

Abstract
p53 mutation remains the most common genetic change identified in human neoplasia. In breast cancer, p53 mutation is associated with more aggressive disease and worse overall survival. The frequency of mutation in p53 is, however, lower in breast cancer than in other solid tumours. Changes, both genetic and epigenetic, have been identified in regulators of p53 activity and in some downstream transcriptional targets of p53 in breast cancers that express wild-type p53. Molecular pathological analysis of the structure and expression of constituents of the p53 pathway is likely to have value in diagnosis, in prognostic assessment and, ultimately, in treatment of breast cancer.
Keywords: breast cancer, p53, tumour suppressor gene
Introduction
First described in 1979, and initially believed to be an oncogene, p53 was the first tumour suppressor gene to be identified. p53 functions to eliminate and inhibit the proliferation of abnormal cells, thereby preventing neoplastic development. Abrogation of the negative growth regulatory functions of p53 occurs in many, perhaps all, human tumours. The p53 signalling pathway is in 'standby' mode under normal cellular conditions. Activation occurs in response to cellular stresses, and several independent pathways of p53 activation have been identified that appear to be dependent on distinct upstream regulatory kinases [1] (Fig. 1). These include an ataxia-telangectasia mutated (ATM)/human homologue of Rad53 (Chk2)-dependent pathway activated by DNA double-strand breaks, a second pathway dependent on the alternative product of the INK4 gene, p14ARF (which is activated by expression of oncogenes), and a third pathway whose activity is increased by cytotoxic anti-tumour agents and ultraviolet light, but is independent of ATM, Chk2 and p14ARF. Activation of this pathway may be mediated by other kinases such as the ATM relative ataxia-telangectasia and Rad3-related protein (ATR) [1].
Figure 1.
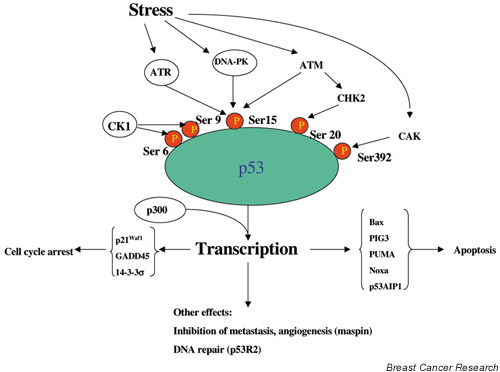
A simplified model of some of the components of p53 signalling. Under normal conditions, the p53 pathway operates on 'standby' mode. Activation occurs in response to a variety of cellular stresses such as DNA damage and expression of activated oncogenes. See [1] for a more detailed description of the pathways activated by specific stresses. Post-translational modifications (such as phosphorylation at the indicated serine residues) activate the protein for DNA binding and transactivation of downstream 'effector' genes that mediate the tumour suppressor actions of p53. The outcome of activation depends on the nature and magnitude of the stress, its transduction via specific upstream kinases, and the resultant programme of p53-dependent gene expression. Transcriptional coactivators such as apoptosis stimulating protein of p53 and BRCA1 (not shown) may further 'fine tune' the response and, in some cases, preferentially promote specific cellular responses such as apoptosis. Many of the components of this signalling pathway are targets for genetic and/or epigenetic changes in breast cancer as described in the text. Not shown is the induction of MDM2, which acts as a negative feedback regulator of the pathway by promoting the degradation of p53. Because of space limitations, other important constituents of the pathway have had to be omitted.
Activation results in an increase in the levels of p53 protein due to reduced mouse double minute 2 (MDM2)-dependent proteolytic degradation, and increased affinity of p53 for DNA. Whereas phosphorylation of the N-terminus may affect the stability of p53, lysine acetylation and/or serine phosphorylation in the C-terminus of the protein promotes DNA binding. As a result of activation, the wild-type protein acquires sequence-specific DNA binding activity, and an increasing number of genes are being identified as transcriptional targets of wild-type p53 [2]. These can be placed into a number of classes according to their functions.
Inhibitors of cell cycle
These targets include the cyclin-dependent kinase inhibitor p21Waf1, 14-3-3sigma (σ), which tethers cyclin B1–CDK1 complexes in the cytoplasm, contributing to maintenance of G2 cell cycle arrest, and GADD45 and BTG.
Apoptosis regulators
Proapoptotic proteins directly induced by p53 include, among others, Bax, Apaf 1, PUMA, p53AIP1, PIDD and NOXA.
