

Abstract
An estimated 8 million people are infected each year with the pathogen, Mycobacterium tuberculosis, and over 2 million die annually1. Yet only about 10% of those infected develop tuberculosis. Genetic variation within host populations is known to play a significant role in humans and animals 2,3, but the nature of genetic control of host resistance to tuberculosis remains poorly understood. Previously we mapped a new genetic locus on mouse chromosome 1, designated sst1 (for supersusceptibility to tuberculosis1)4. Here we demonstrate in sst1 congenic mouse strains that this locus mediates innate immunity, and identify a candidate gene, Intracellular Pathogen Resistance 1 (Ipr1), within the sst1 locus. The Ipr1 gene is upregulated in the sst1 resistant macrophages upon activation and infection, but is not expressed in the sst1 susceptible macrophages. Expression of the Ipr1 transgene in the sst1 susceptible macrophages limits multiplication not only of MTB but also Listeria monocytogenes and switches a cell death pathway of the infected macrophages from necrosis to apoptosis. Our data suggest that the Ipr1 gene product may play a novel role in integrating signals generated by intracellular pathogens with mechanisms controlling innate immunity, cell death and pathogenesis.
Keywords: Bacterial, Lung, Inflammation, Innate Immunity, Tuberculosis, Rodent
It is estimated that approximately one third of the human population on the planet has been infected by virulent Mycobacterium tuberculosis (MTB)1,5. Susceptibility to clinical tuberculosis is known to be influenced by the environmental factors, such as stress, malnutrition, concomitant infections (HIV) or senescence6,7. While genetic variation within host populations is also known to play a role in resistance and susceptibility, individual genes responsible for innate immunity to the pathogen have been elusive. In susceptible individuals, progression of lung tuberculosis often leads to formation of characteristic necrotic ‘cavities’ that destroy significant portions of the lung. Beyond their life-threatening clinical consequences, these lesions are essential for efficient transmission of MTB via aerosol. Since in humans tuberculosis is transmitted primarily via the respiratory route, the ability to cause lung disease is considered a key aspect of the pathogen’s virulence strategy, which ensures its evolutionary success. Therefore, understanding pathogenic mechanisms that are employed by virulent MTB during lung tuberculosis in susceptible individuals is essential for developing effective prevention and treatment strategies8,9. However, detailed mechanistic studies of pathogenesis of lung tuberculosis and its genetic control have been limited by the fact that in mouse models of MTB infection, necrotic lesions in the lungs are rarely found unless the mouse is rendered systemically immunodeficient.
C3HeB/FeJ inbred mice are extremely susceptible to MTB and develop dramatic lung pathology, which leads to their rapid death after infection with virulent MTB10,11. We generated a congenic mouse strain C3H.B6-sst1 (sst1R) that carries the C57BL/6J-derived resistant allele at the sst1 locus on the C3HeB/FeJ genetic background. The survival time of the sst1R congenic mice infected either intravenously (i.v.) with a high dose of MTB (Fig. 1a), or with a low dose of MTB via respiratory route (Fig. 1b), relative to their sst1S counterparts is significantly lengthened, indicating a profound effect of the locus on anti-tuberculosis immunity. However, the shorter survival of the C3H.B6-sst1 (sst1R) mice, as compared to the resistant parental strain C57BL/6J (B6), clearly indicates that the sst1 locus is responsible for a significant portion, but not the whole, of the tuberculosis resistance phenotype of the B6 mice.
Figure 1. The sst1 locus mediates innate immunity to tuberculosis.
