

Abstract
The past decade has seen the definition of key signalling pathways downstream of receptor tyrosine kinases (RTKs) in terms of their components and the protein-protein interactions that facilitate signal transduction. Given the strong evidence that links signalling by certain families of RTKs to the progression of breast cancer, it is not surprising that the expression profile of key downstream signalling intermediates in this disease has also come under scrutiny, particularly because some exhibit transforming potential or amplify mitogenic signalling pathways when they are overexpressed. Reflecting the diverse cellular processes regulated by RTKs, it is now clear that altered expression of such signalling proteins in breast cancer may influence not only cellular proliferation (eg Grb2) but also the invasive properties of the cancer cells (eg EMS1/cortactin).
Keywords: breast cancer, SH2 domain, SH3 domain, signal transduction, tyrosine kinase
Introduction
Ligand binding to RTKs induced receptor dimerization, leading to activation of the intracellular kinase domain and autophosphorylation of specific tyrosine residues. This creates binding sites for phosphotyrosine binding modules on signalling proteins, such as SH2 and phosphotyrosine-binding domains [1]. SH2 domains are modules of approximately 100 amino acids that recognize phosphotyrosines within specific sequence contexts. This is achieved through a bipartite binding site that consists of a phosphotyrosine binding pocket and a pocket or groove for specific residues that are carboxyl-terminal to the phosphotyrosine. Phosphotyrosine-binding domains are conserved sequences of 100-150 amino acids, a subset of which recognize the consensus sequence NPXpY [1].
In addition to SH2 domains, many signalling proteins also contain SH3 and/or pleckstrin homology domains. SH3 domains consist of 50-75 residues and bind proline-rich motifs with the core sequence PXXP. Pleckstrin homology domains bind to specific polyphosphoinositides and inositol polyphosphates, and thus regulate membrane association and/or responses to the generation of specific second messengers [1].
SH2 domain-containing proteins are divided into two classes (Fig. 1a) [2]: class I proteins possess enzymatic activity and include c-Src and phospholipase C (PLC)-γ1; and class II SH2 proteins, such as Grb2, do not have a known catalytic activity and function as adaptor proteins, linking receptors or cytoplasmic tyrosine kinases to downstream effectors.
Figure 1.
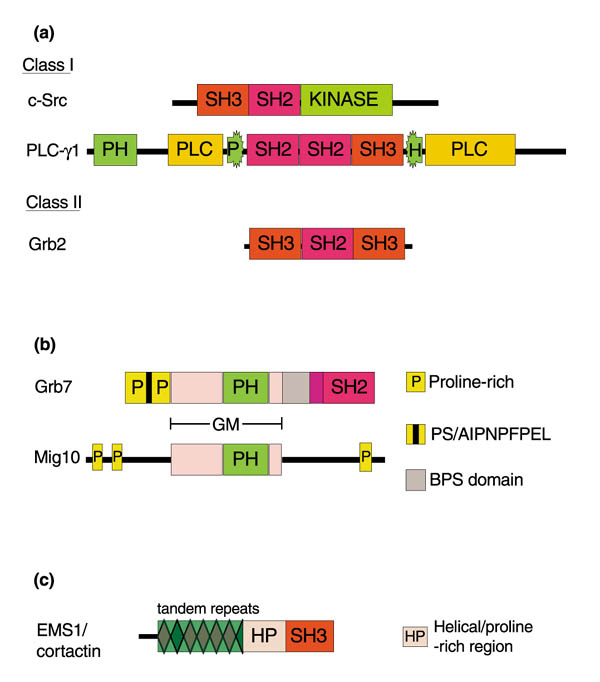
Structure of signalling intermediates that exhibit aberrant expression in human breast cancer. (a) Schematic representation of the structures of c-Src and PLC-γ1 (class 1 SH2 domain-containing proteins) and Grb2 (a class II protein). PLC-γ1 has two regions (designated PLC in the figure) that constitute its catalytic domain, and the second pleckstrin homology (PH) domain is interrupted by SH2 and SH3 modules. The different proteins and their domains are not drawn to scale; for details refer to text. (b) Comparison of the molecular architectures of Grb7 and Caenorhabditis elegans mig10. These two proteins possess a homologous central region (termed the Grb-Mig or GM domain), which contains a PH domain. Other than the presence of proline-rich regions, however, the remainder of their structures are dissimilar. (c) Schematic representation of EMS1/cortactin. The filamentous actin-binding repeat region is separated from the carboxyl-terminal SH3 module by a predicted helical domain followed by a stretch of amino acids rich in serine, threonine and proline residues (together designated HP in the figure).
