

Abstract
Two recent papers provide new evidence relevant to the role of the breast cancer susceptibility gene BRCA2 in DNA repair. Moynahan et al provide genetic data indicating a requirement for BRCA2 in homology-dependent (recombinational) repair of DNA double-strand breaks. The second paper, by Davies et al, begins to address the mechanism through which BRCA2 makes its contribution to recombinational repair. BRCA2 appears to function in recombination via interactions with the major eukaryotic recombinase RAD51 [1,2,3]. We briefly review the context in which the two studies were carried out, we comment on the results presented, and we discuss models designed to account for the role of BRCA2 in RAD51–mediated repair.
Keywords: breast cancer, homologous recombination
BRCA2
BRCA2 was the second breast cancer susceptibility gene to be discovered, and was isolated through positional cloning using data from families with inherited breast cancer [4]. Cells with mutant BRCA2 protein are, like many cancer cells, genetically unstable and accumulate gross chromosomal rearrangements [5,6]. The sequence of this large protein (3418 amino acids) offers very little clue to its function, although there are eight repeated segments (termed BRC repeats) in the middle of the protein that are highly conserved among mammalian orthologs [7,8].
Breast cancer susceptibility genes and DNA repair
Following the landmark discovery by Scully et al that the homologous recombinase RAD51 colocalizes at subnuclear sites with BRCA1 [9], a number of additional results have provided evidence that both BRCA1 and BRCA2 are involved in recombinational repair of DNA damage. BRCA1 and BRCA2 form discrete nuclear foci during the S phase and after exposure to DNA damaging agents [9,10,11]. These foci are probably sites of repair of spontaneous and induced DNA damage [12,13,14]. Cell lines defective in either BRCA1 or BRCA2 are sensitive to damaging agents that form double-strand breaks (DSBs), as are other cell lines defective in recombinational repair (reviewed in [15]). BRCA2 interacts with the RAD51 recombinase via direct protein-protein contacts [16,17,18,19]. Biochemical analysis also showed interaction between BRCA1 and RAD51, although these detected interactions may have been indirect [9]. The BRC repeats of BRCA2 are responsible for direct RAD51 interaction. Cells lacking BRCA1/2 fail to form damage-induced subnuclear RAD51 foci with normal efficiency, suggesting that these proteins are required for the formation of recombinase complexes at the sites of DNA damage [20,21]. Finally, genetic measurements of recombination frequency have shown that Brca1-/- embryonic stem (ES) cells are deficient in recombinational DSB repair [22,23]. The similarity between phenotypes associated with BRCA1 and BRCA2 deficiency, together with data showing a similar effect of DNA damage on distribution of BRCA1 and BRCA2 in repair-proficient cells, led to the hypothesis that BRCA2, like BRCA1, is required for efficient recombinational repair. The paper by Moynahan et al [24] provides important support for this hypothesis.
Measuring DSB-induced recombination frequency in BRCA2-defective cells
Pierce et al have designed a set of recombination substrates for measuring the level of homologous recombination in vivo (Fig. 1) [25]. The DNA substrate contains a pair of mutated GFP genes (GFP encodes the easily detected green fluorescent protein), one of which contains a restriction site for I-SceI, a yeast intron encoded endonuclease with an 18 base pair recognition site. Transient transfection of an I-SceI expression vector results in the production of a DSB in the first mutated copy of GFP. One or both DNA ends formed by the break invade(s) the homologous sequence in the second mutant GFP copy, resulting in repair of the DSB via a homology-mediated gene conversion event. The configuration of the GFP construct is such that homology-mediated repair often results in the formation of a functional copy of GFP. Such events can be detected by fluorescence-activated cell sorting analysis by virtue of their expression of GFP.
Figure 1.