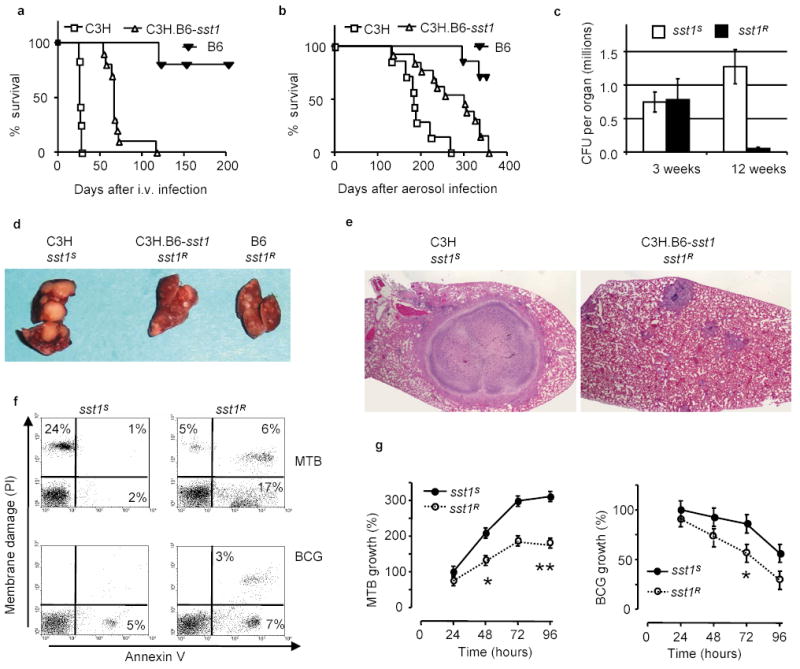
a, b, Survival of C3H, B6, and the C3H.B6-sst1 (sst1R) mice after i.v. (a) or aerosol (b) infection with MTB; c, MTB bacterial loads in the lungs of the sst1 congenic mice after the aerosol infection; d, e Tuberculosis lung lesions 25 days after i.v. infection (d) and 12 weeks after aerosol infection, H&E, 40X original magnification (e); f, FACS analysis of mechanism of cell death of the sst1 congenic macrophages infected with MTB (top panels) or BCG (bottom panels) in vitro; g, Multiplication of MTB (left panel) or M. bovis BCG (right panel) in the sst1 congenic macrophages in vitro (*p<0.01, **p<0.001). Error bars represent 95% confidence intervals.
The specific effect of the sst1 locus on progression of tuberculosis was related to more efficient control of MTB multiplication primarily in the lungs after both respiratory challenge by aerosolized MTB (Fig. 1c), and systemic i.v. infection (Supplementary Fig.2a). The development of large necrotic lung lesions within 4 weeks after i.v. infection characteristic of sst1S mice was prevented in the presence of the sst1R allele (Fig. 1d). After a low dose aerosol infection, chronic tuberculosis infection ensued, and the sst1S mice developed encapsulated necrotic lung lesions, in some cases reaching approximately one third of the lung lobe (Fig. 1e), which resembled tuberculosis cavities in human lungs. Mycobacteria were present both extracellularly within necrotic central areas surrounded by the fibrotic capsule as well as within macrophages of the granuloma wall (Supplementary Fig.1). In the sst1R mice, lung lesions were much smaller and contained fewer macrophages infected with MTB.
Although the most dramatic effect of the sst1 polymorphism on progression of tuberculosis was observed in the lungs; bone marrow transplantation experiments demonstrated that bone marrow-derived, but not lung cells, were responsible for the effect of the sst1 locus (Supplementary Fig.2b). It is known that T lymphocytes and macrophages play a major role in host resistance to tuberculosis. We have found that, while T lymphocytes are functionally unaffected by the sst1 polymorphism (Jobe, in preparation), the sst1 disparate macrophages display considerable differences in their ability to control MTB in vitro. The rate of MTB multiplication was significantly higher in the sst1S macrophages (Fig. 1g, left panel). There was also a clear distinction in mechanism of macrophage cell death after the infection: the sst1S macrophages showed characteristic necrosis, while the sst1R macrophages underwent apoptosis (Fig.1f, upper left and upper right panels, respectively). The effect of the sst1 locus was much more pronounced upon macrophage infection with virulent MTB, since avirulent vaccine strain of M. bovis BCG failed to multiply in (Fig.1g, right panel) and to induce necrosis of the sst1S macrophages (Fig.1f, lower left panel).
In vivo, many cells within the tuberculosis lung granulomas of the sst1R mice contained TUNEL-positive apoptotic nuclei and no necrosis was observed, while the apoptotic nuclei were largely absent from the necrotic lesions of the sst1S mice (see Supplementary Note). Thus our studies, both in vivo and in vitro, indicated that the extreme susceptibility to virulent MTB of sst1S mice was associated with necrotic death of the susceptible macrophages. Virulent MTB was shown to cause necrosis of the infected epithelial cell lines 12. This necrosis-inducing propensity is specific for virulent MTB and is lost in RD1 mutants of MTB that have also dramatically decreased virulence12,13. It appears from our studies that, in addition to virulence determinants of the pathogen itself, mechanisms of host cell death depend on the host polymorphic gene(s) encoded within the sst1 locus.
