

Abstract
The accumulation of mutations is a feature of all normal cells. The probability of any individual gene in any cell acquiring a mutation is, however, low. Cancer is therefore a rare disease in comparison with the number of susceptible cells. Mutations in normal tissue are stochastic, vary widely among cells and are therefore difficult to detect using standard methods because each change is so rare. If, however, a tissue such as the breast undergoes considerable clonal expansion, particularly if relatively late in life, normal tissue may have accumulated many thousands of detectable mutations. Since breast cancers are clonal and have almost certainly undergone many more cell divisions than normal cells, each tumour may have many millions of mutations, most of which are entirely innocent and some of which have accumulated in the cell of origin prior to tumorigenesis. Despite some claims to the contrary, even at normal mutation rates, clonal expansion within a tumour is quite sufficient to account for the mutations of five or six genes that are generally supposed necessary for carcinogenesis to occur. Hypermutability does, however, contribute to the pathogenesis of many cancers and, although evidence is indirect in breast cancer, may take forms such as karyotypic instability via centrosome amplification.
Keywords: genomic instability, mutation, selection, tumorigenesis
Introduction
A few years ago, a fascinating article by Deng et al [1] showed loss of heterozygosity (LOH) on chromosome 3p in apparently normal breast tissue adjacent to a carcinoma. Evidence was presented to show that the carcinoma also showed 3p loss. The importance accorded to these findings was reflected by their publication in Science. Readers inferred that 3p loss may sometimes be an early event in breast tumorigenesis that might have produced a selective advantage and clonal expansion, but led to no change in morphology. This model of tumorigenesis was therefore subtly different from that proposed for the pathogenesis of colorectal tumours, in which even the earliest lesions were morphologically distinct from normal tissue [2].
Lakhani et al [3] studied short-term cultures of luminal epithelial and myoepithelial breast cells derived from macroscopically normal fresh breast tissue adjacent to carcinoma. This method was used to exclude the possibility that the 'normal' tissue studied by Deng et al [1] might actually have been heavily contaminated by cancer cells that had undergone Pagetoid spread. Microdissected normal duct-lobular units were also analysed, thus providing a direct comparison with the study of Deng et al [1]. Five of 13 cases showed some LOH in normal tissue, although this was variable in its origin (microdissected or cultured, adjacent to cancer or distant from cancer). Loss was seen in normal tissue, most often at markers near BRCA2 on chromosome 13q and rarely at markers on chromosome 3p near those studied by Deng et al [1]. Loss in normal tissue was not always observed in any adjacent carcinoma.
No large sample series has ever confirmed the existence of detectable mutations in apparently normal breast tissue (perhaps no one has tried) and each of the two studies above had potential technical problems, which were almost unavoidable. The ability of polymerase chain reaction to produce consistent, artefactual allelic loss, especially in archival samples, is becoming increasingly well documented. In vitro selection may be strong in short-term culture and result in outgrowth of occasional cells with mutations unrelated to tumorigenesis in vivo. It would nevertheless be wrong to dismiss the findings of Deng et al [1] or those of Lakhani et al [3] on the grounds that they are uninteresting, unimportant or simply incorrect. On the contrary, the topic of mutations in normal tissues not only is associated with precursor lesions for breast tumours, but also is potentially important for studies of patch size and the presence of normal, clonal cell populations in the breast, and for the number and significance of mutations subsequently found in breast tumours that develop from those tissues.
Allelic loss and clonal origins of normal breast
An important consideration when analysing mutations in normal tissue using molecular techniques is that any allelic loss in normal tissue must either have led to some expansion of that clone or must have occurred before normal clonal expansion; otherwise, the genetic change would not be detectable using standard methods. Strictu sensu allelic loss can thus not truly be detected in normal tissue unless the progenitor cell has undergone clonal expansion. The topic of clonal expansion in normal breast, and hence patch size, is a vexed one. Given that breast proliferation occurs relatively late in development, human breast tissue may ultimately be derived from few stem cells, leading to exceptionally large patch size and consequent problems for studies of monoclonality in breast tumours. Moreover, any genetic change that occurred in the breast prior to puberty would probably be manifest as a detectable (clonal) mutation in normal adult breast tissue, and an unwarranted significance might be ascribed to that mutation. There is unfortunately very little evidence concerning patch size in the normal breast, owing to the difficulties inherent in such an analysis [4]. Distinguishing between this explanation for the data of Deng et al [1] and the alternative explanation (that the 'normal' tissue is actually an early neoplasm) is not currently possible.