DNA repair
Wild-type p53 directly activates a number of genes that function in pathways of DNA repair, including a ribonucleotide reductase gene, p53R2, involved in repair of DNA damage.
Inhibitors of angiogenesis and metastasis
Perhaps the most interesting member of this class of p53-inducible genes is mammary serine protease inhibitor (maspin), a serine protease inhibitor that inhibits angiogenesis, invasion and metastasis. A second recognised metastasis suppressor protein, KAI1, has also been reported to be regulated by p53.
p53 is inactivated by multiple mechanisms in cancer
p53 remains the most commonly mutated gene in many common human cancers, with mutations (principally, but not exclusively, missense) estimated to occur in 50% of all cancers. Mutant proteins are almost always defective for sequence-specific DNA binding, and thus for transactivation of genes upregulated by the wild-type protein [3]. Interestingly, the proportion of missense mutations in p53 is higher than that seen in other tumour suppressor genes, suggesting that expression of p53 mutants may confer selective advantage over and above loss of wild-type function [4].
Consistent with this hypothesis, many human tumour-associated p53 mutants possess a number of properties absent from the wild-type protein [3]. In a high proportion of cancers lacking mutations, p53 function is compromised by other recognised mechanisms [1]. In virus-associated cancers, this may occur via interaction with virally encoded proteins resulting in sequestration or enhanced degradation of p53. MDM2 binds to p53 and promotes the ubiquitination of the C-terminus of p53 and subsequent degradation. p14ARF interacts with MDM2, preventing association of p53 and MDM2, and thereby stabilising p53. Degradation of p53 may therefore be inappropriately stimulated by overexpression of MDM2 or by deletion or epigenetic silencing of p14ARF. Loss of this protein has been reported in several common human cancers, particularly (but not exclusively) those in which the p53 gene is wild-type. Yet another mechanism of inactivation involves cytoplasmic sequestration of p53 protein, preventing nuclear localisation of the protein and thus inhibiting its activity.
Mechanisms of inactivation of p53 in breast cancer
p53 mutation in breast cancer
Germ-line mutations in p53 occur in a high proportion of individuals with the Li–Fraumeni cancer susceptibility syndrome, which confers an increased risk of breast cancer [5]. This implies an important role for p53 inactivation in mammary carcinogenesis, and the structure and expression of p53 has been widely studied in breast cancer. In early studies, expression of mutant p53 was demonstrated in breast cancer cell lines [6]. Loss of heterozygosity (LOH) in the p53 gene was shown to be a common event in primary breast carcinomas [7] and this is accompanied by mutation of the residual allele in some cases. In colon carcinomas the residual p53 allele is mutant in the vast majority of cases, but in breast cancer at least 60% of cases with LOH retain a wild-type p53 allele [7].
Nevertheless, numerous studies have identified coding mutations in p53 in breast cancer and this is now recognised as a common, but by no means ubiquitous, somatic genetic change in breast cancer. Indeed, a comprehensive meta-analysis revealed that only approximately 20% of all cases express mutant p53 [8]. Several studies have sought to identify the stage of breast tumourigenesis at which p53 mutation occurs. Careful studies of microdissected tumour material show that low-grade ductal carcinoma in situ (DCIS) is essentially devoid of mutations, whereas mutations are more common in high-grade DCIS [9].
Although the overall frequency of p53 mutation in breast cancer is approximately 20% [8], certain types of the disease are associated with higher frequencies. For example, a number of studies have identified an increased rate of p53 mutations in cancers arising in carriers of germ-line BRCA1 and BRCA2 mutations [10,11]. Moreover, a distinct spectrum of p53 mutations occurs in such carcinomas [12]. Strikingly, in typical medullary breast carcinomas, p53 mutation occurs in 100% of cases [13]. This is of particular interest, since it is now well recognised that medullary breast cancers share clinicopathological similarities with BRCA1-associated cases. Indeed, methylation-dependent silencing of BRCA1 expression occurs commonly in medullary breast cancers [14].
The p53 pathway in breast cancers lacking p53 mutations
Although the structure of p53 has been extensively studied, the absolute frequency of mutations in breast cancer is significantly lower than that in many other common cancers. What are the molecular mechanisms by which cancers lacking mutations subvert the tumour suppressor properties of wild-type p53? Studies of this question have shed fascinating light into some of the myriad regulatory pathways of p53 function.