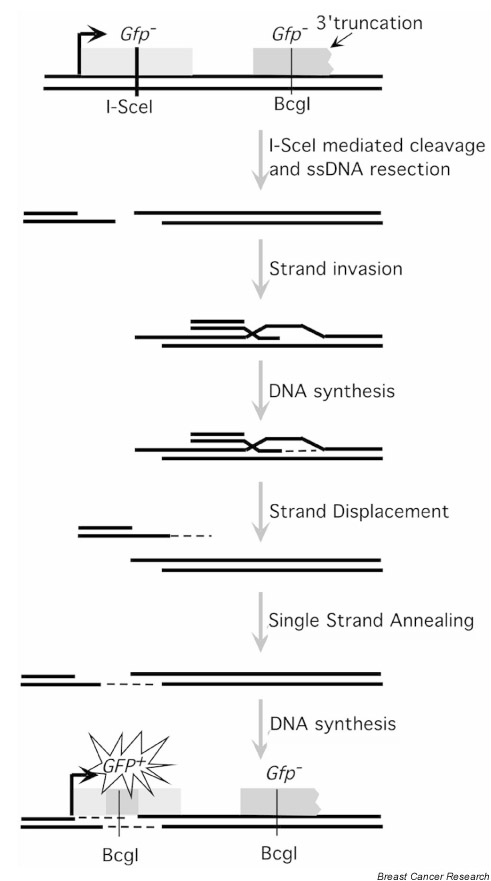
Recombination substrates used for assaying homology-directed repair. Cutting at the I-SceI site within the mutant GFP (SceGFP) results in a double-strand break that can be repaired through homologous gene conversion using a 3'-truncated copy of GFP as sequence donor. The mechanism results in the formation of a functional copy of the GFP gene. The model shown assumes gene conversion occurs via the synthesis-dependent annealing mechanism.
Moynahan et al [24] have used such a GFP recombination substrate to demonstrate that cells with defective BRCA2 protein are deficient in their ability to repair the I-SceI-induced DSB through homologous recombination. Expression of I-SceI resulted in 1 out of 1400 cells producing GFP via homologous recombination in the human pancreatic tumor cell line CAPAN-1. CAPAN-1 cells carry a deletion of BRCA2 on one homolog and codes for a protein truncated at amino acid 1981 on the other homolog. The authors indicate that the level of I-SceI-induced recombination in CAPAN-1 is over 100-fold less than that seen using other (BRCA2+) human tumor cell lines. The different lines examined, however, are very likely to differ genetically from CAPAN-1 cells not only at the BRCA2 locus, but also at a very large number of additional loci. This raises the possibility that some or even all of the recombinational repair defect seen in CAPAN-1 could be due to mutations at non-BRCA2 loci.
While Moynahan et al were careful to point out this problem, two sets of results argue against the possibility that the recombinational repair deficiency of CAPAN-1 cells is completely independent of its defect in BRCA2. First, in the same study [24], recombination in a mouse ES cell line that expresses only truncated BRCA2 protein was measured. This line was found to have lower recombination efficiency than isogenic cells expressing full-length BRCA2 (the defect observed was about fivefold to sixfold). Second, in an independent study, Powell was able to compare derivatives of CAPAN-1 that differed only in their ability to express full-length BRCA2 protein (S. Powell, personal communication, 2001). In these experiments, the derivative expressing full-length BRCA2 yielded 10-fold more recombinants than controls expressing only truncated BRCA2 (S. Powell, personal communication, 2001). While the experimental design of these experiments was somewhat different from that of Moyna-han et al [24], the results raise the possibility that CAPAN-1 cells have more than one mutation that lowers the efficiency of recombinational repair relative to that observed in other human cell lines. Conversely, the BRCA2 defect in CAPAN-1 could be fully responsible for the 100-fold defect in recombination if the level of BRCA2 complementation observed by Powell was incomplete. Furthermore, the discrepancy between the 100-fold difference between CAPAN-1 and the other human lines as compared with the fivefold to sixfold difference between the Brca2lex1/lex2cells and Brca2+/+ ES cells might simply be accounted for by the fact that Brca2lex1/lex2 is not a null allele. Additional studies are likely to shed light on the efficiency of BRCA2-independent recombination pathways. Taken together, the results indicate that BRCA2 is indeed required for high levels of recombinational repair in both human and mouse.