Mutations in normal breast tissue
The results of Deng et al [1] serve another useful purpose, which is to highlight the often neglected fact that lots of mutations occur continuously in all normal cells at normal rates of mutation. How many mutations might occur in the normal breast epithelial stem cell in a 10-year period? Assuming that the normal mutation rate is 1.4 × 10-9 per nucleotide per cell division, that a diploid genome has 1010 bases and that the cell undergoes 50 divisions per year, then 7000 mutations would occur. Almost all of these mutations will have no functional effect, because they occur in 'junk' DNA (or, more rarely, diploidy protects against functional consequences). Tumours thus arise only exceptionally because at least 'two hits' usually have to occur somatically in the same gene, and the chances of this are very small. If a breast tumour does happen to arise, however, it starts to grow with many thousands of mutations that have already been acquired by the cell of origin during normal tissue development and maintenance. Recent surprise that cancers have as many as 10,000 mutations [5,6] is thus entirely misplaced: the tumour progenitor cell may already have this many mutations before any genetic changes occur during tumorigenesis.
How many mutations in a cancer?
It is often claimed that normal mutation rates are too low to account for changes in the five or six genes generally supposed to be mutated, as a minimum, for a cancer to occur [7]. It is thus argued that all cancers must have some form of genomic instability, probably from an early stage. This reasoning is based on a simple calculation. Loeb [7] stated that "if the mutation rate is normally 1.4 × 10-9 per nucleotide per cell division and that per gene is 2.0 × 10-7, a [stem] cell with a genome of 70,000 genes will accumulate one mutant gene during an individual's lifetime … assuming 100 cell divisions in a normal lifespan".
Let us take Loeb's calculation as stated, even though some stem cells almost certainly divide many thousands of times in a 65-year period, Loeb's statement ignores the crucial question of how the cell acquires genomic instability. To inactivate three tumour suppressor genes and to activate three oncogenes requires a total of nine mutations. Making no allowance for the fact that LOH is probably more frequent than small intragenic mutations, simple calculations show that the probability of a cell acquiring any one of these mutations is about 4 × 10-5 and that the probability of acquiring all nine mutations is about 10-40.
Even if there are many millions of stem cells in any tissue and the lifetime risk of breast cancer is 10%, the probability of developing a cancer would thus be minuscule according to this model. Crucially, however, the simple model takes no account of clonal expansion [8]. The simple calculation assumes that the stem cell simply waits to acquire mutations. The best evidence suggests, however, that a stepwise model of tumorigenesis, in which successive advantageous mutations lead to successive rounds of clonal expansion, is a much more likely scenario [9]. The probability of a cancer acquiring sufficient mutations is thus much higher than that in Loeb's calculation, because all the stem cell's progeny in the expanding tumour clone have a chance of acquiring mutations. Clonal expansion is not simply a facet of tumorigenesis that allows mutations to occur; on the contrary, clonal expansion is tumorigenesis, driven by selection.
A simulation model of stepwise tumorigenesis illustrates how powerful clonal selection can be by, in effect, raising the mutation rate through increasing population size. In this simulation, it is assumed that a stem cell has already acquired 'two hits' at a tumour suppressor locus. From Loeb's parameters, the probability of this happening for any individual cell during an individual lifetime is about 5 × 10-10. This probability may seem low for a common cancer, but it should not be forgotten that the probability of a stem cell acquiring genomic instability by, for example, inactivating a DNA repair gene, is similarly low. Moreover, each tissue may have many millions of stem cells and there may be several different genes that can cause cancer when mutated. Hence, the effective mutation rate may actually be much higher than that calculated from Loeb's parameters.
The mutant clone with only 'two hits' initially expands relatively slowly. It is assumed that mutation of three oncogenes and one further tumour suppressor are required to give a cancer; the incorporation of extra genes makes the model impracticably complex. Each activation of an oncogene or inactivation of the tumour suppressor gene confers an additional selective advantage, as long as the necessary mutations earlier in the pathway have already occurred in that cell. Although a detailed analysis is inappropriate here, typical results of the simulation are shown in Figure 1. It is clear that clonal expansion results in the growing tumour having more than sufficient time to acquire all the necessary mutations (three oncogenes and two more at a tumour suppressor locus) in this model within a realistic time. Given that the tumour is very large, the extra mutations required in Loeb's suggested six-locus model will also almost certainly occur within a realistic time. The first mutations in tumorigenesis are indeed the rate-limiting step here.
Figure 1.