An early insight into mechanisms of p53 inactivation in breast cancer was afforded by a study of breast cancers of varying p53 mutation status. It was shown that, in a proportion of cases with wild-type p53, there was only cytoplasmic protein staining [15]. Exclusion of the wild-type protein from the nucleus thus represents a potential mechanism for p53 inactivation independent of mutation. Subsequently, alterations have been identified both in upstream regulatory proteins and in downstream p53-induced proteins that may disable or compromise the pathway in breast cancers lacking mutations.
Changes in upstream regulators of p53
Aside from the two well-recognised breast cancer susceptibility genes BRCA1 and BRCA2, it is an attractive hypothesis that mutations in other genes with lower penetrance may account for a significant proportion of hereditary breast cancers. One such gene may be ATM, the gene mutated in ataxia-telangectasia (A-T). The link between ATM and p53 was suggested in early studies revealing defective induction of p53 following irradiation of A-T cells. It has subsequently been established that phosphorylation of both p53 and BRCA1 in response to γ-irradiation occurs via ATM.
A-T patients have a high incidence of cancer, and some develop breast carcinomas [16]. A recent study examined the entire ATM coding sequence in a large series of breast cancer patients and identified heterozygosity for truncating mutations in approximately one in 50 patients, consistent with the hypothesis that A-T heterozygotes are more common in breast cancer patients than in the general population [17]. It has been hypothesised that inactivation of ATM may be an alternative to p53 mutation in leukaemia. There is evidence that low or absent expression of ATM occurs commonly in sporadic breast cancer [18]. Interestingly, some cancers in this study had both low ATM expression and p53 mutation, suggesting that the inactivation of the two genes is not necessarily exclusive.
A second protein operating upstream to transduce DNA damage to phosphorylation of p53 is Chk2. Chk2 is activated (by phosphorylation of threonine 68) by ATM in response to double strand breaks and, in turn, catalyses phosphorylation of p53 at serine 20. Analysis of Chk2 sequence in Li–Fraumeni families lacking p53 mutations identified heterozygous germ-line mutations in some cases [19], suggesting that Chk2 is a human tumour suppressor gene and implying that loss of function in Chk2 might be equivalent to p53 mutation. Several studies have subsequently examined Chk2 germ-line sequence in families with hereditary breast cancer. In all cases, germ-line mutations in Chk2 were uncommon [20,21]. Chk2 mutations in sporadic breast cancers are also rare, but a significant proportion of such cases exhibit reduced or absent expression of Chk2 [22]. Interestingly, cases with loss of Chk2 expression were both wild type and mutant for p53.
The steady-state level of p53 mRNA is lower in many breast cancers than in normal breast epithelium [23]. Detailed analysis of the p53 promoter has revealed the presence of several consensus-binding sites for the homeobox protein HoxA5, and these are responsive to HoxA5. In a high proportion of primary breast carcinomas, expression of HoxA5 is significantly reduced. This is attributable to aberrant methylation of the Hox A5 promoter [23]. By limiting basal expression of p53 mRNA, epigenetic silencing of HoxA5 thus represents a novel and important mechanism by which p53 signalling is attenuated in breast cancer. It will be of interest to determine whether expression of other components of the pathway is also subject to similar regulation.
Changes in p53 transcriptional target genes
Perhaps surprisingly, relatively few studies have investigated the status of these genes in human cancer in general, and in breast cancer specifically. One such gene of particular interest is σ, which was originally identified in squamous epithelium and shown to be downregulated in a small number of breast cancer cell lines. It was subsequently shown that σ is a direct transcriptional target for p53 and that it mediates maintenance of a G2 checkpoint. Analysis in primary breast carcinomas revealed that, despite the absence of intragenic mutation and LOH, σ is subject to methylation-dependent silencing in a very high proportion of cases [24]. Moreover, loss of expression is present in a significant number of cases of DCIS [25]. It will be interesting to determine whether any relationship exists between p53 status and silencing of σ in breast cancer.
A second key gene whose expression is directly upregulated by wild-type p53 is MDM2. Amplification and overexpression of MDM2 is a recognised mechanism of p53 inactivation, but although one study detected MDM2 amplification in a small proportion of cases [26], amplification of MDM2 is an infrequent event in breast cancer [27]. A second mechanism to promote MDM2-dependent p53 degradation involves loss of p14ARF either by mutation, deletion or epigenetic silencing. Mutations and deletions in p14ARF are uncommon in breast cancer, but absent or reduced expression occurs in a subset of cases and this is associated with aberrant hypermethylation of the p14ARFpromoter [28]. Inactivation is frequently seen in cases with p53 mutation, implying that loss of p14ARF expression is not functionally equivalent to mutation of p53. Nevertheless, these results suggest a role for p14ARFinactivation in breast cancer.
p21Waf1 (also known as Cip1) is an inhibitor of the cyclin-dependent kinases and is directly induced by p53. The Waf1 gene is not a frequent target for mutational inactivation in breast (or other) cancers [29]. Moreover, p21Waf1 expression is not significantly associated with clinical outcome in breast cancer [30].