To identify the critical gene(s), we employed a positional cloning strategy (see Supplementary Data and Supplementary Fig.3 for details). First, the sst1 minimal region was reduced to an interval between D1Mit439 and D1Mit49 markers on mouse chromosome 1 (Supplementary Fig.3a). This region contains a so-called HSR (for homogeneously stained region) repeat (Fig.2a). The HSR repeat region is, arguably, the largest repetitive region in the mouse genome14,15. Its size in inbred mouse strains, is estimated to be between 3.5 and 6 Mb15,16 and it remains unfinished by both mouse genome projects. After identifying and testing progeny of additional recombinants within this interval (Fig.2a), we concluded that the sst1 candidate region encompasses part of the HSR repeat region and a region of mouse chromosome 1 immediately downstream of the repeat, i.e. between the repeat region and the NppC gene. A total of 22 known and predicted genes are encoded within the sst1 critical region according to Ensembl and Celera databases of the mouse genome (Supplementary Table 1). It was impossible further to reduce the sst1 critical region by genetic recombination. Therefore, in the next step we tested expression of each of the sst1-encoded candidate genes in the lungs during tuberculosis infection in vivo and in macrophages infected with MTB in vitro using RT-PCR and RACE. The Ifi75 gene appeared from our studies as the most likely candidate (Supplementary Fig.3b and e).
Figure 2. Identification of the sst1candidate gene.
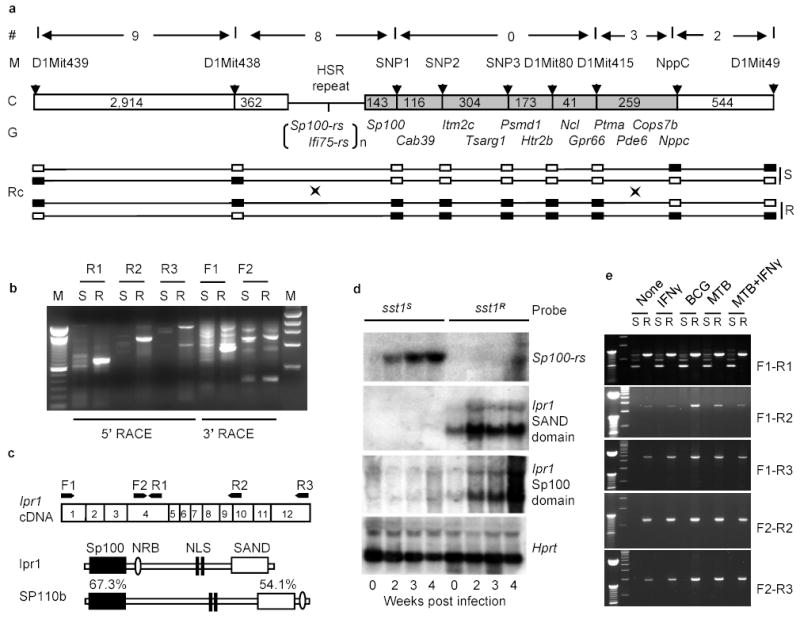
a, Physical map of the sst1 minimal region. (#)- number of recombination evens, (M) – polymorphic markers, (C) – chromosome with distances between the markers (kb); (G) – known genes; (RC) - recombinant chromosomes containing the sst1 resistant (R) or susceptible (S) alleles, genotypes for each marker are represented by solid (B6) and opened (C3H) boxes; b, Analysis of the Ifi75-rs expression in the tuberculosis lung lesions of the sst1 congenic mice by RACE. c, Domain structure of the Ipr1 and its human homolog SP110b and location of the PCR primers. d, Ipr1 and Sp100-rs gene expression in the lungs during MTB infection (Northern blot); e, Ipr1 gene expression in sst1S (S) or sst1R (R) macrophages infected MTB, BCG or activated with IFN-γ in vitro.