Overexpression of class I SH2 proteins may lead to signal amplification through enhanced recruitment of the enzyme to activated receptors. Class II SH2 proteins, however, must bind to effector proteins with enzymatic activity in order to generate a signalling cascade. In this case, the effect of overexpression will depend on the ratio of catalytic subunits to adaptors. If the adaptor is limiting, then overexpression will result in increased complex formation between adaptor and effector, as well as increased association of this complex with the receptor. Importantly, over-expression of a particular signalling intermediate may amplify signalling downstream of multiple growth factor receptors if they couple to this pathway.
Altered expression of signalling intermediates in human breast cancer
c-Src
The protooncogene c-src (Fig. 1a) is a nonreceptor tyrosine kinase that is regulated by intramolecular interactions involving the SH2 and SH3 domains, the former binding phosphorylated Tyr530 at the carboxyl-terminus (for review [3].)
Evidence implicating a role for c-Src in breast cancer emerged when two studies [4,5] demonstrated enhanced c-Src kinase activity in breast tumours relative to that in normal breast tissue. Furthermore, increased cytosolic tyrosine kinase activity, largely attributable to c-Src, correlated with early systemic relapse in this disease [6,7]. Increases in both the expression and specific activity of the enzyme in breast cancers have been observed [5,7,8]. One mechanism for activation of c-Src in breast cancer is through SH2-domain-mediated association with the epidermal growth factor (EGF) receptor and erbB2, which both recruit this enzymes [9] and are commonly overexpressed in this disease [10]. An alternative mechanism, however, is through increased expression and/or activity of a Tyr530 phosphatase. Egan et al [11**] detected high levels of Src kinase activity in four breast cancer cell lines compared with in normal human foreskin fibroblasts and a normal breast epithelial cell line. This correlated with reduced phosphorylation of Tyr530 and increased membrane-associated protein tyrosine phosphatase activity, which preferentially dephosphorylated a synthetic Src family carboxyl-terminal phosphopeptide. Interestingly, this activity did not correlate with the expression or chromatographic profile of several known protein tyrosine phosphatases.
Coupling of the EGF receptor to Src is considered in more detail elsewhere in this thematic review [12], as is evidence from transgenic models for a role of Src in mammary tumourigenesis [13].
Phospholipase C-γ1
PLC-γ1 (Fig. 1a) cleaves phosphatidylinositol (4,5) bisphosphate into the second messengers inositol-1,4,5-trisphosphate and diacylglycerol, leading to calcium mobilization and activation of protein kinase C, respectively. PLC-γ1 tyrosine phosphorylation follows its interaction with activated EGF and erbB2 receptors in vivo, which stimulates its enzymatic activity [14].
Arteaga et al [15] demonstrated by both immunohistochemistry and immunoblotting that PLC-γ1 was expressed at higher levels in breast tumours relative to its expression in normal tissue. Furthermore, PLC-γ1 was tyrosine phosphorylated in the majority of the overexpressing tumours, and this correlated with the presence of high levels of the EGF receptor or erbB2. PLC-γ1 expression was also associated with oestrogen receptor (ER) negativity and a higher histological grade.
The significance of PLC-γ1 overexpression in breast cancer is unclear, because studies investigating the role of this enzyme in growth factor-stimulated proliferation have yielded conflicting results [14]. It should be noted, however, that PLC-γ1 also regulates cytoskeletal reorganization and hence cell motility (eg via indirect effects on the activity of actin-modifying proteins). Indeed, Chen et al [16] demonstrated that activation of PLC-γ1 was required for EGF-mediated motility, but not for mitogenesis. Consequently, overexpression of this enzyme may also influence cellular invasion and/or metastasis.