One of the most commonly deleted chromosomal regions in breast cancer is 11q23-q25, which contains a number of putative tumour suppressor loci, including ATM, CHK1, PPP2R1B and PIG8. A recent study of the structure of these genes in early onset breast cancer determined that the gene most frequently mutated in this region was PIG8, a gene induced by p53 and a putative mediator of p53-dependent apoptosis [31]. Loss of PIG8 function via inactivating mutations thus represents a further potential mechanism by which p53-dependent apoptosis can be impaired in breast cancer.
Changes in p53 coactivators
In addition to proteins such as ATM, ATR and Chk2 that regulate the stability and function of p53 through phosphorylation, a second, functionally distinct, group of proteins is now emerging that appear to operate as cofactors stimulating one or more of the wild-type properties of p53. One such family with possible involvement in breast cancer is the apoptosis stimulating protein of p53 (ASPP). Two members of this family (ASPP1 and ASPP2), encoded by separate genes, have recently been described [32].
Expression of either ASPP1 or ASPP2 stimulates the proapoptotic function of wild-type p53 by increasing p53-dependent induction of apoptotic effectors such as Bax and PIG3, whereas expression of nonapoptotic proteins such as p21Waf1 was much less affected. In primary breast cancers lacking p53 mutation, expression of both ASPP1 and ASPP2 was reduced. These observations are supported by an earlier report that, using microarray methodology, also identified p53 BP2 (ASPP2) downregulation in breast cancer [33]. Taken together, these studies suggest that downregulation of ASPP proteins attenuates p53-dependent apoptosis, thus conferring a selective advantage to breast carcinomas with intact p53.
Another transcriptional coactivator for p53 is BRCA1. It is, of course, well known that germ-line mutations in BRCA1 predispose carriers to breast and ovarian cancer. BRCA1 is phosphorylated after DNA damage by ATM, ATR and Chk2. BRCA1 associates with the C-terminus of wild-type p53 and stimulates transcription from p53-responsive promoters, whereas tumour-associated mutants of BRCA1 are deficient in coactivating activity [34]. Although somatic mutations in BRCA1 have not been described in sporadic breast cancer, expression of BRCA1 is decreased in the majority of cases, implying a further mechanism whereby p53 function can be inhibited in breast carcinomas. Downregulation of BRCA1 expression is attributable to methylation-dependent silencing in a small proportion of cases [35], but the mechanism underlying the low level of expression seen in many sporadic breast cancers awaits clarification.
The DNA binding and therefore the transcriptional activating function of p53 is potentiated by acetylation of lysine residues in the C-terminus of the protein. This is accomplished by the histone acetyltransferase p300. Truncating mutations in p300 have been described in breast cancer cell lines and primary breast cancer, and a missense mutation in a further breast cancer cell line [36]. In some cases, mutations in p300 and p53 are not mutually exclusive, suggesting that mutation of p300 does not abrogate selective pressure for p53 mutation.
p53 status may be predictive of outcome in breast cancer
The association between p53 alterations and clinical outcome in breast cancer has been the subject of numerous investigations. The possibility that p53 status influences biological behaviour was raised in an early study in which the presence of p53 mutations in aggressive breast cancer was demonstrated [37], and the majority of studies support an association between worse survival and the presence of p53 mutations. This association was confirmed in a comprehensive meta-analysis of the effect of somatic p53 mutations on prognosis in breast cancer [8].
Potential correlations between the type of p53 mutation and the clinical phenotype in breast cancer have been described [38]. In this study, it was shown that mutations affecting amino acids critical for DNA binding were associated with very aggressive cancers, whereas null mutations and other missense mutations were associated with an indeterminate clinical phenotype. A recent study suggests that p53 mutation may be an important molecular genetic correlate of breast cancer progression [39]. In a further study of primary breast carcinomas, expression of the angiogenic vascular endothelial growth factor was shown to correlate with poor prognosis and with mutation in p53 [40].
