

Abstract
Voltage-gated K+ channel activation is proposed to result from simultaneous bending of all S6 segments away from the central axis, enlarging the aperture of the pore sufficiently to permit diffusion of K+ into the water-filled central cavity. The hinge position for the bending motion of each S6 segment is proposed to be a Gly residue and/or a Pro-Val-Pro motif in Kv1–Kv4 channels. The KCNQ1 (Kv7.1) channel has Ala-336 in the Gly-hinge position and Pro-Ala-Gly. Here we show that mutation of Ala-336 to Gly in KCNQ1 increased current amplitude and shifted the voltage dependence of activation to more negative potentials, consistent with facilitation of hinge activity that favors the open state. In contrast, mutation of Ala-336 to Cys or Thr shifted the voltage dependence of activation to more positive potentials and reduced current amplitude. Mutation of the putative Gly hinge to Ala in KCNQ2 (Kv7.2) abolished channel function. Mutation-dependent changes in current amplitude, but not kinetics, were found in heteromeric KCNQ1/KCNE1 channels. Mutation of the Pro or Gly of the Pro-Ala-Gly motif to Ala abolished KCNQ1 function and introduction of Gly in front of the Ala-mutations partially recovered channel function, suggesting that flexibility at the PAG is important for channel activation.
INTRODUCTION
X-ray crystallographic studies of several bacterial K+ channels have provided valuable insights into the structural basis of K+ channel function. These channel structures revealed the first glimpses of the molecular configuration of the closed (KcsA) and open (MthK) states of ion channels (1,2). The x-ray and electron microscopic structures of KvAP, an archaebacterial channel (3), led to the proposal of a novel and controversial (4–6) mechanism of voltage sensor movement in a voltage-gated K+ (Kv) channel. However, introduction of a His into the S4-segment of Shaker channels can generate a proton channel, arguing against a paddle model (7). More recently, based on site-directed spin labeling and electron parametric resonance spectroscopy experiments, Cullo et al. (8) proposed a model of Shaker B in the open state which includes membrane spanning kinked S3 and S4 α-helical formation. In addition, several open and closed state models have been generated based on experimental data that also refute the paddle model (9–11).
Comparison of the KcsA and MthK x-ray structures strongly suggested that channel opening involves movement of the inner helices (equivalent to the S6 domains in a six-transmembrane domain Kv channel) away from the central cavity via a hinge-like motion centered at a Gly residue that is highly conserved in K+ channels (Fig. 1 A). Indeed alignment of 112 out of 116 K channel sequences revealed the presence of a Gly in a homologous position (12). However, the universality of the Gly-hinge hypothesis is challenged by some biophysical studies and the fact that a few channels have an Ala at the hinge position, a residue that usually stabilizes the structure of an α-helix. For example, KCNQ1 (but not KCNQ2-5) and Drosophila EAG channels have an Ala in the hinge position.
FIGURE 1.

Putative hinge and bend points in the S6 domain of a Kv channel. (A) Sequence alignment of several potassium channels. The selectivity filter, gating hinge (boxed red), and PAG motif (marked by §) are noted. (B) Pore loop and S6 α-helical segment of KcsA and MthK are overlaid (left). Homology models of hKCNQ1 based on the solved structures of KcsA and MthK of the same region and a KcsA-based model with deviated S6 at the PAG-motif are shown in the right-hand panel. (C) Variable flexibility of the gating hinge could lead to a variable aperture of the intracellular pore.
The alternative model for Kv channel activation is based on biophysical studies that demonstrate gated access and binding of intracellularly applied Cd2+ to engineered Cys residues (13,14). This model proposes that the upper half of the S6 domain containing the highly conserved Gly is relatively immobile and that bending or twisting of S6 nearer the intracellular end of the pore mediates channel opening and closing (Fig. 1 B). The location of the S6 bend in this model is a Pro-Val(Ile)-Pro motif that is highly conserved in Kv1-KvX channels but is Ile-Phe-Gly in EAG and ERG channels and Pro-Ala-Gly in KCNQ channels (Fig. 1).
Here, we used a site-directed mutagenesis approach to assay the relative importance of the putative gating hinge (Ala-336) and the putative Pro-Ala-Gly bend of the S6 domain in the activation of channels formed by assembly of KCNQ1 subunits alone or in combination with KCNE1 β-subunits. Mutation of Ala-336 affected current amplitude, single channel properties, and voltage-dependent gating. Mutation to Ala of the Pro or Gly in the Pro-Ala-Gly motif abolished KCNQ1 channel function. Our findings suggest that the Pro-Ala-Gly motif is more important than the putative Gly-gating hinge in the S6 domain for activation of KCNQ1 channels.
MATERIALS AND METHODS
Molecular biology
The molecular biological procedures were the same as previously described (15). Human KCNQ1 and human KCNQ2 were mutated at positions Ala-336 (KCNQ1) and Gly-301 (KCNQ2) and at sites 342–347 in KCNQ1 by polymerase-chain-reaction-based site-directed mutagenesis with cloned Pfu-polymerase. All constructs were confirmed by automated DNA sequencing.