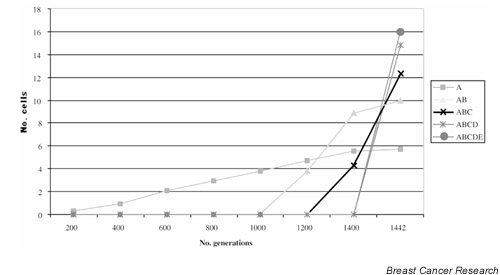
Model of clonal evolution and mutation in cancer, run as a simulation with a typical result shown. For the starting point of the model, a single cell is assumed to have acquired two mutations at a tumour suppressor locus (A), and thereby to have acquired a small replicative advantage (here, 1.01 per generation). The simulation subsequently allows mutation to occur at random in each tumour cell at oncogene loci B, D and E and at tumour suppressor locus C, at a constant rate of 2 × 10-7 per gene per generation. The selective advantage associated with activation of B is 1.05. The advantage associated with biallelic inactivation of C is 5.0, as long as B is already mutated. The advantage associated with D activation is 20.0, as long as B and C are mutated. The advantage associated with activation of E is 100, as long as B, C and D are all already mutated. These advantages are multiplicative. The lines show numbers of tumour cells at each generation with the 'genotypes' A mutant only, A and B both mutant, A-C mutant, A-D mutant, and A-E all mutant. The results show that a tumour of (nominal) size 1016 cells (y axis is log scale) with all loci mutated is readily achieved within 1500 cell generations (i.e. 30 years if, conservatively, 50 stem cell generations occur per year). Note: This model greatly understates the case for the ability of cancers to develop at a normal mutation rate because it assumes only a single genetic pathway of tumorigenesis and hence a much lower effective mutation rate than exists in reality. Moreover, action of extrinsic carcinogens may also cause the mutation rate to be raised above the 'normal' level.
Normal mutation rates are thus quite sufficient to explain the crucial mutations needed to drive tumorigenesis, but how many mutations exist within a typical cancer? Even at normal mutation rates, it can be calculated that a cancer will contain many billions of mutations (data not shown), and that millions of these mutations will be present in a large enough proportion of the tumour to be readily detectable using standard molecular techniques. Most mutations will not involve coding genes and will have no selective effect.
The predicted number of mutations in the typical cancer seems huge, but it can be accounted for by three factors. First, most cancers take many years to grow and they therefore almost certainly undergo many more cell divisions than the theoretical minimum required to produce a tumour of the size typically removed by surgery. Second, there is a tendency to underestimate the huge number of background mutations, because these rarely occur in regions of the genome analysed by molecular techniques and they are not observed consistently in different tumours of the same type. Finally, the genetic mutations that already exist in the tumour cell of origin are sometimes forgotten.
The role of genomic instability
Although unnecessary for tumorigenesis, there is evidence that many cancers are genetically unstable [10]. There are also good reasons to expect genomic instability to be disadvantageous, at least early in tumorigenesis, because intact cell cycle checkpoints tend to push cells with DNA damage into apoptosis [11]. Later in tumorigenesis, however, the advantages of genomic instability may outweigh the disadvantages. First, instability might have occurred by chance, 'hitch-hiking' with advantageous mutations. Second, instability might be directly advantageous. In a large cancer, for example, nutrient supply may be rate-limiting for growth, which might be almost static; considerable numbers of cells (up to 50% of all daughters) would therefore die in each generation. Under this scenario, genomic instability might confer a net selective advantage, by compensating for the lack of clonal expansion and producing novel combinations of mutations. This production may lead, for example, to invasion of surrounding tissues or metastasis, while being of relatively little selective disadvantage, especially if a cell had acquired cell cycle checkpoint mutations by this stage. Third, a possibility [11], for which circumstantial evidence is accruing [12], is that a single mutation (such as a mutation in p53 or APC) may have pleiotropic effects, providing a cancer cell with both a replicative advantage and an increased mutation rate. Many connections between DNA repair, chromosome segregation and the cell cycle have been discovered, and it is easy to see how a defective cell cycle checkpoint could lead both to faster replication and to tolerance of accumulated mutations.
Types of genomic instability are best characterized in col-orectal cancer, where they include microsatellite instability, a poorly defined phenomenon of 'chromosomal instability' and a methylator phenotype. Other types of instability have been hypothesised, including a 'translocator phenotype' and a 'mitotic recombination phenotype'. Many breast cancers are aneuploid/polyploid and thus possibly chromosomally unstable, and other cancers (largely lobular and grade I ductal) are near-diploid. Nevertheless, little is known about the presence and type of genetic instability in breast tumours. There is some evidence of centrosome amplification, either as a cause of or consequence of chromosomal instability [13,14,15], but there is no evidence of true microsatellite instability (despite some reports) [16,17] nor, as yet, of a methylator phenotype.