The serpin family member maspin is an inhibitor of angiogenesis, invasion and metastasis. A step-wise decrease in the expression of maspin in the sequence DCIS > invasive cancer > lymph node metastasis has been described, strongly supporting an important role in breast cancer progression [41]. Maspin is directly transcriptionally induced by wild-type p53, thus providing an interesting connection between p53 and progression in ductal breast carcinomas [42]. It will clearly be of interest to determine how expression of maspin relates to p53 status in breast cancer.
Studies of the effect of p53 mutations on chemosensitivity of human tumours have produced conflicting results. In breast cancer, there is evidence that specific mutations correlate with primary resistance to doxorubicin and that the presence of such mutations may be predictive of early relapse [43]. This hypothesis was further supported by a later study from the same group [44]. In another study, cancers with p53 mutation were more likely to respond to paclitaxel [45].
A number of recent reports have described the detection of tumour-specific DNA in plasma from patients with breast carcinomas. p53 mutations can be detected in peripheral blood in a significant proportion of patients whose primary tumours contain mutations. Furthermore, the presence of p53 mutations in plasma DNA is strongly correlated with various clinicopathological parameters and is a significant prognostic factor [46]. p53 autoantibodies are also detectable in patients with breast cancer. These were reported to occur in 15% of patients but the presence of such antibodies had no relationship to disease status [47].
p53 family members in breast cancer
Two structural and partial functional homologues of p53 (p63 and p73) have been described. Mutations in p73 are uncommon in human neoplasia, overexpression of p73 being the most frequent abnormality of p73. A subset of cases in breast cancer overexpress p73, and in one study this was associated with lymph node metastasis, vascular invasion and high-grade malignancy [48]. Analysis of p63 in breast tissuerevealed that expression, specifically of the transdominant DNp63, is restricted exclusively to myoepithelial cells. Indeed, p63 has been proposed as a specific and sensitive marker for myoepithelial cells [49].
Conclusions
Abundant data from mechanistic, molecular pathological and transgenic animal studies support an important role for p53 in mammary carcinogenesis. However, despite the convincing evidence implicating loss of function of p53 in breast neoplasia, mutations in the gene occur at a significantly lower frequency than in other common solid tumours. Over the past few years, knowledge of the upstream pathways regulating p53 activity has increased greatly and numerous transcriptional targets for p53 have been described. These discoveries have allowed analysis of the molecular mechanisms, in addition to mutation, by which p53 is disabled in breast cancer and have provided new insights into pathways of neoplasia in the breast. Molecular pathological analysis of specific components of the p53 pathway is likely to have diagnostic and prognostic utility in breast cancer. Moreover, a number of innovative strategies have been proposed to restore p53 function to tumours [50]. It will be of great interest to observe how these and other novel therapeutic approaches targeted to the p53 pathway impact on clinical outcome in breast cancer.
Abbreviations
ASPP = apoptosis stimulating protein of p53; A-T = ataxia-telangectasia; ATM = ataxia-telangectasia mutated; ATR = ataxia-telangectasia and Rad3-related protein; Chk2 = human homologue of Rad53; DCIS = ductal carcinoma in situ; LOH = loss of heterozygosity; maspin = mammary serine protease inhibitor; MDM2 = mouse double minute 2; p14ARF = alternative product of the INK4 gene; σ = 14-3-3sigma.