Western blot
To identify the fraction of KCNQ1 channel proteins inserted in the plasma membrane, surface proteins were tagged with Sulfo-NHS-LC-Biotin and isolated by NeutrAvidin-mediated precipitation of the biotinylated protein. Briefly, intact oocytes were incubated in 1 mg/ml Sulfo-NHS-LC-Biotin (Pierce Chemical, Rockford, IL) for 30 min at room temperature. After five 10-min washes in normal frog Ringer's solution, intact oocytes were homogenized with a Teflon pestle in H-buffer (20 μl/oocyte; 100 mM NaCl, 20 mM Tris-HCl, pH 7.4, 1% Triton X-100, 1 mM phenylmethylsulphonyl fluoride plus a mixture of protease inhibitors (Complete tablets, Boehringer, Ingelheim, Germany) and were kept at 4°C for 1 h on a rotator. After centrifugation for 60 s at 16,000 × g, the supernatants were supplemented with 20 μl washed immobilized NeutrAvidin Biotin-Binding Protein (Pierce, IL) and incubated at 4°C for 3 h on the rotator. The beads were then pelleted by a 120 s spin at 1600 × g and washed three times in H-buffer. The final pellets were boiled in 40 μl sodium dodecylsulphate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer (0.8 M β-mercaptoethanol, 6% SDS, 20% glycerol, 25 mM Tris-HCl, pH 6.8, 0.1% bromphenol blue). The samples were Western blotted and probed with an affinity-purified goat polyclonal KCNQ1 antibody directed against a peptide mapping at the carboxy terminus of KCNQ1 (sc-10646, Santa Cruz Biotechnology, Santa Cruz, CA).
Oocyte expression
Xenopus laevis frogs were anesthetized by immersion in a 0.2% tricaine solution and Stage V and VI oocytes collected, injected with ∼50 nl of cRNA, and incubated at 17°C for 3–4 days as previously described (15). Individual oocytes were injected with ether 5 ng of KCNQ1 cRNA alone or with 5 ng KCNQ1 cRNA plus 1 ng of KCNE1 cRNA. For weakly expressing KCNQ1 mutant (L-342A and A-344C) channels, oocytes were injected with 10 ng KCNQ1 cRNA with or without 2 ng of KCNE1 cRNA.
Electrophysiology
Standard two-electrode voltage-clamp techniques (16) were used to record whole cell currents in Xenopus oocytes at room temperature (22°C–23°C). Data were acquired and analyzed using pCLAMP 8.0 software (Axon Instruments, Sunnyvale, CA). Graphic analyses were made using Origin 6.0 software (Microcal-Additive, Friedrichsdorf/Ts, Germany). For voltage clamp experiments, oocytes were bathed in ND96 solution containing (in mM): 96 NaCl, 4 KCl, 1.8 MgC12, 0.1 CaC12, 5 HEPES; pH 7.6. Pipettes were filled with 3 M KCl and had resistances of 0.5–1 MΩ.
For patch clamp measurements in the cell-attached mode, the vitelline layer was removed from the oocytes after short exposure to a hyperosmotic shrinking solution. The cell-attached current measurements were performed as described before (19) using an EPC9 amplifier and Pulse software (HEKA, Lambrecht, Germany). The patch pipettes had resistances between 0.5 and 2 MΩ when filled with a solution containing (in mM): 10 KCl, 90 K-glutamate, 5 MgSO4, 5 Hepes; pH 7.3. The extracellular solution contained (in mM): 10 KCl, 90 K-glutamate, 2 EGTA, 1 MgSO4, 5 Hepes; pH 7.3. In this solution, the resting potential of the oocytes was near 0 mV. Recordings were made at room temperature 3–4 days after injection of oocytes with cRNA. Currents were low-pass filtered at 2.2 kHz and digitized at 6.7 kHz.
Data analysis
Tail current analyses at a potential of −120 mV was used to assess the voltage dependence of KCNQ1 channel activation and inactivation (17,18). Tail currents were fitted to a biexponential equation of the form
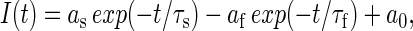 |
with a fast time constant τf and a slow time constant τs, their respective amplitudes, as and af, and a steady-state current, a0. A relatively larger fast component, af, indicates the presence of a more pronounced “hook” in the tail current, representing recovery of channels from inactivation (19) and was of variable magnitude for mutant KCNQ1 channels. The slower component was extrapolated to the start of the tail pulse, and the estimated amplitude was used to determine the voltage dependence of channel activation.
The activation curves obtained by plotting normalized tail current amplitudes (Itail) versus test potential were fitted to a standard Boltzmann equation of the form
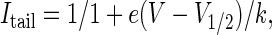 |
where V1/2 is the voltage of half-maximal activation and k is the slope factor.
Nonstationary noise analysis was used to estimate single channel conductance (20). From a holding potential of −70 mV, test pulses were applied to +60 mV for 400 ms, followed by a repolarizing pulse to −100 mV for 800 ms. Tail currents during deactivation were analyzed using custom software (available on website www.ge.cnr.it/ICB/conti_moran_pusch/programs-pusch/software-mik.htm). Variance-current relationships were fitted to
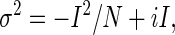 |
where σ2 is the variance, I is the macroscopic current, N is the number of channels, and i is the single channel current amplitude. Open state probability (po) was calculated according to
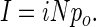 |
Data are presented as mean ± SE.
Molecular modeling
The Kv1.2 structure (17) was retrieved from the NCBI Protein Data Bank (2A79). A three-dimensional (3D) structural model of the S5/H5/S6 domains of KCNQ1 was constructed on homology. The KCNQ1 homology model was generated using Swiss-Model (http://www.expasy.org/swissmod/SWISS-MODEL.html). Energy minimization was performed with GROMOS96 force field.