Grb2
Grb2 is an adaptor protein that consists of a SH2 domain flanked by two SH3 domains (Fig. 1a) [1,14]. Genetic and biochemical evidence indicates that Grb2 serves to link RTKs to the Ras pathway through binding of its SH3 domains to proline-rich regions in the GDP-GTP exchange factor for ras, Sos.
In a study of Grb2 expression among breast cancer cell lines [17], overexpression of Grb2 protein was observed in MCF-7, MDA-MB-361 and -453 cells relative to normal breast epithelial cells, and this was accompanied by a small amplification of the GRB2 gene locus. Also, increased Grb2 expression enhanced complex formation with Sos. Recently, we have extended this analysis to primary breast cancers (Yip et al, manuscript submitted), and demonstrated that approximately 50% of those tested exhibited a twofold or greater overexpression of GRB2 mRNA. Verbeek et al [18] found that Grb2 protein was overexpressed in all primary breast cancers compared with normal tissue, which suggests that a further upregulation of Grb2 expression may also occur at the translational level. Interestingly, in our study breast cancers exhibiting low levels of the EGF receptor expressed significantly higher GRB2 mRNA than high EGF receptor-expressing breast cancers, suggesting selection for a mechanism that amplifies signalling downstream from the receptor.
Although Grb2 does not have transforming potential when overexpressed [19], early studies demonstrated that Grb2 overexpression could amplify activation of Ras or mitogen-activated protein kinase (MAPK) in response to particular growth factors [19,20,21]. More recently, these analyses have been extended to mouse mammary tumourigenesis in response to expression of polyomavirus middle T (PyV MT) antigen, which couples to the Ras pathway via recruitment of a Shc-Grb2-Sos complex. Mice that were transgenic for PyV MT and heterozygous for GRB2 gene disruption had a delayed onset of mammary tumours induced by PyV MT relative to wild-type animals, indicating that the GRB2 gene dosage was limiting for tumourigenesis [22**]. It is therefore likely that upregulation of Grb2 expression functions in breast cancer progression to amplify signalling via the Ras pathway downstream of particular tyrosine kinases.
The Grb7 family
The Grb7 family of adaptors consists of three proteins (Grb7, Grb10 and Grb14), which display a conserved structure (Fig. 1b; for review [23]). Recently, a novel region was identified between the pleckstrin homology and SH2 domains (the BPS domain), which, in the case of Grb10 and Grb14, mediates binding to the activated insulin receptor [24*, 25*].
Stein et al [26] demonstrated that, due to the close proximity of these genes on chromosome 17q, GRB7 is frequently coamplified and coexpressed with ERBB2 in breast cancer cell lines and tumours. Furthermore, because Grb7 binds tightly to erbB2, this suggests that amplification of a novel erbB receptor signalling pathway occurs in a subset of breast cancers. We have recently extended this analysis and demonstrated that Grb7 is also recruited by erbB3 and erbB4 upon heregulin stimulation of breast cancer cells [27]. Because Grb7 is also bound by tyrosine phosphorylated Shc [26,27], this indicates that Grb7 is a major component of the erbB receptor signalling cascade in breast cancer cells.
Grb10 and Grb14 are both recruited in vivo by the insulin receptor [23,25*], although contrasting results have been presented for the ability of Grb10 to coimmunoprecipitate with the insulin-like growth factor (IGF)-I receptor in vivo [23]. Grb10 is expressed by most breast cancer cell lines [28], whereas GRB14 mRNA is differentially expressed among a panel of such lines, expression correlating with ER positivity [29]. We have yet to extend this analysis of Grb14 expression to primary breast cancers.