References
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2001;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- el-Deiry WS. Regulation of p53 downstream genes. Semin Cancer Biol. 1998;8:345–357. doi: 10.1006/scbi.1998.0097. [DOI] [PubMed] [Google Scholar]
- Sigal A, Rotter V. Oncogenic mutations of the p53 tumor suppressor: The demons of the guardian of the genome. Cancer Res. 2000;60:6788–6793. [PubMed] [Google Scholar]
- Hussain SP, Harris CC. Molecular epidemiology and carcino-genesis: endogenous and exogenous carcinogens. Mutat Res. 2000;462:311–322. doi: 10.1016/s1383-5742(00)00015-6. [DOI] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JF, Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, Friend SH. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- Bartek J, Iggo R, Gannon J, Lane DP. Genetic and immunocytochemical analysis of mutant p53 in human breast cancer. Oncogene. 1990;5:893–899. [PubMed] [Google Scholar]
- Davidoff AM, Humphrey PA, Iglehart JD, Marks JR. Genetic basis for p53 overexpression in human breast cancer. Proc Natl Acad Sci USA. 1991;88:5006–5010. doi: 10.1073/pnas.88.11.5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharaoh PD, Day NE, Caldas C. Somatic mutations in the p53 gene and prognosis in breast cancer: a meta-analysis. Br J Cancer. 1999;80:1968–1973. doi: 10.1038/sj.bjc.6690628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Done SJ, Eskandarian S, Bull S, Redston M, Andrulis IL. p53 mis-sense mutations in microdissected high-grade ductal carcinoma in situ of the breast. J Natl Cancer Inst. 2001;93:700–704. doi: 10.1093/jnci/93.9.700. [DOI] [PubMed] [Google Scholar]
- Smith PD, Crossland S, Parker G, Osin P, Brooks L, Waller J, Philp E, Crompton MR, Gusterson BA, Allday MJ, Crook T. Novel p53 mutants selected in BRCA-associated tumours which dissociate transformation suppression from other wild-type p53 functions. Oncogene. 1999;18:2451–2459. doi: 10.1038/sj/onc/1202565. [DOI] [PubMed] [Google Scholar]
- Phillips KA, Nichol K, Ozcelik H, Knight J, Done SJ, Goodwin PJ, Andrulis IL. Frequency of p53 mutations in breast carcinomas from Ashkenazi Jewish carriers of BRCA1 mutations. J Natl Cancer Inst. 1999;91:469–473. doi: 10.1093/jnci/91.5.469. [DOI] [PubMed] [Google Scholar]
- Greenblatt MS, Chappuis PO, Bond JP, Hamel N, Foulkes WD. TP53 mutations in breast cancer associated with BRCA1 or BRCA2 germ-line mutations: distinctive spectrum and structural distribution. Cancer Res. 2001;61:4092–4097. [PubMed] [Google Scholar]
- De Cremoux P, Salomon AV, Liva S, Dendale R, Bouchind'homme B, Martin E, Sastre-Garau X, Magdelenat H, Fourquet A, Soussi T. p53 mutation as a genetic trait of typical medullary breast carcinoma. J Natl Cancer Inst. 1999;91:641–643. doi: 10.1093/jnci/91.7.641. [DOI] [PubMed] [Google Scholar]
- Esteller M, Silva JM, Domingue G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, Gabrielson E, Schutte M, Baylin SB, Herman JG. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2001;92:564–569. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- Moll UM, Riou G, Levine AJ. Two distinct mechanisms alter p53 in breast cancer: mutation and nuclear exclusion. Proc Natl Acad Sci USA. 1992;89:7262–7266. doi: 10.1073/pnas.89.15.7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankovic T, Kidd AM, Sutcliffe A, McGuire GM, Robinson P, Weber P, Bedenham T, Bradwell AR, Easton DF, Lennox GG, Haites N, Byrd PJ, Taylor AM. ATM mutations and phenotypes in ataxia-telangiectasia families in the British Isles: expression of mutant ATM and the risk of leukemia, lymphoma, and breast cancer. Am J Hum Genet. 1998;62:334–345. doi: 10.1086/301706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dork T, Bendix R, Bremer M, Rades D, Klopper K, Nicke M, Skawran B, Hector A, Yamini P, Steinmann D, Weise S, Stuhrmann M, Karstens JH. Spectrum of ATM gene mutations in a hospital-based series of unselected breast cancer patients. Cancer Res. 2001;61:7608–7615. [PubMed] [Google Scholar]