As shown in Fig.2b, the 5’ RACE products of the Ifi75 in the lungs of tuberculosis-infected mice was strikingly different between the sst1 congenic strains: a major single band was amplified from the lungs of the sst1 resistant mice, whereas this band was absent from the lungs of the sst1 susceptible strain and, instead, multiple weak products were obtained. Although some aberrant transcripts were present in the lung tissue of the sst1R animals as well, the majority of the Ifi75- related transcripts in their tuberculosis lung lesions were represented by a single isoform, which we named as Ipr1 (for intracellular pathogen resistance) to differentiate it from other Ifi75-related sequences (Ifi75-rs) identified by RACE and, perhaps, also encoded within the HSR repeat. The predicted Ipr1 protein is 92% identical to the Mus caroli Ifi75. It contains an Sp100-like domain in its N-terminus, LXXLL-type nuclear receptor binding motif (NRB), bipartite nuclear localization signal (NLS), and a SAND domain in its C-terminus (Fig.2c).
Using DNA probes specific for the Sp100 and SAND domains of the Ipr1, we have analyzed the kinetics of its expression by Northern hybridization in the lungs of the sst1 congenic mouse strains during progression of tuberculosis (Fig.2d). Expression of the Ipr1 gene was detectable in the lungs of the naive sst1R mice, and its expression increased significantly 2 weeks after intravenous infection with MTB and remained at elevated levels at later time points. However, expression of the Sp100 and SAND domain-containing Ifi75-rs in the lungs of the sst1 susceptible C3HeB/FeJ mice remained below the level of detection by Northern blot hybridization. Instead, the level of transcripts of another gene encoded within the HSR repeat region, Sp100-rs, was elevated in the lungs of the sst1S mice (Fig.2d).
To investigate expression of the Ipr1 gene in macrophages, we used five overlapping combinations of the Ipr1-specific PCR primers that cover the full length Ipr1 transcript (Fig.2e). The Ipr1 gene was expressed in non-activated sst1R macrophages and the level of its expression increased after infection with both avirulent BCG and virulent MTB. No expression of the full length transcript of the Ipr1 gene was seen in the sst1S macrophages under any stimulation conditions. Macrophages isolated from the tuberculosis lung lesions of the sst1R mice also expressed the full length Ipr1 transcript, while those from the sst1S mice did not (data not shown). We were unable to detect additional Ipr1-related transcripts induced in macrophages upon infection in vivo and in vitro or stimulation with IFN-γ. Despite the fact that the Ipr1 gene is encoded within the HSR repeat region, our data suggest that a single major isoform of this gene is expressed in sst1R macrophages either before or during tuberculosis infection and this isoform is not expressed in the sst1S C3HeB/FeJ mice.
The C3HeB/FeJ substrain is unique among all other substrains of C3H mice in terms of its extreme susceptibility to tuberculosis10,11. The C3HeB/FeJ mice die abruptly within 3.5–4 weeks after the infection displaying severe lung pathology. In our experiments, the survival time of other substrains of C3H was considerably longer and similar to the sst1R congenic strain C3H.B6-sst1 (Fig.3a). The bacterial loads in the lungs of the C3HeB/FeJ mice at 3 weeks post infection were 50 to 100 fold higher as compared to other substrains of C3H and the sst1R congenics (Fig.3b), suggesting a unique allele at the sst1 locus. We compared expression of the sst1-encoded candidate genes in the lungs of mice of four C3H substrains and the sst1R congenics and found that the lack of expression of the Ipr1 gene differentiated the C3HeB/FeJ from all other substrains of C3H (Fig.3c). Since all the C3H substrains originate from a common ancestor17, it is likely that they are genetically identical within the sst1 region and a unique de novo mutation has led to the defect of the Ipr1 gene expression in a C3HeB/FeJ mice and is responsible for a severe defect in their tuberculosis resistance.
Figure 3. Lack of the Ipr1 gene expression in the C3HeB/FeJ substrain correlates with its extreme susceptibility to MTB infection.
