since the behavior of the membrane Young's modulus does not seem to depend on the side indented at high salt concentration, Enat was logically set as a global parameter, giving a value of Enat = 28 MPa. This value is however an estimate, first because the highest salt concentration studied is still ∼10 times less than that of the natural medium concentration for the bacterium and also due to the very large uncertainty in the moduli obtained from force curve fitting. It is difficult to select the right region of the curve investigated to fit the model. To be valid, the indentation curve should be fit between the double layer electrostatic region and the substrate region where the indentation reaches its maximum, δ = δmax (see Fig. 5). At high salt concentration, the membrane stiffens and it becomes more difficult to determine the transition between the indentation region and the substrate region; this problem is the main factor of uncertainty. The uncertainty arising from the moduli calculated over the different curves obtained in one buffer is negligible (<5%). The model used assumes a linear deformation of the indented layer (Hooke deformation), which may not be true at low salt concentration. However, since the force curves used for fitting were identical in extension and retraction, the deformation of this membrane is elastic. The model used also takes into account a hard substrate underneath (Emica = 15–25 GPa), and the use of small forces (<2 nN) gives consistent results in comparison with surface force apparatus measurements on phospholipid bilayers (60,61). The systematic 35% overestimation of Young's modulus using the Sneddon's modified Hertzian theory is consistent with the observations made by Dimitriadis et al. (62) when comparing the two theories used here for thin film indentation.
Changing the pH of the buffer solution does not seem to modify the stiffness of PM. The EC and CP Young's moduli obtained by fitting the corresponding force curves provided no evidence for the membrane to be asymmetrically affected by pH changes within the range measured (Fig. 7, A–C).
Finally, the evolution of the maximal indentation δmax with changing KCl concentration (Fig. 6 B) is consistent with a qualitative observation of the force curves. Both theories used for fitting gave a similar value for δmax within 5%.
CONCLUSIONS
We have studied salt and pH effects on cohesion, stiffness, and mobility in both leaflets of PM independently, concentrating on intertrimer lipids and protein-lipid interactions. AFM imaging allowed a direct observation of how PM is asymmetrically affected by salt removal. Side-specific indentation of the membrane provided a quantification of the changes in stiffness, using a novel method for distinguishing the EC and the CP sides which exploits the phase shift information available when the AFM is operated in AC mode. This method, based on PM natural asymmetry, does not require high resolution imaging. Fitting the force curves separately obtained for the EC and the CP side of PM, we have calculated the evolution of PM normal Young's modulus with increase of the buffer pH and salt concentration and carried out an estimation of PM modulus under natural saline concentration. NMR measurements offer an independent way of observing the phospholipid mobility and order, showing an increased mobility of the headgroups on salt removal. All the experiments tend to show that PM can be seen as a structure held together by specific protein-lipid and lipid-lipid interactions within the EC leaflet. This scaffolding seems static and is very resistant to pH or salt concentration changes of the surrounding buffer, maintaining the well-known stability of PM. The CP leaflet, on the other hand, contains very mobile intertrimer phospholipids largely affected by salt and pH changes. The mobility is due to nonspecific long range electrostatic forces characterizing the CP leaflet. Further AFM experiments on PM and on asymmetric membranes in general should take into account the different behavior of the leaflets since results obtained from both sides may not be similar.
Acknowledgments
The authors thank Prof. G. Turner (University of Miami School of Medicine) for the generous gift of M163C-bR mutant and Dr. Maurits de Planque for useful suggestions in the interpretation of the data.
This work has been supported by the Biotechnology and Biological Sciences Research Council, the Engineering and Physical Sciences Research Council, the Medical Research Council, and the Ministry of Defense through the Bionanotechnology Interdisciplinary Research Collaboration. K.V. acknowledges support by the Lord Florey scholarship.
Miya Kamihira's present address is Dept. of Food and Nutritional Sciences, University of Shizuoka, 52-1 Yada, Suruga, Shizuoka 422-8526, Japan.
References
- 1.Houslay, M. D., and K. K. Stanley. 1982. Dynamics of Biological Membranes. Wiley Interscience, New York.