RESULTS
To determine the potential importance of Ala-336 in gating of KCNQ1 channels, we first mutated this residue to Gly, the amino acid that is present in most other Kv channels, including KCNQ2-5 (Kv7.2–5). The A-336G KCNQ1 channel current was larger than the wild type (WT) KCNQ1 channel current at all test potentials (Fig. 2, A and B). In contrast, mutation of Ala-336 to the larger residues Cys or Thr reduced current magnitude relative to WT channels. Tail current analysis was used to estimate the voltage dependence of channel activation. The activation curve of A-336G channels had a steeper slope and a more negative half-point (V1/2) than WT, A-336C, or A-336T channels (Fig. 2 C). The current magnitude at the highest voltage examined (+70 mV) was inversely related to the volume of the amino acid side chain (Fig. 2 D). The time constants of activation were also dependent on the residue at position 336 (Fig. 2, E and F). The rank order for the time constant of activation was Gly < Ala < Cys = Thr. The rate of deactivation for WT and mutant channels was similar at all potentials except −40 mV, where A-336C and A-336T KCNQ1 channels were slower than WT or A-336G channels (Fig. 2 G). Together these data indicate that current magnitude and the voltage dependence and kinetics of KCNQ1 channel gating is dependent on side-chain volume of the residue located in position 336. The smaller residues Gly and Ala enabled faster gating than the larger residues Cys or Thr. These findings indicate that the presence of a Gly in this key position can facilitate channel gating and current amplitude.
FIGURE 2.

Mutation of Ala-336 alters properties of KCNQ1 channel currents. (A) KCNQ1 currents recorded from Xenopus oocytes. Currents in the upper panel were elicited from a holding potential of −80 mV with pulses applied in 10 mV increments to potentials ranging from −120 to +80 mV. Currents in the lower panel were elicited from a holding potential of −80 mV with pulses to 40 mV and then deactivated by pulses from −140 to −40 mV applied in 20-mV increments. Lower traces were scaled to the same size to facilitate comparison. A-336P resulted in very small currents (∼0.2 μA at 40 mV), which could not be kinetically analyzed. (B) Current-voltage (I-V) relationship for WT and mutant channels. Currents were measured at the end of 2-s pulses and mean values ± SE are depicted (n = 11–13). (C) Voltage dependence of KCNQ1 channel activation determined by tail current analysis. The data were fit to the Boltzmann equation and values were KCNQ1 A-336G: V1/2 = −17.9 ± 0.6, k = 13.2 ± 0.5; KCNQ1 A-336: V1/2 = −3.7 ± 1–2, k = 21.1 ± 1.1; KCNQ1 A-336C: V1/2 = −8.8 ± 1.6, k = 20.4 ± 1.6, and KCNQ1 A-336T: V1/2 = 64.9 ± 13.2, k = 42.7 ± 4.8 (n = 11–13). (D) Currents measured at +70 mV are plotted versus the side-chain volume of the residues at KCNQ1 position 336 (n = 11–13). (E and F) Time constants for activation of KCNQ1 currents. The activating phase of currents was fit to two exponentials, and the fast and slow components were plotted versus voltage (n = 7–13). (G) Effect of mutation of residue Ala-336 on time constant of deactivation (τdeactivation) as a function of voltage (n = 3–13).
To further test the importance of Ala-336 as a gating hinge we mutated this residue to Pro, a change expected to introduce a joint-like hinge into the α-helix. Pro has a side-chain volume that is similar to Thr (Pro: 42 Å3, Thr: 45 Å3) but its side chain is less flexible. The mutant A-336P was nonfunctional (Fig. 2 A), suggesting that the presumed introduced flexure of the S6 α-helix was not conducive to KCNQ1 channel activation.
KCNQ2-5 channels have a Gly in the position equivalent to Ala-336 of KCNQ1. G301A KCNQ2 channel currents could not be detected over a voltage range from −120 to +60 mV (Fig. 3 A). Therefore, we constructed two chimeric channels by introducing the S6 segment from KCNQ2 into KCNQ1 (KCNQ1S6Q2) or the S6 from KCNQ1 into KCNQ2 (KCNQ2S6Q1). KCNQ1S6Q2 channels were functional and had a voltage dependence of activation comparable to WT KCNQ2 (Fig. 3, A and C). However, unlike WT KCNQ1 and WT KCNQ2, these chimeric channels activated faster, inactivated by ∼90% at the end of a 2 s pulse to +40 mV and had a current size comparable to WT KCNQ1 (Fig. 3, A, B, and D). KCNQ2S6Q1 channel currents were large, had a voltage dependence of activation comparable to WT KCNQ1, activated and deactivated faster than WT KCNQ1 and WT KCNQ2, and did not exhibit macroscopic inactivation (Fig. 3, A–E). Finally, introduction of the A-336G equivalent mutation into the KCNQ2S6Q1 chimera enhanced the rates of activation and deactivation but was otherwise similar to the chimeric channel without the putative hinge mutation (Fig. 3, A–E). These data indicate that the putative gating hinge function of this key residue may be dependent on other interactions determined by the entire S6 domain.
FIGURE 3.