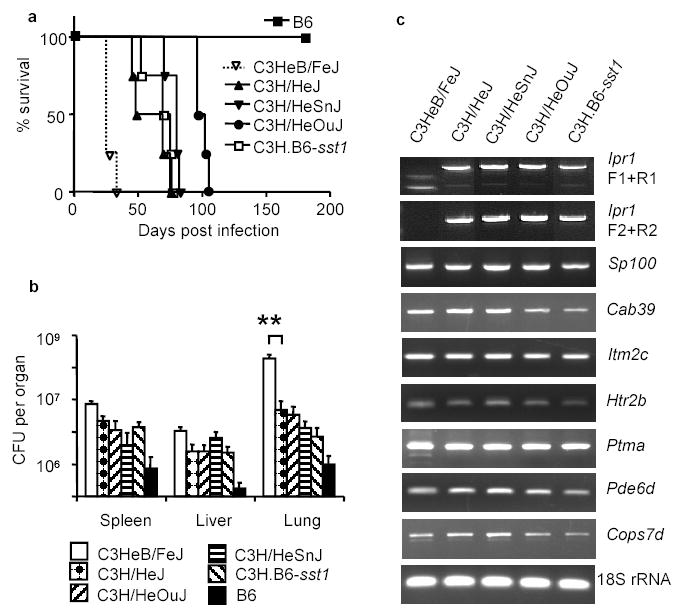
a, Survival after the intravenous infection with MTB; b, MTB bacterial loads three weeks after the infection (4 mice per strain, ** p<0.001, error bars represent standard deviation); c, Analysis of the sst1-encoded candidate gene expression in the tuberculosis lung lesions by RT-PCR three weeks after the infection.
We generated transgenic mice that expressed a full length copy of the Ipr1 cDNA on the susceptible C3HeB/FeJ background in a macrophage-specific manner under control of the human scavenger receptor A promoter (SR-A). Mature bone marrow-derived macrophages, as well as resident peritoneal macrophages obtained from those mice, expressed the Ipr1 transgene (Fig.4a). Despite the fact that the regulation of Ipr1 gene expression in the transgenic macrophages was clearly less efficient from the SR-A promoter than from the endogenous Ipr1 promoter (Fig.4a), when the Ipr1 transgenic mice were infected with virulent MTB, a statistically significant difference in the bacterial loads between the sst1S (Tg −/−) and the Ipr1 transgenic (Tg +/−) animals was observed in the lungs (Fig.4b). In vitro, the Ipr1 transgenic macrophages also controlled multiplication of MTB more effectively (Fig.4c) and turned on the apoptotic pathway of cell death upon interaction with virulent MTB (Fig.4d, right panels). The growth of another intracellular pathogen, L. monocytogenes, was dramatically suppressed in the Ipr1 transgenic macrophages by 50 to 100 fold (Fig.4e). Similar to the MTB infection, necrotic death accompanied infection of the sst1S macrophages with virulent L. monocytogenes (Fig.4f, left panel), while the Ipr1 transgenic macrophages displayed markers of apoptotic death (Fig.4f, right panel).
Figure 4. Expression of the Ipr1 transgene in the sst1S macrophages confers resistance to intracellular pathogens MTB and L. monocytogenes.