- Angele S, Treilleux I, Taniere P, Martel-Planche G, Villaume M, Bailly C, Bremond A, Montesano R, Hall J. Abnormal expression of the ATM and TP53 genes in sporadic breast cancer. Clin Cancer Res. 2000;6:3536–3544. [PubMed] [Google Scholar]
- Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, Birch JM, Li FP, Garber JE, Haber DA. Heterozygous germ line hCHK2 mutations in Li–Fraumeni syndrome. Science. 1999;286:2528–2531. doi: 10.1126/science.286.5449.2528. [DOI] [PubMed] [Google Scholar]
- Allinen M, Huusko P, Mantyniemi S, Launonen V, Wingvist R. Mutation analysis of the CHK2 gene in families with hereditary breast cancer. Br J Cancer. 2001;85:209–212. doi: 10.1054/bjoc.2001.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahteristo P, Tamminen A, Karvinen P, Eerola H, Eklund C, Aalto-nen LA, Blomqvist C, Aittomaki K, Nevanlinna H. p53, CHK2, and CHK1 genes in Finnish families with Li–Fraumeni syndrome: further evidence of CHK2 in inherited cancer predisposition. Cancer Res. 2001;61:5718–5722. [PubMed] [Google Scholar]
- Sullivan A, Yuille M, Repellin C, Reddy A, Olivier Reelfs O, Bell A, Dunne B, Gusterson BA, Osin P, Farrell PJ, Yulug I, Evans A, Ozcelik T, Gasco M, Crook T. Concomitant inactivation of p53 and Chk2 in breast cancer. Oncogene. 2002. [DOI] [PubMed]
- Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E, Marks J, Sukumar S. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature. 2000;405:974–978. doi: 10.1038/35016125. [DOI] [PubMed] [Google Scholar]
- Ferguson AT, Evron E, Umbricht CB, Pandita TK, Chan TA, Hermeking H, Marks JR, Lambers AR, Futreal PA, Stampfer MR, Sukumar S. High frequency of hypermethylation at the 14-3-3σ locus leads to gene silencing in breast cancer. Proc Natl Acad Sci USA. 2000;97:6049–6054. doi: 10.1073/pnas.100566997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbricht CB, Evron E, Gabrielson E, Ferguson A, Marks J, Sukumar S. Hypermethylation of 14-3-3 sigma (stratifin) is an early event in breast cancer. Oncogene. 2001;20:3348–3353. doi: 10.1038/sj.onc.1204438. [DOI] [PubMed] [Google Scholar]
- Ho GH, Calvano JE, Bisogna M, Abouezzi Z, Borgen PI, Cordon-Cardo C, van Zee KJ. Genetic alterations of the p14ARF-hdm2-p53 regulatory pathway in breast carcinoma. Breast Cancer Res Treat. 2001;65:225–232. doi: 10.1023/a:1010686518990. [DOI] [PubMed] [Google Scholar]
- Quesnel B, Preudhomme C, Fournier J, Fenaux P, Peyrat JP. MDM2 gene amplification in human breast cancer. Eur J Cancer. 1994;30A:982–984. doi: 10.1016/0959-8049(94)90128-7. [DOI] [PubMed] [Google Scholar]
- Silva J, Dominguez G, Silva JM, Garcia JM, Gallego I, Corbacho C, Provencio M, Espana P, Bonilla F. Analysis of genetic and epigenetic processes that influence p14ARF expression in breast cancer. Oncogene. 2001;20:4586–4590. doi: 10.1038/sj.onc.1204617. [DOI] [PubMed] [Google Scholar]
- Lukas J, Groshen S, Saffari B, Niu N, Reless A, Wen WH, Felix J, Jones LA, Hall FL, Press MF. WAF1/Cip1 gene polymorphism and expression in carcinomas of the breast, ovary and endometrium. Am J Pathol. 1997;150:167–175. [PMC free article] [PubMed] [Google Scholar]
- Diab SG, Yu YY, Hilsenbeck SG, Allred DC, Elledge RM. WAF1/CIP1 protein expression in human breast tumours. Breast Cancer Res Treat. 1997;43:99–103. doi: 10.1023/a:1005752829260. [DOI] [PubMed] [Google Scholar]
- Gentile M, Ahnstrom M, Schon F, Wingren S. Candidate tumour suppressor genes at 11q23-q24 in breast cancer: evidence of alterations in PIG8, a gene involved in p53-induced apoptosis. Oncogene. 2001;20:7753–7760. doi: 10.1038/sj/onc/1204993. [DOI] [PubMed] [Google Scholar]
- Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L, Crook T, Lu X. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell. 2001;8:781–794. doi: 10.1016/s1097-2765(01)00367-7. [DOI] [PubMed] [Google Scholar]
- Sgroi DC, Teng S, Robinson G, LeVangie R, Hudson JR, Jr, Elkahloun AG. In vivo gene expression profile analysis of human breast cancer progression. Cancer Res. 1999;59:5656–5661. [PubMed] [Google Scholar]