KCNQ1-KCNQ2 chimera activate rapidly, inactivate to a different degree, and gate faster with a Gly at the gating hinge position. (A) Shown are current traces from KCNQ1, KCNQ2, KCNQ2 point mutant KCNQ2-G-301A, and KCNQ1/KCNQ2-chimera. The chimera KCNQ1S6Q2 was constructed by splicing the S6 segment from KCNQ2 (black region) into the homologous position in KCNQ1. The chimera KCNQ2S6Q1 was constructed by splicing the S6 segment from KCNQ1 into the homologous position in KCNQ2 resulting in an inverse chimera. Further, in this chimera the Ala at the putative hinge position was mutated to Gly as indicated by the circle in the lower cartoon. Representative current traces of the constructs measured with the same pulse protocols as in Fig. 2 are shown below the cartoon. The horizontal scale bars indicate 1 s, the vertical 5 μA. (B) Peak current amplitudes of KCNQ1 (circles), KCNQ2 (squares), KCNQ1S6Q2 (triangles), Q2-S6Q1 (solid diamonds), and Q2-A-336GS6Q1 (open diamonds) channels were measured and plotted versus voltage. (C) Initial tail current amplitudes were determined and normalized to the maximal initial tail current amplitude. The data were fit to the Boltzmann equation and values were KCNQ2: V1/2 = −23.0 ± 1.0, k = 14.2 ± 0.9, n = 10; Q1S6Q2: V1/2 = −24.8 ± 3.1, k = 10.8 ± 2.7, n = 10; Q2S6Q1: V1/2 = −11.8 ± 1.7, k = 22.3 ± 1.7, n = 7, and Q2S6Q1-A-336G: V1/2 = 0.8 ± 1.7, k = 25.4 ± 1.8, n = 7. Symbols are used as in panel B. (D) The rising/activating phase of KCNQ1, KCNQ2, Q2S6Q1, and Q2A-336GS6Q1 current traces at depolarizing pulses were fit to a single exponential function, and the time constants of channel activation (τactivation) are shown. The rising and decaying phases of KCNQ1S6Q2 current traces at depolarizing pulses were fit to a double exponential. The values of the fast increasing phase describe channel activation. The time constants of activation (τactivation) are plotted versus test pulse voltages. Symbols used as in panel B. (E) The deflection of tail current traces upon channels' closure were fit to a single exponential function, and the time constants (τdeactivation) of channel deactivation are shown. Usage of symbols is as in panel B. Mean data are shown with error bars as mean ± SE.
The observed effects on current amplitudes could be due to an increased incorporation of A-336G KCNQ1 and decreased incorporation of A-336C and A-336T KCNQ1 channels into the plasma membrane. This hypothesis was tested by Western blot analysis of surface KCNQ1 protein. No obvious differences in expression levels of KCNQ1 membrane protein were observed (Fig. 4 A). Therefore, the changes in current density caused by mutation of Ala-336 were more likely caused by mutation-induced alterations in single channel properties. We performed nonstationary noise analysis on WT and A-336G KCNQ1 channels. By analyzing the mean-variance plot of 150 consecutively recorded depolarizing pulses (Fig. 4 B), the A-336G mutant KCNQ1 channels were found to have a significantly increased open probability (0.73, n = 6) compared to WT channels (0.49, n = 9) (Fig. 4 C). The single channel amplitude (i) declined slightly, but not significantly, from 0.46 ± 0.04 pA for WT to 0.32 ± 0.05 pA for A-336G (Fig. 4 D).
FIGURE 4.

Western blot demonstrating surface protein expression and noise analysis of the KCNQ1 channels. (A) Samples including controls from uninjected oocytes were separated on an SDS gel, Western-blotted, and probed with an affinity-purified goat polyclonal KCNQ1 antibody directed against a peptide mapping at the carboxy terminus of KCNQ1. KCNQ1 protein has an apparent molecular weight of ∼78 kDa. Panel shows a Western blot of KCNQ1 membrane protein separated by biotinylation. (B) Plot of mean current versus variance for WT KCNQ1 tail currents measured in cell-attached macropatches. (C and D) Open probability (po) and single channel current amplitude (i) estimated by noise analysis. *P < 0.05.
Coexpression of KCNQ1 with the β-subunit KCNE1 produced a slow delayed rectifier K+ current, IKs, with increased current magnitude, a slowed rate of activation, no apparent inactivation, and an increased single channel conductance (21–24). We tested the hypothesis that some of the effects mediated by KCNE1 were due to its interaction with the putative gating hinge of KCNQ1. WT and mutant KCNQ1 channels were coexpressed with KCNE1 and the resulting IKs was recorded using 7-s pulses to potentials ranging from −70 to +70 mV. The slow kinetics of activation of the mutant IKs channels were similar, but the magnitude of the heteromeric WT and mutant IKs channel currents exhibited the same rank order as that observed for homotetrameric KCNQ1 channels: A-336G > WT > A-336C > A-336T (Fig. 5, A and B). The increase in current as a function of the volume of the residue side-chain volume for IKs (Fig. 5 C) was similar to the relationship noted for KCNQ1 alone (Fig. 2 D). The increase in current magnitude induced by coexpression with KCNE1 was ∼10-fold for WT and the mutant channels (Fig. 5 D). Coexpression of KCNE1 shifts the voltage dependence of KCNQ1 activation to more positive potentials, and this gating effect was not markedly altered by mutation of A-336 (Fig. 5 E). Thus, coexpression with KCNE1 did not alter the effects on KCNQ1 channel function associated with mutation of the putative gating hinge residue.