We hypothesize that members of the Grb7 family have a dual role in tyrosine kinase signalling. First, there is probably an adaptor function mediated by the amino-terminal and GM regions, which may be related to regulation of cell migration, because increased expression of a Grb7 variant correlates with the invasive and metastatic potential of oesophageal cancers, and downregulation of Grb7 in an oesophageal cancer cell line inhibits cell invasion [30*]. Whether this is a reflection of their homology to mig-10, which is required for long-range migration of certain cell types during Caenorhabditis elegans development [23], is unclear, and the effector molecules involved are currently unknown. The second role is as repressors of signalling by specific RTKs. For example, overexpression of Grb14 reduces insulin-induced DNA and glycogen synthesis [25*]. These results are compatible with the Grb10/14 BPS-SH2 region binding the IGF-I receptor/insulin receptor kinase domain and inhibiting substrate recruitment and/or phosphorylation [24*, 25*]. This repressor role is also supported by the recent identification of GRB10 as a candidate gene for Silver-Russell syndrome [31].
These two functions may reflect a need to coordinate cell division with cell migration, and the net effect of overexpression of a Grb7 family protein may be determined by the particular family member, the RTK system under study, and the expression levels of receptor and adaptor. Consequently, overexpression of Grb7 in breast cancer may lead to a more invasive phenotype due to upregulation of the adaptor (cell migration) function, whereas the loss of Grb14 expression in ER-negative cell lines [29] can be reconciled with the loss of a repressor of IGF signalling, because the IGFs are important mitogens for breast cancer cells (for review [32]). Clearly these issues need to be addressed by further research at both a basic and translational level.
EMS1/cortactin
EMS1 is the human homologue of the v-Src substrate cortactin, and is a 80-85 kDa multidomain protein that colocalizes with cortical actin [33] (Fig. 1c). Tyrosine phosphorylation of EMS1/cortactin is increased by a variety of stimuli, including growth factor receptor activation or integrin-mediated cell adhesion, and is tightly linked to the activatioin of Src family kinases [34,35*]. Recombinant cortactin binds and crosslinks actin filaments in vitro via the repeat region, and this is inhibited by Src-mediated tyrosine phosphorylation [34]. Overexpression of cortactin increases cell motility in both fibroblasts [36] and endothelial cells [35*], and this is dependent on the presence of the sites of tyrosine phosphorylation by Src [35*].
The gene encoding EMS1 localizes to chromosome 11q13, which is amplified in approximately 13% of breast tumours, this correlating with ER positivity [37]. 11q13 is a large amplicon of 2.5-5 Mb that contains several potential oncogenes in CCND1, INT-2/FGF3, HST-1/FGF4 and EMS1. Because the INT-2 and HST-1 genes are rarely expressed in tumours with 11q13 amplification [37], the candidate oncogenes are CCND1 and EMS1. A recent study [38**] investigated the frequency of amplification of EMS1 relative to other 11q13 markers in breast cancer patients and the association of these events with particular disease phenotypes. Analysis of approximately 1000 primary breast cancers revealed that EMS1 was amplified in approximately 15% of the samples, and in 7% of the patients this amplification was independent of CCND1 or INT-2. Interestingly, in common with CCND1, EMS1 amplification was associated with increased risk of relapse in lymph node-negative disease. Increased EMS1 gene copy number was associated with increased risk of relapse and death in patients with ER-negative tumours, but was without effect in ER-positive patients. In contrast, CCND1 amplification was associated with poor prognosis in the ER-positive, but not ER-negative subset. A further study that examined EMS1 mRNA expression in a different patient cohort also detected an association between EMS1 overexpression and reduced disease-free survival in ER-negative patients [39]. Consequently, not only are CCND1 and EMS1 amplified independently, but they are also associated with different disease phenotypes.
Considering the biological properties of EMS1, it is likely that overexpression increases the invasive and/or metastatic potential of the cancer cells. What are the possible underlying mechanisms for the poor prognosis specifically associated with EMS1-amplified, ER-negative tumours? One possibility is enhanced tyrosine phosphorylation of EMS1 due to activation of Src by the EGF receptor and erbB2, the expression of which are inversely related to ER status [10]. Tyrosine phosphorylation of EMS1/cortactin is required for the increased motility of cells engineered to overexpress this protein [35*]. However, serum or growth factor treatment of EMS1-amplified cells leads to a mobility shift of EMS1/cortactin from 80 to 85 kDa, which corresponds to increased serine/threonine phosphorylation, and a redistribution of the protein to cell-substratum contact sites [40]. We have recently demonstrated [41*] that the EGF-induced mobility shift is MAPK kinase dependent and identified the MAPKs as candidates for phosphorylation of the EMS1 helical-proline-rich region. Consequently, this represents a further mechanism whereby EGF receptor signalling may modulate EMS1 function in ER-negative breast cancer cells.