- Zhang H, Somasundaram K, Peng Y, Tian H, Zhang H, Bi D, Weber BL, El-Deiry WS. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene. 1998;16:1713–1721. doi: 10.1038/sj/onc/1201932. [DOI] [PubMed] [Google Scholar]
- Catteau A, Harris WH, Xu CF, Solomon E. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene. 1999;18:1957–1965. doi: 10.1038/sj/onc/1202509. [DOI] [PubMed] [Google Scholar]
- Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin S-F, Daigo Y, Russell P, Wilson A, Sowter HM, Delhanty JDA, Ponder BAJ, Kouzarides T, Caldas C. Mutations truncating the EP300 acetylase in human cancers. Nat Genet. 2000;24:300–303. doi: 10.1038/73536. [DOI] [PubMed] [Google Scholar]
- Mazars R, Spinardi L, BenCheikh M, Simony-Lafontaine J, Jeanteur P, Theillet C. p53 mutations occur in aggressive breast cancer. Cancer Res. 1992;52:3918–3923. [PubMed] [Google Scholar]
- Alsner J, Yilmaz M, Guldberg P, Hansen LL, Overgaard J. Heterogeneity in the clinical phenotype of TP53 mutations in breast cancer. Clin Cancer Res. 2000;6:3923–3931. doi: 10.1186/bcr109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg T, Klaar S, Karf G, Nordgren H, Holmberg L, Bergh J. Increased p53 mutation frequency during tumor progression – results from a breast cancer cohort. Cancer Res. 2001;61:8317–8321. [PubMed] [Google Scholar]
- Linderholm BK, Lindahl T, Holmberg L, Klaar S, Lennerstrand J, Henriksson R, Bergh J. The expression of vascular endothelial growth factor correlates with mutant p53 and poor prognosis in human breast cancer. Cancer Res. 2001;61:2256–2260. [PubMed] [Google Scholar]
- Maass N, Teffner M, Rosel F, Pawaresch R, Jonat W, Nagasaki K, Rudolph P. Decline in the expression of the serine proteinase inhibitor maspin is associated with tumour progression in ductal carcinomas of the breast. J Pathol. 2001;195:321–326. doi: 10.1002/path.948. [DOI] [PubMed] [Google Scholar]
- Zou Z, Gao C, Nagaich AK, Connell T, Saito S, Moul JW, Seth P, Appella E, Srivastava S. p53 regulates the expression of the tumor suppressor gene maspin. J Biol Chem. 2000;275:6051–6054. doi: 10.1074/jbc.275.9.6051. [DOI] [PubMed] [Google Scholar]
- Aas T, Borresen AL, Geisler S, Smith-Sorensen B, Johnsen H, Varhaug JE, Akslen LA, Lonning PE. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat Med. 1996;2:811–814. doi: 10.1038/nm0796-811. [DOI] [PubMed] [Google Scholar]
- Geisler S, Lonning PE, Aas T, Johnsen H, Fluge O, Haugen DF, Lillehaug JR, Akslen LA, Borresen-Dale AL. Influence of TP53 gene alterations and c-erbB-2 expression on the response to treatment with doxorubicin in locally advanced breast cancer. Cancer Res. 2001;61:2505–2512. [PubMed] [Google Scholar]
- Kandioler-Eckersberger D, Ludwig C, Rudas M, Kappel S, Janschek E, Wenzel C, Schlagbauer-Wadl H, Mittlbock M, Gnant M, Steger G, Jakesz R. TP53 mutation and p53 overexpression for prediction of response to neoadjuvant treatment in breast cancer patients. Clin Cancer Res. 2000;6:50–56. [PubMed] [Google Scholar]
- Shao ZM, Wu J, Shen ZZ, Nguyen M. p53 mutation in plasma DNA and its prognostic value in breast cancer patients. Clin Cancer Res. 2001;7:2222–2227. [PubMed] [Google Scholar]
- Metcalfe S, Wheeler TK, Picken S, Negus S, Milner AJ. p53 autoantibodies in 1006 patients followed up for breast cancer. Breast Cancer Res. 2000;2:438–443. doi: 10.1186/bcr91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez G, Silva JM, Silva J, Garcia JM, Sanchez A, Navarro A, Gallego I, Provencio M, Espana P, Bonilla F. Wild type p73 over-expression and high-grade malignancy in breast cancer. Breast Cancer Res Treat. 2001;66:183–190. doi: 10.1023/a:1010624717311. [DOI] [PubMed] [Google Scholar]
- Barbareschi M, Pecciarini L, Cangi MG, Macri E, Rizzo A, Viale G, Doglioni C. p63, a p53 homologue is a selective marker of myoepithelial cells of the human breast. Am J Surg Pathol. 2001;25:1054–1060. doi: 10.1097/00000478-200108000-00010. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Achilles' heel of cancer? Nature. 2001;412:865–866. doi: 10.1038/35091170. [DOI] [PubMed] [Google Scholar]