- 2.Op den Kamp, J. A. F. 1979. Lipid asymmetry in membranes. Annu. Rev. Biochem. 48:47–71. [DOI] [PubMed] [Google Scholar]
- 3.Rothman, J. E., and J. Lenard. 1977. Membrane asymmetry. Science. 195:743–753. [DOI] [PubMed] [Google Scholar]
- 4.Bergelson, L. D., and L. I. Barsukov. 1977. Topological asymmetry of phospholipids in membranes. Science. 197:224–230. [DOI] [PubMed] [Google Scholar]
- 5.Gale, P., and A. Watts. 1992. Effect of bacteriorhodospin on orientation of headgroup od 1,2-dimyristoyl-sn-glycero-3-phosphocholine in bilayers: a 31P- and 2H-NMR study. Biochim. Biophys. Acta. 1106:317–324. [DOI] [PubMed] [Google Scholar]
- 6.Renthal, R., and C. H. Cha. 1984. Charge asymmetry of the purple membrane measured by uranyl quenching of dansyl fluorescence. Biophys. J. 45:1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Müller, D. J., C. A. Schoenenberger, G. Buldt, and A. Engel. 1996. Immuno-atomic force microscopy of purple membrane. Biophys. J. 70:1796–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cartailler, J. P., and H. Luecke. 2003. X-ray and crystallographic analysis of lipid-protein interactions in the bacteriorhodopsin purple membrane. Annu. Rev. Biophys. Biomol. Struct. 32:285–310. [DOI] [PubMed] [Google Scholar]
- 9.Luecke, H., B. Schobert, H. T. Richter, J. P. Cartailler, and J. K. Lanyi. 1999. Structure of bacteriorhodopsin at 1.55 Å resolution. J. Mol. Biol. 291:899–911. [DOI] [PubMed] [Google Scholar]
- 10.Lanyi, J. K. 1995. Bacteriorhodopsin as a model for proton pumps. Nature. 375:461–463. [DOI] [PubMed] [Google Scholar]
- 11.Krebs, M. P., and T. A. Isenbarger. 2000. Structural determinants of purple membrane assembly. Biochim. Biophys. Acta. 1460:15–26. [DOI] [PubMed] [Google Scholar]
- 12.Lee, A. G. 2003. Lipid-protein interactions in biological membranes: a structural perspective. Biochim. Biophys. Acta. 1612:1–40. [DOI] [PubMed] [Google Scholar]
- 13.Kates, M., S. C. Kushwaha, and G. D. Sprott. 1982. Lipids of purple membrane from extreme halophiles and of methanogenic bacteria. Methods Enzymol. 88:98–111. [Google Scholar]
- 14.Corcelli, A., V. M. T. Lattanzio, G. Mascolo, P. Papadia, and F. Fanizzi. 2002. Lipid-protein stoichiometries in a crystalline biological membrane: NMR quantitative analysis of the lipid extract of the purple membrane. J. Lipid Res. 43:132–140. [PubMed] [Google Scholar]
- 15.Corcelli, A., V. M. T. Lattanzio, G. Mascolo, P. Papadia, and F. Fanizzi. 2002. Lipid-protein stoichiometries in a crystalline biological membrane: NMR quantitative analysis of the lipid extract of the purple membrane. J. Lipid Res. 43:132–140. [PubMed] [Google Scholar]
- 16.Sternberg, B., C. L'Hostis, C. A. Whiteway, and A. Watts. 1992. The essential role of specific Halobacterium halobium polar lipids in 2D-array formation of bacteriorhodopsin. Biochim. Biophys. Acta 1108:21–30. [DOI] [PubMed] [Google Scholar]
- 17.Grigorieff, N., E. Beckmann, and F. Zemlin. 1995. Lipid location in deoxycholate-treated purple membrane at 2.6 Å. J. Mol. Biol. 254:404–415. [DOI] [PubMed] [Google Scholar]
- 18.Pebay-Peyroula, E., G. Rummel, J. P. Rosenbusch, and E. M. Landau. 1997. X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science. 277:1676–1681. [DOI] [PubMed] [Google Scholar]
- 19.Belrhali, H., P. Nollert, A. Royant, C. Menzel, J. P. Rosenbusch, E. M. Landau, and E. Pebay-Peyroula. 1999. Protein, lipid and water organization in bacteriorhodopsin crystals: a molecular view of the purple membrane at 1.9 Å resolution. Structure. 7:909–917. [DOI] [PubMed] [Google Scholar]
- 20.Watts, A. 1995. Bacteriorhodopsin: the mechanism of 2D-array formation and the structure of retinal in the protein. Biophys. Chem. 55:137–151. [DOI] [PubMed] [Google Scholar]