The function of BRCA2-RAD51 interaction
The second paper providing evidence, by Davies et al [26], is a biochemical study of the interaction between the homologous recombinase RAD51 and a peptide consisting of one of the eight BRC repeats from human BRCA2. Expression of a single BRC repeat (BRC4) had previously been reported to act as an inhibitor of DNA repair by Chen et al [27]. These investigators showed that expression of constructs containing the BRC4 repeat in MCF-7 cells enhances the radiosensitivity of cells and blocks both the G2/M delay associated with damage and the ability of the transfected cells to assembly subnuclear RAD51 foci. In the paper by Davies et al, data provide evidence for a plausible mechanism of the dominant negative effect associated with expression of a single BRC repeat. DNA binding assays and electron microscopy methods were used to show that a BRC3 peptide interferes with the ability of RAD51 to assemble into oligomeric filaments on DNA. A BRC4 peptide was also reported to inhibit the RAD51–DNA interaction. These experiments raise the possibility that the full-length BRCA2 protein could act as a negative regulator of inappropriate RAD51–DNA interaction.
The final experiment reported by Davies et al [26] is particularly important with respect to understanding the mechanism(s) through which BRCA2 promotes RAD51-dependent recombination. The localization of RAD51 was examined in CAPAN-1 cells, the same human tumor line examined by Moynahan et al [24]. It had been shown previously that the small amount of truncated BRCA2 protein expressed by CAPAN-1 cells was mislocalized to the cytoplasm, consistent with the fact that the truncation protein lacks a nuclear localization signal [28]. Davies et al showed CAPAN-1 cells to be defective in nuclear localization of RAD51, raising the possibility that RAD51 is normally carried to the nucleus by binding BRCA2. It is possible that RAD51 may alternatively be capable of BRCA2-independent transport, but mislocalization of BRCA2 to the cytoplasm may sequester RAD51 and block its normal mode of transport. Support for the hypothesis that the observed mislocalization of RAD51 is an indirect consequence of the BRCA2 defect requires reintroduction of functional BRCA2 into CAPAN-1.
Given the elaborate nature of the RAD51–BRCA2 interaction, it seems somewhat unlikely that the only role of BRCA2 in promoting recombination is to serve as a specific nuclear transporter of RAD51. An attractive alternative, favored by Davies et al [26], is that the association of RAD51–BRCA2 is required to maintain RAD51 in a form that is in a state of 'readiness' (Fig. 2a). In the absence of this interaction with BRCA2, RAD51 might exist in a form that is not capable of being recruited into a functional repair complex when damage occurs. For example, previous studies indicated that RAD51 shows little binding preference for single-strand DNA (ssDNA) over double-strand DNA (dsDNA) [3,29]. This result could mean that regulatory factors, such as BRCA2, are required to suppress RAD51–dsDNA interactions to prevent sequestration of RAD51 in a nonfunctional form. Alternatively, or in addition, suppression of RAD51–ssDNA interaction might be important for preventing RAD51 from binding to the regions of ssDNA that form during normal DNA replication. BRCA2 may thus promote RAD51 assembly into recombinational repair complexes via a negative regulatory mechanism (i.e. by blocking RAD51–BRCA2 interaction until damage has occurred and factors required for 'productive' assembly of RAD51 at damaged sites are in place). As mentioned previously, the ability of a BRC peptide to repress the RAD51–BRCA2 interaction could reflect a role of full-length protein in suppressing unwanted RAD51–BRCA2 interactions.
Figure 2.
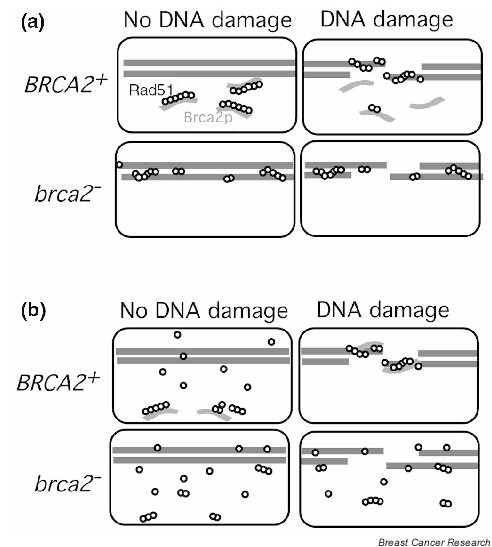
Potential roles of BRCA2 in promoting assembly of Rad51 at sites of DNA damage. Chromosomal DNA is shown as pairs of straight lines, Rad51 as open circles, and BRCA2 as grey bars. (a) Prevention of nonproductive DNA interactions. BRCA2–Rad51 interaction is proposed to suppress RAD51–BRCA2 interactions until DNA damage is present. When damage occurs, Rad51 is recruited to damaged sites where is polymerizes into nucleoprotein filaments. In this model, BRCA2 is not required for assembly of functional complexes at damaged sites, only to prevent a substantial fraction of Rad51 from being sequestered in a nonfunctional form. In a BRCA2-defective cell, mutant Rad51 becomes associated with DNA at random sites and is therefore not readily recruited to sites of damage. (b) Positive regulation. BRCA2 is proposed to be required for Rad51 to assemble into functional recombinational repair complexes at sites of damage. In BRCA-defective cells, Rad51 fails to associate with sites of damage due to lack of an assembly factor.