FIGURE 5.

WT and mutant KCNQ1/KCNE1 (IKs) channel currents. (A) KCNQ1 was coexpressed with KCNE1 in Xenopus oocytes. IKs was elicited from a holding potential of −80 mV by 7-s pulses applied in 10-mV increments to potentials ranging from −120 to +70 mV. Tail currents were elicited by stepping to −120 mV. (B) I-V relationship for peak currents (n = 17–20). (C) Currents at +70 mV plotted versus side-chain volume of residue 336 (n = 17–20). (D) Fold-increase in KCNQ1 current magnitude (+70 mV) induced by coexpression with KCNE1 plotted versus side-chain volume of residue 336 (n = 11–20). (E) Normalized peak tail I-V relationship for IKs (n = 11–25). V1/2 values were A-336G/KCNE1 = 37.6 ± 1.9 mV, n = 11; A-336/KCNE1 = 48.3 ± 1.3 mV, n = 11; A-336C/KCNE1 = 43.1 ± 2.6 mV, n = 8; A-336T/KCNE1 = 41.1 ± 1.8 mV, n = 9.
Another key region identified as important for Kv channel gating is the Pro-Val(Ile)-Pro motif located near the intracellular end of the S6 domain (13,25). In KCNQ1 channels this motif (residues 343–345) is modified to Pro-Ala-Gly (Fig. 6 A). We previously reported that mutation of Pro-343 or Gly-345 to Ala rendered the channels nonfunctional (26). In Fig. 6 B we show that P-343A and G-345A mutant channels are expressed at the surface membrane, strongly suggesting that mutation of these residues induces functional rather than trafficking defects. Mutation of the flanking residues L-342 and I-346 channels were also characterized (Fig. 6 B). I-346A channels were nonfunctional, whereas L-342A channels expressed at about a threefold reduced level. The half point of L-342A channel activation was −43 mV (n = 6), and coexpression with KCNE1 induced the normal changes in biophysical properties, including slowed activation and increased current magnitude and caused a positive shift (∼15 mV) in the voltage dependence of activation (Fig. 6, C and D). A-344C channels also functionally expressed, albeit with very small currents (Fig. 6 E) and coexpression, with KCNE1 also led to the usual changes in biophysical properties characteristic of IKs (Fig. 6 F, n = 6). Thus, mutation of the Pro or Gly but not Ala in the PAG motif of KCNQ1 prevents channel activation.
FIGURE 6.

Mutations of residues in or near the S6 PAG motif of KCNQ1 channels. (A) sequence of the KCNQ1 S6 transmembrane segment, highlighting location of the putative gating hinge and the PAG motif. The crosses indicate positions where mutation to Ala resulted in constructs lacking functional expression in Xenopus oocytes (16). (B) Samples including controls from uninjected oocytes were separated on an SDS gel and KCNQ1 membrane protein separated form intracellular protein by biotinylation and was probed by Western blot under the same conditions as in Fig. 4 A. (C) Current traces for L-342A KCNQ1 alone and coexpressed with KCNE1. The holding potential was −80 mV, and test pulses were to voltages from −100 to +50 mV, applied in 10-mV increments; tail currents were elicited at −60 mV. The horizontal scale bars indicate 1 s; the vertical 0.5 μA. (D) I-V relationship for peak tail currents normalized to the maximal value (n = 5–6). (E) Current traces for A344C KCNQ1 alone and coexpressed with KCNE1. (F) I-V relationship for peak tail currents normalized to the maximal value (n = 3–4).
We introduced Gly next to the P-343A and G-345A mutation to reintroduce a flexible residue next to the mutated Pro/Gly in hKCNQ1 (Fig. 7 A). This approach was used to test if flexibility of the PAB-motif is needed to generate functional channels. The resulting channels KCNQ1-LP342/343GA and SG344/345GA were functional, whereas KCNQ1-AG344/345GA was nonfunctional (Fig. 7 B). The channels KCNQ1-LP342/343GA and SG344/345GA activated with a similar nonsigmoidal shaped activation curve, indicating similar effects on KCNQ1-activation gating (Fig. 7 C). We generated a KCNQ1-3D-model on the basis of the recently solved Kv1.2 crystal structure (17). Inspection of the PAG-region (APAGIL347) suggests intersubunit interactions of Ile-346 with Glu-261 and Leu-347 with Leu-262 and intersubunit interactions of Leu-347 with Leu-342 (Fig. 7 D).
FIGURE 7.

Introduction of glycine can recover part of the function of alanine mutations of residues in PAG motif of KCNQ1 channels. (A) Sequence of the KCNQ1 S6 transmembrane segment, highlighting the PAG region and indicating positions of double point mutations. (B) Current traces for KCNQ1 double mutants and uninjected oocytes. The holding potential was −80 mV and test pulses were to voltages from −120 to 60 mV, applied in 20-mV increments, and tail currents were elicited at −120 mV. Horizontal scale bars indicate 1 s; the vertical 1 μA. (C) I-V relationship for peak tail currents normalized to the maximal value of KCNQ1-LP342/343GA (square) and KCNQ1-AG344/345GA (circle) (n = 18/12). Data did not show a sigmoidal shaped curve and therefore were not fitted to the Boltzmann function. Data as mean and error bars represent mean ± SE. (D) S5 α-helical segment, pore loop, and S6 α-helical segment of two adjacent KCNQ2 subunits. Homology models of hKCNQ1 based on the solved structure of Kv1.2 of the S5–S6 region. Lower panel shows a magnified view on possibly interacting residues. Leu-347 might interact with Leu-342 from an adjacent subunit and with Leu-262 within the same subunit. Ile-346 could interact with Cβ/Cγ of Glu-346. Ala-mutations of the PAG motif might be expected to disrupt these interactions.