Conclusion
Clearly some of the signaling proteins described in this review are worthy of further investigation as potential prognostic indicators, and the protein-protein interactions that underline their function provide attractive targets for therapeutic intervention. Further elucidation of the roles of these proteins in breast tumourigenesis is likely to involve manipulation via gene knockout and transgenic approaches, as recently performed on Grb2 [22**]. However, we would also like to highlight the synergy that can occur between basic and translational research in this area, because our recent clinical studies on EMS1 in breast cancer have provided interesting hypotheses regarding regulation of EMS1 function, which are currently being addressed in the laboratory.
References
- Pawson T, Scott J. Signaling through scaffold, anchoring and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Schlessinger J, Ullrich A. Growth factor signaling by receptor tyrosine kinases. Neuron. 1992;9:383–391. doi: 10.1016/0896-6273(92)90177-f. [DOI] [PubMed] [Google Scholar]
- Mayer B. Clamping down on Src activity. Curr Biol. 1997;7:R295–R298. doi: 10.1016/s0960-9822(06)00141-2. [DOI] [PubMed] [Google Scholar]
- Jacobs C, Rubsamen H. Expression of pp60c-src protein kinase in adult and fetal human tissue: high activities in some sarcomas and mammary carcinomas. Cancer Res. 1983;43:1696–1702. [PubMed] [Google Scholar]
- Rosen N, Bolen JB, Schwartz AM, et al. Analysis of pp60c-src protein kinase activity in human tumor cell lines and tissues. J Biol Chem. 1986;261:13754–13759. [PubMed] [Google Scholar]
- Hennipman A, van Oirschot BA, Smits J, Rijksen G, Staal GEJ. Tyrosine kinase activity in breast cancer, benign breast disease, and normal breast tissue. Cancer Res. 1989;49:516–521. [PubMed] [Google Scholar]
- Ottenhoff-Kalff AE, Rijksen G, van Beurden EACM, et al. Characterisation of protein tyrosine kinases from human breast cancer: Involvement of the c-src oncogene product. Cancer Res. 1992;52:4773–4778. [PubMed] [Google Scholar]
- Verbeek B, Vroom T, Adriaansen-Slot S, et al. c-Src protein expression is increased in human breast cancer. An immunohistochemical and biochemical analysis. J Pathol. 1996;180:383–388. doi: 10.1002/(SICI)1096-9896(199612)180:4<383::AID-PATH686>3.3.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Daly RJ. Take your partners, please: signal diversification by the erbB family of receptor tyrosine kinases. Growth Factors. 1999;16:255–263. doi: 10.3109/08977199909069144. [DOI] [PubMed] [Google Scholar]
- Pinkas-Kramarski R, Alroy I, Yarden Y. ErbB receptors and EGF-like ligands: cell lineage determination and oncogenesis through combinatorial signalling. J Mam Gland Biol Neoplasia. 1997;2:97–107. doi: 10.1023/a:1026343528967. [DOI] [PubMed] [Google Scholar]
- Egan C, Pang A, Durda D, et al. Activation of Src in human breast tumor cell lines: elevated levels of phosphotyrosine phosphatase activity that preferentially recognizes the Src carboxy terminal negative regulatory tyrosine 530. Oncogene. 1999;18:1227–1237. doi: 10.1038/sj/onc/1202233. [DOI] [PubMed] [Google Scholar]
- Biscardi JS, Ishizawar RC, Silva CM, Parsons SJ. Tyrosine kinase signalling in breast cancer: EGF receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000. [DOI] [PMC free article] [PubMed]
- Andrechek ER, Muller WJ. Tyrosine kinase signalling in breast cancer: tyrosine kinase-mediated signal transduction in transgenic mouse models of human breast cancer. Breast Cancer Res. 2000. [DOI] [PMC free article] [PubMed]