- 21.Saito, H., Y. Yamamoto, S. Tuzi, and S. Yamaguchi. 2003. Backbone dynamics of membrane proteins in lipid bilayers: the effect of two-dimensional array formation as revealed by site-directed solid-state 13C NMR studies on [3–13C]Ala-and [1–13]Val-labeled bacteriorhodopsin. Biochim. Biophys. Acta. 1616:127–136. [DOI] [PubMed] [Google Scholar]
- 22.Sato, H., K. Takeda, K. Tani, T. Hino, T. Okada, M. Nakasako, N. Kamiya, and T. Kouyama. 1999. Specific lipid-protein interactions in a novel honeycomb lattice structure of bacteriorhodopsin. Acta Crystallogr. D. 55:1251–1256. [DOI] [PubMed] [Google Scholar]
- 23.Weik, M., H. Patzelt, G. Zaccai, and D. Oesterhelt. 1998. Localization of glycolipids in membranes by in vivo labeling and neutron diffraction. Mol. Cell. 1:411–419. [DOI] [PubMed] [Google Scholar]
- 24.Henderson, R., J. S. Jubb, and S. Whytock. 1978. Specific labelling of the protein and lipid on the extracellular surface of purple membrane. J. Mol. Biol. 123:259–274. [DOI] [PubMed] [Google Scholar]
- 25.Grigorieff, N., T. A. Ceska, K. H. Downing, J. M. Baldwin, and R. Henderson. 1996. Electron-crystallographic refinement of the structure of bacteriorhodopsin. J. Mol. Biol. 259:393–421. [DOI] [PubMed] [Google Scholar]
- 26.Möller, C., M. Allen, V. Elings, A. Engel, and D. J. Müller. 1999. Tapping-mode atomic force microscopy produces faithful high-resolution images of protein surfaces. Biophys. J. 77:1150–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Müller, D. J., J. B. Heymann, F. Oesterhelt, C. Möller, H. Gaub, G. Buldt, and A. Engel. 2000. Atomic force microscopy of native purple membrane. Biochim. Biophys. Acta. 1460:27–38. [DOI] [PubMed] [Google Scholar]
- 28.Subramaniam, S., M. Gerstein, D. Oesterhelt, and R. Henderson. 1993. Electron diffraction analysis of structural changes in the photocycle of bacteriorhodopsin. EMBO J. 12:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seelig, J. 1978. 31P nuclear magnetic resonance and the head group structure of phospholipids in membranes. Biochim. Biophys. Acta. 515:105–140. [DOI] [PubMed] [Google Scholar]
- 30.Watts, A. 1998. Solid-state NMR approaches for studying the interaction of peptides and proteins with membranes. Biochim. Biophys. Acta. 1376:297–318. [DOI] [PubMed] [Google Scholar]
- 31.Ulrich, A. S., M. P. Heyn, and A. Watts. 1992. Structure determination of the cyclohexene ring of retinal in bacteriorhodopsin by solid-state deuterium nmr. Biochemistry. 31:10390–10399. [DOI] [PubMed] [Google Scholar]
- 32.Oesterhelt, D., and W. Stoeckenius. 1974. Isolation of the cell membrane of Halobacterium halobium and its fractionation into red and purple membrane. Methods Enzymol. 31:667–678. [DOI] [PubMed] [Google Scholar]
- 33.Müller, D. J., A. Engel, and M. Amrein. 1997. Adsorption of biological molecules to a solid support for scanning probe microscopy. J. Struct. Biol. 119:172–188. [DOI] [PubMed] [Google Scholar]
- 34.Biscan, J., N. Kallay, and T. Smolic. 2000. Determination of iso-electric point of silicon nitride by adhesion method. Colloids Surf. A. 165:115–123. [Google Scholar]
- 35.Butt, H. J., and M. Jaschke. 1995. Calculation of thermal noise in atomic force microscopy. Nanotechnology. 6:1–7. [Google Scholar]
- 36.Hutter, J. L., and J. Bechhoefer. 1993. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 64:1868–1873. [Google Scholar]
- 37.Sader, J. E. 1995. Parallel beam approximation for V-shaped atomic force microscope cantilevers. Rev. Sci. Instrum. 66:4583–4587. [Google Scholar]
- 38.Israelachvili, J. N. 1991. Intermolecular and Surface Forces. Academic Press, London.