Yet another potential role for BRCA2 in promoting RAD51-dependent recombinational repair is a positive role in assembly of RAD51 at damaged sites (Fig. 2b). Several proteins that interact with RAD51 are thought to 'mediate' assembly of RAD51 at sites of DNA damage (reviewed in [30]). RAD51 paralogs in particular, including XRCC2, XRCC3, RAD51B, RAD51C and RAD51D, may function as a complex that actively promotes RAD51 assembly [31]. BRCA2 could cooperate with RAD51 paralogs in promoting RAD51 assembly, or even provide an alternative assembly pathway. The observation that BRCA2 localizes to damage-induced subnuclear foci with RAD51 seems consistent with the possibility that BRCA2 plays a positive role in assembly of the recombination complex [11].
Finally, it is important to note that the two models for the function of the BRCA2–RAD51 interaction shown in Figure 2 are not mutually exclusive. BRCA2 could prevent inappropriate RAD51 assembly in the absence of damage and promote functional RAD51 assembly at DNA lesions during the damage response. Further experiments are clearly required to clarify the functional interactions between BRCA2 and RAD51 during recombinational repair. It should be relatively straightforward to determine whether the role of BRCA2 is limited to transporting RAD51 to the nucleus. If this were the case, addition of a nuclear localization signal to RAD51 should suppress the RAD51-mediated phenotypes associated with a BRCA2 mutation. However, biochemical experiments are needed to determine whether full-length BRCA2 blocks or promotes RAD51–BRCA2 interaction. Such experiments could also reveal additional roles for BRCA2 in recombinational repair. However, several technical obstacles, including the large size of BRCA2, promise to make biochemical characterization of BRCA2 function difficult.
Abbreviations
DSB = double-strand break; dsDNA = double-strand DNA; ES = embryonic stem; GFP = green fluorescent protein; ssDNA = single-strand DNA.
References
- Shinohara A, Ogawa H, Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992;69:457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science. 1994;265:1241–1243. doi: 10.1126/science.8066464. [DOI] [PubMed] [Google Scholar]
- Baumann P, Benson FE, West SC. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell. 1996;87:757–766. doi: 10.1016/s0092-8674(00)81394-x. [DOI] [PubMed] [Google Scholar]
- Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- Patel KJ, Yu VP, Lee H, Corcoran A, Thistlethwaite FC, Evans MJ, Colledge WH, Friedman LS, Ponder BA, Venkitaraman AR. Involvement of Brca2 in DNA repair. Mol Cell. 1998;1:347–357. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- Yu VP, Koehler M, Steinlein C, Schmid M, Hanakahi LA, van Gool AJ, West SC, Venkitaraman AR. Gross chromosomal rearrangements and genetic exchange between nonhomolo-gous chromosomes following BRCA2 inactivation. Genes Dev. 2000;14:1400–1406. [PMC free article] [PubMed] [Google Scholar]
- Bignell G, Micklem G, Stratton MR, Ashworth A, Wooster R. The BRC repeats are conserved in mammalian BRCA2 proteins. Hum Mol Genet. 1997;6:53–58. doi: 10.1093/hmg/6.1.53. [DOI] [PubMed] [Google Scholar]
- Bork P, Blomberg N, Nilges M. Internal repeats in the BRCA2 protein sequence. Nat Genet. 1996;13:22–23. doi: 10.1038/ng0596-22. [DOI] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- Chen J, Silver DP, Walpita D, Cantor SB, Gazdar AF, Tomlinson G, Couch FJ, Weber BL, Ashley T, Livingston DM, Scully R. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cell. 1998;2:317–328. doi: 10.1016/s1097-2765(00)80276-2. [DOI] [PubMed] [Google Scholar]