- van der Geer P, Hunter T. Receptor protein tyrosine kinases and their signal transduction pathways. Ann Rev Cell Biol. 1994;10:251–337. doi: 10.1146/annurev.cb.10.110194.001343. [DOI] [PubMed] [Google Scholar]
- Arteaga CL, Johnson MD, Todderud G, et al. Elevated content of the tyrosine kinase substrate phospholipase C-γl in primary human breast carcinomas. Proc Natl Acad Sci USA. 1991;88:10435–10439. doi: 10.1073/pnas.88.23.10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Gupta K, Wells A. Cell movement elicited by epidermal growth factor receptor requires kinase and autophosphorylation but is separable from mitogenesis. J Cell Biol. 1994;124:547–555. doi: 10.1083/jcb.124.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly RJ, Binder MD, Sutherland RL. Overexpression of the Grb2 gene in human breast cancer cell lines. Oncogene. 1994;9:2723–2727. [PubMed] [Google Scholar]
- Verbeek B, Adriaansen-Slot S, Rijksen G, Vroom T. Grb2 overexpression in nuclei and cytoplasm of human breast cells: a histochemical and biochemical study of normal and neoplastic mammary tissue specimens. J Pathol. 1997;183:195–203. doi: 10.1002/(SICI)1096-9896(199710)183:2<195::AID-PATH901>3.3.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Suen KL, Bustelo XR, Pawson T, Barbacid M. Molecular cloning of the mouse grb2 gene: differential expression of the Grb2 adaptor protein with epidermal growth factor and nerve growth factor receptors. Mol Cell Biol. 1993;13:5500–5512. doi: 10.1128/mcb.13.9.5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale NW, Kaplan S, Lowenstein EJ, Schlessinger J, Bar-Sagi D. Grb2 mediates the EGF-dependent activation of guanine nucleotide exchange on ras. Nature. 1993;363:88–92. doi: 10.1038/363088a0. [DOI] [PubMed] [Google Scholar]
- Skolnik EY, Batzer A, Li N, et al. The function of GRB2 in linking the insulin receptor to ras signaling pathways. Science. 1993;260:1953–1955. doi: 10.1126/science.8316835. [DOI] [PubMed] [Google Scholar]
- Cheng A, Saxton T, Sakai R, et al. Mammalian Grb2 regulates multiple steps in embryonic development and malignant transformation. Cell. 1998;95:793–803. doi: 10.1016/s0092-8674(00)81702-x. [DOI] [PubMed] [Google Scholar]
- Daly R. The Grb7 family of signalling proteins. Cell Signal. 1998;10:613–618. doi: 10.1016/s0898-6568(98)00022-9. [DOI] [PubMed] [Google Scholar]
- He W, Rose D, Olefsky J, Gustafson T. Grb10 interacts differentially with the insulin receptor, insulin-like growth factor I receptor, and epidermal growth factor receptor via the Grb10 src homology 2 (SH2) domain and a second novel domain located between the pleckstrin homology and SH2 domains. J Biol Chem. 1998;273:6860–6867. doi: 10.1074/jbc.273.12.6860. [DOI] [PubMed] [Google Scholar]
- Kasus-Jacobi A, Perdereau D, Auzan C, et al. Identification of the rat adapter Grb14 as an inhibitor of insulin actions. J Biol Chem. 1998;273:26026–26035. doi: 10.1074/jbc.273.40.26026. [DOI] [PubMed] [Google Scholar]
- Stein D, Wu J, Fuqua SAW, et al. The SH2 domain protein Grb-7 is co-amplified, overexpressed and in a tight complex with HER2 in breast cancer. EMBO J. 1994;13:1331–1340. doi: 10.1002/j.1460-2075.1994.tb06386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiddes RJ, Campbell DH, Janes PW, et al. Analysis of Grb7 recruitment by heregulin-activated erbB receptors reveals a novel target selectivity for erbB3. J Biol Chem. 1998;273:7717–7724. doi: 10.1074/jbc.273.13.7717. [DOI] [PubMed] [Google Scholar]