DISCUSSION
Two different regions of the S6 domain, a Gly hinge and a Pro-Val-Pro motif, have been proposed to be of central relevance in Kv channel gating. We investigated the homologous regions in KCNQ1 and KCNQ2. Mutation of Ala-336 to a Gly residue enhances channel opening of KCNQ1 channels, measured as a leftward shift in the voltage dependence of activation and kinetics of whole-cell currents, and an increased open probability of single channels estimated by nonstationary noise analysis. These findings are consistent with residue 336 acting as a hinge point for outward splaying of S6 during channel activation. Although mutation of Ala-336 to a Cys or Thr hampered channel opening, it was surprising that these channels were functional at all, given the proposed requirement for bending of S6 at this position. The presence of a Cys or Thr would prevent helix distortion or bending of the S6 domain. Thus, although hinging of S6 at the precise position 336 can facilitate opening, it is not required for KCNQ1 channel function. Instead we find a dependence of macroscopic currents on the side-chain volume (Figs. 2 D and 5 C) and indeed a kink might even disrupt normal activation gating when the volume of the kink residue is large. This is indicated by the nonfunctionality of mutant A-336P, which can be expected to force a rigid artificial kink into the S6-helix while introducing a relatively large Pro. However, based on our data it cannot be excluded that the S6-helix can bend over an extended area without a precise hinge at residue 336. Mutation of the residue analogous to Ala-336 of KCNQ1 in Shaker and G-protein activated GIRK K+ channels, and the bacterial Na+ channel NaChBac yielded channels with strongly open-state shifted open/close state equilibria (27–30). Interestingly, the recently published Kv1.2 crystal structure exhibits an open channel conformation without any notable kink at the Gly hinge (31). Therefore, KCNQ1 might open like Kv1.2, without the need of a gating hinge.
KCNQ2-5 channels have a Gly at the putative hinge position (Fig. 1 A). Introduction of the closely related KCNQ2 S6 sequence into the homologous region of KCNQ1 produced channels with faster activation and deactivation kinetics, largely resembling the phenotype obtained by introduction of Gly at position Ala-336. It is likely the KCNQ2-S6 segment surrounded by KCNQ1 channel protein also introduces the same flexibility at the gating hinge as the Gly in A-336G KCNQ1 channels. Furthermore, the chimeric channel current exhibited strong inactivation. KCNQ1 channels were proposed to inactivate by a mechanism involving a fast flickering block event of the second of two open states (32). Because fast gating involves the selectivity filter, an increased fast flickery block of the second open state in the chimera channel could represent a functional link between the S6 segment and the selectivity filter.
KCNQ2 did not tolerate the relatively small change in structure associated with mutation of the Gly hinge to an Ala residue, suggesting an unexpected but important difference in the mechanism of gating between KCNQ1 and other KCNQ channels that may also involve inactivation. Inserting the KCNQ1 S6 region into a KCNQ2 backbone resulted in noninactivating channels with large currents. These chimeric channels activated and deactivated faster when the putative hinge residue was a Gly. These findings underscore the idea that not only the specific residue at the putative gating hinge position, but also its immediate environment determines the importance of a hinge in KCNQ channel gating. Perhaps the coupling between voltage sensor movement and channel opening is tighter for KCNQ1 than KCNQ2. Although KCNQ2 did not tolerate mutation of the gating hinge Gly to Ala, other channels are like KCNQ1 in that they can either tolerate mutation of the Gly to Ala (e.g., hERG, 33) or have an Ala as the native residue in the hinge position (e.g., Drosophila EAG). The Gly hinge in the S6 domain is also a critical component of normal gating of Na+ channels. Mutation of Gly-219 to Pro in the sixth-transmembrane domain of the bacterial Na+ channel NaChBac stabilizes the open state as indicated by a far more negative voltage range for activation and greatly impaired inactivation (30). Mutation of Gly-219 in NaChBac to an Ala had relatively minor effects on channel activation, similar to our observation of Gly versus Ala in the hinge position of KCNQ1. Together, these studies emphasize that despite the fact that the Gly hinge is highly conserved, the qualitative importance of the S6 hinge at this position is variable.
In a recent study it was proposed that a Gly hinge could be of some relevance for the functional interaction of KCNQ1 α-subunits with KCNE1 β-subunits (34). However, IKs induced by coassembly of A-336X KCNQ1 and WT KCNE1 subunits exhibited only minor changes in the voltage dependence or kinetics compared to WT IKs, suggesting that KCNE1 subunits altered channel gating by a mechanism independent of a putative gating hinge.