- Nelms BE, Maser RS, MacKay JF, Lagally MG, Petrini JH. In situ visualization of DNA double-strand break repair in human fibroblasts. Science. 1998;280:590–592. doi: 10.1126/science.280.5363.590. [DOI] [PubMed] [Google Scholar]
- Raderschall E, Golub EI, Haaf T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc Natl Acad Sci USA. 1999;96:1921–1926. doi: 10.1073/pnas.96.5.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- Zheng L, Li S, Boyer TG, Lee WH. Lessons learned from BRCA1 and BRCA2. Oncogene. 2000;19:6159–6175. doi: 10.1038/sj/onc/1203968. [DOI] [PubMed] [Google Scholar]
- Mizuta R, LaSalle JM, Cheng HL, Shinohara A, Ogawa H, Copeland N, Jenkins NA, Lalande M, Alt FW. RAB22 and RAB163/mouse BRCA2: proteins that specifically interact with the RAD51 protein. Proc Natl Acad Sci USA. 1997;94:6927–6932. doi: 10.1073/pnas.94.13.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem. 1997;272:31941–31944. doi: 10.1074/jbc.272.51.31941. [DOI] [PubMed] [Google Scholar]
- Chen PL, Chen CF, Chen Y, Xiao J, Sharp ZD, Lee WH. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc Natl Acad Sci USA. 1998;95:5287–5292. doi: 10.1073/pnas.95.9.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein LY, Ouchi T, Aaronson SA. The BRCA2 gene product functionally interacts with p53 and RAD51. Proc Natl Acad Sci USA. 1998;95:13869–13874. doi: 10.1073/pnas.95.23.13869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J Biol Chem. 2000;275:23899–23903. doi: 10.1074/jbc.C000276200. [DOI] [PubMed] [Google Scholar]
- Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999;59:3547–3551. [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- Snouwaert JN, Gowen LC, Latour AM, Mohn AR, Xiao A, DiBiase L, Koller BH. BRCA1 deficient embryonic stem cells display a decreased homologous recombination frequency and an increased frequency of non-homologous recombination that is corrected by expression of a brca1 transgene. Oncogene. 1999;18:7900–7907. doi: 10.1038/sj/onc/1203334. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, West SC. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell. 2001;7:273–282. doi: 10.1016/s1097-2765(01)00175-7. [DOI] [PubMed] [Google Scholar]
- Chen CF, Chen PL, Zhong Q, Sharp ZD, Lee WH. Expression of BRC repeats in breast cancer cells disrupts the BRCA2–Rad51 complex and leads to radiation hypersensitivity and loss of G(2)/M checkpoint control. J Biol Chem. 1999;274:32931–32935. doi: 10.1074/jbc.274.46.32931. [DOI] [PubMed] [Google Scholar]
- Spain BH, Larson CJ, Shihabuddin LS, Gage FH, Verma IM. Truncated BRCA2 is cytoplasmic: implications for cancer-linked mutations. Proc Natl Acad Sci USA. 1999;96:13920–13925. doi: 10.1073/pnas.96.24.13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung P, Robberson DL. DNA strand exchange mediated by a RAD51–ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell. 1995;82:453–461. doi: 10.1016/0092-8674(95)90434-4. [DOI] [PubMed] [Google Scholar]
- Gasior SL, Olivares H, Ear U, Hari DM, Weichselbaum R, Bishop DK. Assembly of RecA-like recombinases: Distinct roles for mediator proteins in mitosis and meiosis. Proc Natl Acad Sci USA. 2001. [DOI] [PMC free article] [PubMed]
- Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, Thompson LH, Takeda S. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol. 2001;21:2858–2866. doi: 10.1128/MCB.21.8.2858-2866.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]