- 39.Müller, D. J., D. Fotiadis, S. Scheuring, S. A. Müller, and A. Engel. 1999. Electrostatically balanced subnanometer imaging of biological specimens by atomic force microscope. Biophys. J. 76:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.San Paulo, A., and R. Garcia. 2001. Amplitude, deformation and phase shift in amplitude modulation atomic force microscopy: a numerical study for compliant materials. Surf. Sci. 471:71–79. [Google Scholar]
- 41.García, R., J. Tamayo, and A. San Paulo. 1999. Phase contrast and surface energy hysteresis in tapping mode scanning force microscopy. Surf. Interf. Anal. 27:312–316. [Google Scholar]
- 42.Cleveland, J. P., B. Anczykowski, A. E. Schmid, and V. B. Elings. 1998. Energy dissipation in tapping-mode atomic force microscopy. Appl. Phys. Lett. 72:2613–2615. [Google Scholar]
- 43.Stark, M., C. Moller, D. J. Muller, and R. Guckenberger. 2001. From images to interactions: high-resolution phase imaging in tapping-mode atomic force microscopy. Biophys. J. 80:3009–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neugebauer, D. C., H. P. Zingsheim, and D. Oesterhelt. 1983. Biogenesis of purple membrane in halobacteria. Methods Enzymol. 97:218–226. [DOI] [PubMed] [Google Scholar]
- 45.Fisher, K. A., K. Yanagimoto, and W. Stoeckenius. 1978. Oriented adsorption of purple membrane to cationic surfaces. J. Cell Biol. 77:611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zaccai, G. 1987. Structure and hydration of purple membranes in different conditions. J. Mol. Biol. 194:569–572. [DOI] [PubMed] [Google Scholar]
- 47.Gale, P., and A. Watts. 1991. Characterization of phospholipid compositions and physical properties of DMPC/bacteriorhodopsin vesicles produced by a detergent-free method. Biochem. Biophys. Res. Commun. 180:939–944. [DOI] [PubMed] [Google Scholar]
- 48.Blaurock, A. E., and W. Stoeckenius. 1971. Structure of the purple membrane. Nat. New Biol. 233:152–155. [DOI] [PubMed] [Google Scholar]
- 49.Henderson, R., and P. N. Unwin. 1975. Three-dimensional model of purple membrane. Nature. 257:28–32. [DOI] [PubMed] [Google Scholar]
- 50.Essen, L. O., R. Siegert, W. D. Lehmann, and D. Oesterhelt. 1998. Lipid patches in membrane protein oligomers: crystal structure of the bacteriorhodopsin-lipid complex. Proc. Natl. Acad. Sci. USA. 95:11673–11678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Antoranz Contera, S., V. Lemaître, M. R. R. de Planque, A. Watts, and J. F. Ryan. 2005. Unfolding and extraction of a transmembrane α-helical peptide: dynamic force spectroscopy and molecular dynamics simulations. Biophys. J. 89:3129–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blaurock, A. E., and G. I. King. 1977. Asymmetric structure of the purple membrane. Science. 196:1101–1104. [DOI] [PubMed] [Google Scholar]
- 53.Cornell, B. A., C. A. Morris, V. L. B. Braach-Maksvytis, and F. Separovic. 1987. The effect of bacteriorhodopsin on the dynamics of lipid bilayers: a study by solid state 13-C nuclear magnetic resonance. In Retinal Proteins: Proceedings of the International Conference, USSR, 1986. Yu. A. Ovchinnikov, editor. Brill Academic Publishers, Leiden, The Netherlands. 285–293.
- 54.Chadwick, R. S. 2002. Axisymmetric indentation of a thin incompressible elastic layer. SIAM J. Appl. Math. 62:1520–1530. [Google Scholar]
- 55.Haltia, T., and E. Freire. 1995. Forces and factors that contribute to the structural stability of membrane proteins. Bioenergetics. 1228:1–27. [DOI] [PubMed] [Google Scholar]
- 56.Lopez, F., S. Lobasso, M. Colella, A. Agostiano, and A. Corcelli. 1999. Light-dependent and biochemical properties of two different bands of bacteriorhodopsin isolated on phenyl-sepharose CL-4B. Photochem. Photobiol. 69:599–604. [Google Scholar]
- 57.Hertz, H. 1882. Über die Berührung Fester Elasticher Körper. Journal für die reine und angewandte Mathematik. 92:156–171. [Google Scholar]
- 58.Sneddon, I. N. 1965. The relation between load and penetration in the axisymmetric Boussinesq problem for a punch of arbitrary profile. Internat. J. Engin. Sci. 3:47–57. [Google Scholar]
- 59.Domke, J., and M. Radmacher. 1998. Measuring the elastic properties of thin polymer films with the atomic force microscope. Langmuir. 14:3320–3325. [Google Scholar]
- 60.Benz, M., T. Gutsmann, N. Chen, R. Tadmor, and J. Israelachvili. 2004. Correlation of AFM and SFA measurements concerning the stability of supported lipid bilayers. Biophys. J. 86:870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cevc, G., and D. Marsh. 1987. Phospholipid Bilayers: Physical Principles and Models. Wiley, New York.
- 62.Dimitriadis, E. K., F. Horkay, J. Maresca, B. Kachar, and R. S. Chadwick. 2002. Determination of elastic moduli of thin layers of soft material using the atomic force microscope. Biophys. J. 82:2798–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]