Although we did not determine the mechanism of altered current amplitudes resulting from mutation of KCNQ1 Ala-336 in detail, Western blot analysis of surface protein levels suggested a small change in surface expression of mutant versus WT KCNQ1 channels. Alterations in open probability po were suggested in the case of KCNQ1 A-336G compared to WT KCNQ1. Variable flexibility of S6 at the gating hinge caused by mutation of Ala-336 could lead to differences in the rate of opening and the diameter of the intracellular aperture of the pore in the open state (Fig. 1 C). Voltage-dependent channel opening from the energetically favored closed state to the open state conformation is enabled by positive work of the voltage sensor which might disrupt a hydrogen bonding network (11,35). Ala-336 might stabilize the hydrogen bonding network, thereby increasing the energy required for the closed state to open state transition as suggested from a modeling approach (11). Using Brownian dynamics on a simplified model of the KcsA structure, Chung et al. (36) predicted that reduction of the size of the cytosolic entrance to the central cavity would lead to a reduced conductance. These model predictions are in agreement with our experimental data and would help to explain the differences in current magnitude of the mutant KCNQ1 channels. However, the current amplitude of the KCNQ2 chimera with the S6 from KCNQ1 (KCNQ2S6Q1) was not altered when the putative gating hinge residue was mutated from Ala to Gly. Mutation of the Pro or Gly residues in the PAG motif of KCNQ1 prevented channel activation. Pro residues are known to induce a kink and/or swivel point in transmembrane helices (37), and our findings are consistent with the proposal that KCNQ1 channel activation, like many other Kv channels (14), involves a distortion of S6 at or near the Pro residue located near the intracellular end of the pore. 3D-homology modeling suggests inter- and intrasubunit interactions around the PAG motif which might be disrupted by mutation in and around the PAG and might explain the dramatically reduced function of L-342A, P-343A, G-345A, I-346A, and L-347A (see model in Fig. 7 D). The partial regain of function of the mutants KCNQ1-P-343A and G-345A by introduction of the flexible amino acid Gly shows that flexibility of the PAG motive is important for channel function. Maybe the reintroduced flexibility by the Gly-mutations enables an approximate interaction of the residues identified in the model (Fig. 7 D). An extensive study by Labro et al. (38) evaluated the importance of Pro residues in the gating of Kv1.5 channels and demonstrated that Pro residues located at several positions in the S6 domain could substitute for the native PVP motif. Labro et al. (38) also showed that mutation of the second Pro in the Kv1.5 PVP motif to a Gly or Ala residue caused a +58 and +104 mV shift, respectively, in the voltage dependence of channel activation. Thus, although an Ala in the third position of the PVP motif is tolerated in Kv1.5, this mutation greatly impairs normal activation, indicating an important difference between the molecular mechanism of Kv1.5 and KCNQ1 channel activation. Moreover, the central importance of the Gly hinge in Shaker channels was challenged by formation of metal bridges in positions which suggest a much less wide opening of the bundle crossing but high importance of the PVP motif (14). These results are in good agreement with our findings for KCNQ1 channels.
In summary, our findings indicate that the presence of a Gly in the S6 domain is important for the gating of KCNQ2 but not KCNQ1 channels, which has an Ala residue (Ala-336) in the equivalent position. The activation of KCNQ1 appears to be more dependent on the PAG motif located closer to the C-terminus of the S6 domain by acting as a bending point as previously proposed for Shaker channels (10).
Acknowledgments
We gratefully acknowledge support from a Heisenberg-fellowship (to H.L.) and from grant DFG-Le1030/9-1 (to F.L.).
References
- 1.Doyle, D. A., C. J. Morais, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- 2.Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- 3.Jiang, Y., A. Lee, J. Chen, V. Ruta, M. Cadene, B. T. Chait, and R. MacKinnon. 2003. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- 4.Ahern, C. A., and R. Horn. 2004. Specificity of charge-carrying residues in the voltage sensor of potassium channels. J. Gen. Physiol. 123:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang, Q. X., D. N. Wang, and R. MacKinnon. 2004. Electron microscopic analysis of KvAP voltage-dependent K+ channels in an open conformation. Nature. 430:806–810. [DOI] [PubMed] [Google Scholar]
- 6.Laine, M., D. M. Papazian, and B. Roux. 2004. Critical assessment of a proposed model of Shaker. FEBS Lett. 564:257–263. [DOI] [PubMed] [Google Scholar]
- 7.Starace, D. M., and F. Bezanilla. 2004. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature. 427:548–553. [DOI] [PubMed] [Google Scholar]
- 8.Cuello, L. G., D. M. Cortes, and E. Perozo. 2004. Molecular architecture of the KvAP voltage-dependent K+ channel in a lipid bilayer. Science. 306:491–495. [DOI] [PubMed] [Google Scholar]