Conclusions
All cells in all organisms are prone to mutations. Presumably, in the soma, the normal frequency of mutation has arisen as an evolutionary trade-off between the probability of diseases such as cancer and the costs of an even more faithful system of replicating DNA and chromosomes. Normal cells accumulate large numbers of mutations, but these are rarely of functional importance and rarely detected unless single-cell methods are used. There is a tendency for cancer biologists to underestimate the 'power' of the normal mutation rate, and to assume that cancers cannot develop in the absence of genomic instability. This is not the case and many early tumours, in particular, show no evidence of a raised mutation rate. It is nevertheless indisputable that many cancers do have some form of genomic instability, derived from the disruption of certain cellular processes, such as DNA repair or centrosome formation. This genomic instability should arguably be regarded not as the main driving force of tumorigenesis, but as an adaptation to specific environmental conditions or, intriguingly, as a pleiotropic effect of mutations selected primarily for their effects on cell replication and/or death.
Abbreviations
LOH = loss of heterozygosity.
References
- Deng G, Lu Y, Zlotnikov G, Thor AD, Smith HS. Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science. 1996;274:2057–2059. doi: 10.1126/science.274.5295.2057. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Lakhani SR, Chaggar R, Davies S, Jones C, Collins N, Odel C, Stratton MR, O'Hare MJ. Genetic alterations in 'normal' luminal and myoepithelial cells of the breast. J Pathol. 1999;189:496–503. doi: 10.1002/(SICI)1096-9896(199912)189:4<496::AID-PATH485>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Garcia SB, Novelli M, Wright NA. The clonal origin and clonal evolution of epithelial tumours. Int J Exp Pathol. 2000;81:89–116. doi: 10.1046/j.1365-2613.2000.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoler DL, Chen N, Basik M, Kahlenberg MS, Rodriguez-Bigas MA, Petrelli NJ, Anderson GR. The onset and extent of genomic instability in sporadic colorectal tumor progression. Proc Natl Acad Sci USA. 1999;96:15121–15126. doi: 10.1073/pnas.96.26.15121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland CR, Ricciardiello L. How many mutations does it take to make a tumor? Proc Natl Acad Sci USA. 1999;96:14675–14677. doi: 10.1073/pnas.96.26.14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb LA. A mutator phenotype in cancer. Cancer Res. 2001;61:3230–3239. [PubMed] [Google Scholar]
- Nowell PC. The clonal nature of neoplasia. Cancer Cells. 1989;1:29–30. [PubMed] [Google Scholar]
- Tomlinson IP, Novelli MR, Bodmer WF. The mutation rate and cancer. Proc Natl Acad Sci USA. 1996;93:14800–14803. doi: 10.1073/pnas.93.25.14800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill DP, Kinzler KW, Vogelstein B, Lengauer C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. 1999;9:M57–M60. [PubMed] [Google Scholar]
- Tomlinson I, Bodmer W. Selection, the mutation rate and cancer: ensuring that the tail does not wag the dog. Nat Med. 1999;5:11–12. doi: 10.1038/4687. [DOI] [PubMed] [Google Scholar]
- Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH, Clevers H. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3:433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- Sen S, Zhou H, White RA. A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene. 1997;14:2195–2200. doi: 10.1038/sj/onc/1201065. [DOI] [PubMed] [Google Scholar]
- Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, Brinkley BR, Sen S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–193. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- Miyoshi Y, Iwao K, Egawa C, Noguchi S. Association of centrosomal kinase STK15/BTAK mRNA expression with chromosomal instability in human breast cancers. Int J Cancer. 2001;92:370–373. doi: 10.1002/ijc.1200. [DOI] [PubMed] [Google Scholar]
- Siah SP, Quinn DM, Bennett GD, Casey G, Flower RL, Suthers G, Rudzki Z. Microsatellite instability markers in breast cancer: a review and study showing MSI was not detected at 'BAT 25' and 'BAT 26' microsatellite markers in early-onset breast cancer. Breast Cancer Res Treat. 2000;60:135–142. doi: 10.1023/a:1006315315060. [DOI] [PubMed] [Google Scholar]
- Caldes T, Perez-Segura P, Tosar A, De La Hoya M, Diaz-Rubio E. Low frequency of microsatellite instability in sporadic breast cancer. Int J Oncol. 2000;16:1235–1242. doi: 10.3892/ijo.16.6.1235. [DOI] [PubMed] [Google Scholar]