- Dong L, Du H, Porter S, et al. Cloning, chromosome localization, expression and characterization of a Src homology 2 and pleckstrin homology domain-containing insulin receptor binding protein hGrb10γ. J Biol Chem. 1997;272:29104–29112. doi: 10.1074/jbc.272.46.29104. [DOI] [PubMed] [Google Scholar]
- Daly RJ, Sanderson GM, Janes PJ, Sutherland RL. Cloning and characterization of Grb14, a novel member of the Grb7 gene family. J Biol Chem. 1996;271:12502–12510. doi: 10.1074/jbc.271.21.12502. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Mori M, Akiyoshi T, et al. A novel variant of human Grb7 is associated with invasive esophageal carcinoma. J Clin Invest. 1998;102:821–827. doi: 10.1172/JCI2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi N, Kuroiwa Y, Kohda T, et al. Identification of the Meg1/Grb10 imprinted gene on mouse proximal chromosome 11, a candidate for the Silver-Russell syndrome gene. Proc Natl Acad Sci USA. 1998;95:1102–1107. doi: 10.1073/pnas.95.3.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yee D. Tyrosine kinase signalling in breast cancer: insulin-like growth factors and their receptors in breast cancer. Breast Cancer Res. 2000. [DOI] [PMC free article] [PubMed]
- Schuuring E, Vernoeven E, Litvinov S, Michalides RJAM. The product of the EMS1 gene, amplified and overexpressed in human carcinomas, is homologous to a v-src substrate and is located in cell substratum contact sites. Mol Cell Biol. 1993;13:2891–2898. doi: 10.1128/mcb.13.5.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Ni Y, Wang T, et al. Down-regulation of the filamentous actin cross-linking activity of cortactin by Src-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:13911–13915. doi: 10.1074/jbc.272.21.13911. [DOI] [PubMed] [Google Scholar]
- Huang C, Liu J, Haudenschild C, Zhan X. The role of tyrosine phosphorylation of cortactin in the locomotion of endothelial cells. J Biol Chem. 1998;273:25770–25776. doi: 10.1074/jbc.273.40.25770. [DOI] [PubMed] [Google Scholar]
- Patel A, Schechter G, Wasilenko W, Somers K. Overexpression of EMS1/cortactin in NIH3T3 fibroblasts causes increased cell motility and invasion in vitro. Oncogene. 1998;16:3227–3232. doi: 10.1038/sj/onc/1201850. [DOI] [PubMed] [Google Scholar]
- Fantl V, Smith R, Brookes S, Dickson C, Peters G. Chromosome 11q13 abnormalities in human breast cancer. Cancer Surv. 1993;18:77–94. [PubMed] [Google Scholar]
- Hui R, Campbell D, Lee C, et al. EMS1 amplification can occur independently of CCND1 or INT2 amplification at 11q13 and may identify different phenotypes in primary breast cancer. Oncogene. 1997;15:1617–1623. doi: 10.1038/sj/onc/1201311. [DOI] [PubMed] [Google Scholar]
- Hui R, Ball J, Macmillan R, et al. EMS1 gene expression in primary breast cancer: relationship to cyclin D1 and oestrogen receptor expression and patient survival. Oncogene. 1998;17:1053–1059. doi: 10.1038/sj/onc/1202023. [DOI] [PubMed] [Google Scholar]
- van Damme H, Brok H, Schuuring-Scholtes E, Schuuring E. The redistribution of cortactin into cell-matrix contact sites in human carcinoma cells with 11q13 amplification is associated with both overexpression and post-translational modification. J Biol Chem. 1997;272:7374–7380. doi: 10.1074/jbc.272.11.7374. [DOI] [PubMed] [Google Scholar]
- Campbell D, Sutherland R, Daly R. Signaling pathways and structural domains required for phosphorylation of EMS1/cortactin. Cancer Res. 1999;59:5376–5385. [PubMed] [Google Scholar]