- 9.Durell, S. R., I. H. Shrivastava, and H. R. Guy. 2004. Models of the structure and voltage-gating mechanism of the Shaker K+ channel. Biophys. J. 87:2116–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shrivastava, I. H., S. R. Durell, and H. R. Guy. 2004. A model of voltage gating developed using the KvAP channel crystal structure. Biophys. J. 87:2255–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Treptow, W., B. Maigret, C. Chipot, and M. Tarek. 2004. Coupled motions between pore and voltage-sensor domains: a model for Shaker B, a voltage-gated potassium channel. Biophys. J. 87:2365–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shealy, R. T., A. D. Murphy, R. Ramarathnam, E. Jakobsson, and S. Subramaniam. 2003. Sequence-function analysis of the K+-selective family of ion channels using a comprehensive alignment and the KcsA channel structure. Biophys. J. 84:2929–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.del Camino, D., M. Holmgren, Y. Liu, and G. Yellen. 2000. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 403:321–325. [DOI] [PubMed] [Google Scholar]
- 14.Webster, S. M., D. del Camino, J. P. Dekker, and G. Yellen. 2004. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 428:864–868. [DOI] [PubMed] [Google Scholar]
- 15.Seebohm, G., C. R. Scherer, A. E. Busch, and C. Lerche. 2001. Identification of specific pore residues mediating KCNQ1 inactivation. A novel mechanism for long QT syndrome. J. Biol. Chem. 276:13600–13605. [DOI] [PubMed] [Google Scholar]
- 16.Stühmer, W. 1992. Electrophysiological recording from Xenopus oocytes. Methods Enzymol. 207:319–339. [DOI] [PubMed] [Google Scholar]
- 17.Pusch, M. 1998. Increase of the single-channel conductance of KvLQT1 potassium channels induced by the association with minK. Pflugers Arch. 437:172–174. [DOI] [PubMed] [Google Scholar]
- 18.Tristani-Firouzi, M., and M. C. Sanguinetti. 1998. Voltage-dependent inactivation of the human K+ channel KvLQT1 is eliminated by association with minimal K+ channel (minK) subunits. J. Physiol. 510:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pusch, M., R. Magrassi, B. Wollnik, and F. Conti. 1998. Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 75:785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinemann, S. H., and F. Conti. 1992. Nonstationary noise analysis and application to patch clamp recordings. Methods Enzymol. 207:131–148. [DOI] [PubMed] [Google Scholar]
- 21.Barhanin, J., F. Lesage, E. Guillemare, M. Fink, M. Lazdunski, and G. Romey. 1996. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 384:78–80. [DOI] [PubMed] [Google Scholar]
- 22.Sanguinetti, M. C., M. E. Curran, A. Zou, J. Shen, P. S. Spector, D. L. Atkinson, and M. T. Keating. 1996. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 384:80–83. [DOI] [PubMed] [Google Scholar]
- 23.Sesti, F., and S. A. Goldstein. 1998. Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J. Gen. Physiol. 112:651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang, Y., and F. J. Sigworth. 1998. Single-channel properties of IKs potassium channels. J. Gen. Physiol. 112:665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holmgren, M., K. S. Shin, and G. Yellen. 1998. The activation gate of a voltage-gated K+ channel can be trapped in the open state by an intersubunit metal bridge. Neuron. 21:617–621. [DOI] [PubMed] [Google Scholar]
- 26.Seebohm, G., J. Chen, N. Strutz, C. Culberson, C. Lerche, and M. C. Sanguinetti. 2003. Molecular determinants of KCNQ1 channel block by a benzodiazepine. Mol. Pharmacol. 64:70–77. [DOI] [PubMed] [Google Scholar]
- 27.Jin, T., L. Peng, T. Mirshahi, T. Rohacs, K. W. Chan, R. Sanchez, and D. E. Logothetis. 2002. The (beta)gamma subunits of G proteins gate a K(+) channel by pivoted bending of a transmembrane segment. Mol. Cell. 10:469–481. [DOI] [PubMed] [Google Scholar]
- 28.Magidovich, E., and O. Yifrach. 2004. Conserved gating hinge in ligand- and voltage-dependent K+ channels. Biochemistry. 43:13242–13247. [DOI] [PubMed] [Google Scholar]
- 29.Sadja, R., K. Smadja, N. Alagem, and E. Reuveny. 2001. Coupling Gbetagamma-dependent activation to channel opening via pore elements in inwardly rectifying potassium channels. Neuron. 29:669–680. [DOI] [PubMed] [Google Scholar]
- 30.Zhao, Y., V. Yarov-Yarovoy, T. Scheuer, and W. A. Catterall. 2004. A gating hinge in Na+ channels; a molecular switch for electrical signaling. Neuron. 41:859–865. [DOI] [PubMed] [Google Scholar]
- 31.Long, S. B., E. B. Campbell, and R. MacKinnon. 2005. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 309:897–903. [DOI] [PubMed] [Google Scholar]
- 32.Seebohm, G., M. C. Sanguinetti, and M. Pusch. 2003. Tight coupling of rubidium conductance and inactivation in human KCNQ1 potassium channels. J. Physiol. 552:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitcheson, J. S., J. Chen, M. Lin, C. Culberson, and M. C. Sanguinetti. 2000. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. USA. 97:12329–12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melman, Y. F., S. Y. Um, A. Krumerman, A. Kagan, and T. V. McDonald. 2004. KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron. 42:927–937. [DOI] [PubMed] [Google Scholar]
- 35.Yifrach, O., and R. MacKinnon. 2002. Energetics of pore opening in a voltage-gated K(+) channel. Cell. 111:231–239. [DOI] [PubMed] [Google Scholar]
- 36.Chung, S. H., T. W. Allen, and S. Kuyucak. 2002. Modeling diverse range of potassium channels with Brownian dynamics. Biophys. J. 83:263–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cordes, F. S., J. N. Bright, and M. S. Sansom. 2002. Proline-induced distortions of transmembrane helices. J. Mol. Biol. 323:951–960. [DOI] [PubMed] [Google Scholar]
- 38.Labro, A. J., A. L. Raes, I. Bellens, N. Ottschytsch, and D. J. Snyders. 2003. Gating of Shaker-type channels requires the flexibility of S6 caused by prolines. J. Biol. Chem. 278:50724–50731. [DOI] [PubMed] [Google Scholar]