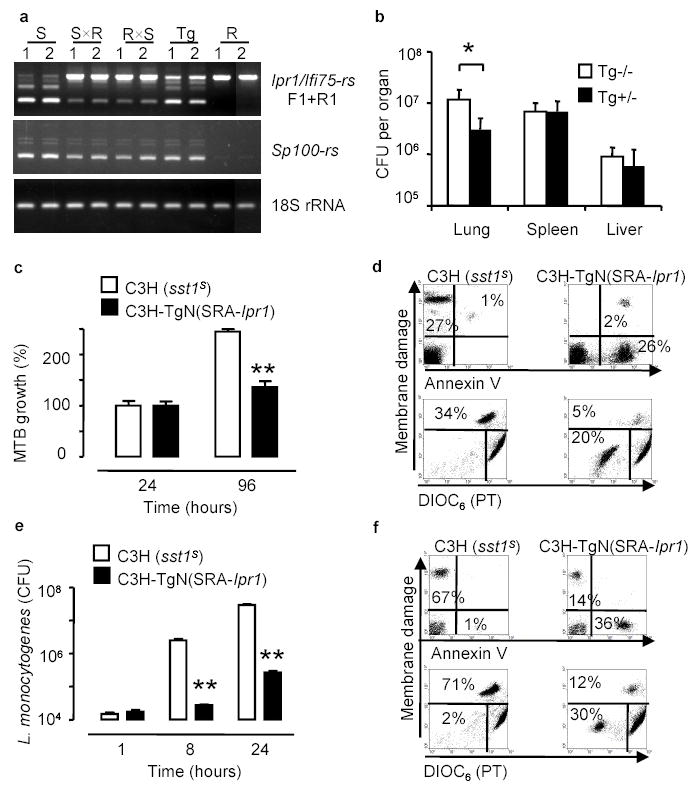
a, RT-PCR of Ipr1 and Sp100-rs in macrophages isolated from the sst1S (S), sst1R(R) mice, their F1 hybrids (SxR, RxS) and the Ipr1 transgenic (Tg) mice, 1 - IFNγ -stimulated, 2 - MTB-infected b, MTB bacterial loads in the Ipr1 transgenic (Tg+/−) and control (Tg−/−) mice after infection with MTB (7 mice/strain, * p<0.05); c, e growth of MTB (c) and L.monocytogenes (e) in the Ipr1 transgenic and control (sst1S) macrophages (three experiments were performed in triplicates, *p<0.01, **p<0.001, error bars represent 95% confidence interval).
Thus expression of a single gene, Ipr1, in the sst1S macrophages restored key functions related to pathogenesis of tuberculosis, which are encoded within the sst1 locus: greater control of multiplication of virulent MTB in vivo and in vitro as well as an apoptotic mechanism of MTB-induced macrophage cell death. Moreover, the Ipr1 gene mediated macrophage resistance to another intracellular pathogen, L. monocytogenes, suggesting that the Ipr1 product controls a common mechanism of innate resistance against several intracellular pathogens.
The closest homologue of the predicted Ipr1 protein in humans (41% of identity) is SP110b18, which localizes to a region of human chromosome 2 sythenic with the sst1 minimal region on mouse chromosome 1. Both the Ipr1 and the human SP110 proteins contain motifs that are involved in protein-protein interactions (Sp100 domain)19,20, chromatin binding (SAND domain)21,22, nuclear localization signal (NLS) and the nuclear receptor binding (NRB) motif LXXLL. Recent evidence suggests that human SP110 protein, may function as a nuclear hormone receptor transcriptional cofactor18 and directly bind the retinoic acid receptor23. Signaling through nuclear receptors, such as the corticosteroid receptor, retinoic acid receptor, PPARs and vitamin D plays an important role in control of various aspects of the macrophage life cycle, including differentiation, activation, response to pathogens and apoptosis24. Expression of both the Ipr1 gene and its human homologue SP110, is regulated by interferons 25, additionally implicating a role in immunity in both species. Moreover, polymorphisms in SP110 gene have been associated with susceptibility to the Hepatitis C virus 26, and the SP110b protein has been shown to physically interact with viral proteins, such as Epstein-Barr virus SM protein and Hepatitis C virus core protein 23,27. It is an intriguing possibility that the Ipr1 and SP110 proteins mediate cross talk between nuclear receptors, interferon signaling and pathogens. The viruses and, perhaps, intracellular pathogens might have evolved mechanisms to interfere with or exploit the Ipr1/SP110 function. Taken together, these data suggest that in mammals, since no Ipr1 homologues were found in yeasts or insects, the Ipr1-related proteins may play a novel role in integrating signals generated by intracellular pathogens or viruses with mechanisms regulating activation, gene expression and cell death of host cells28. Therefore, the SP110 may be an excellent candidate gene for testing for association with susceptibility to tuberculosis in human populations.
Methods:
Animals.
C57BL/6J, C3HeB/FeJ, C3H/HeJ, C3H/HeOuJ, and C3H/HeSnJ inbred mice were obtained from The Jackson Laboratory (Bar Harbor, Maine). The congenic C3H.B6-sst1 (sst1R), B6.C3H-sst1 and transgenic C3H-TgN(SRA-Ipr1) mouse strains were generated in our laboratory. The C3H.B6-sst1 congenic mice were obtained by introgression of an approximately 20 cM segment of B6-derived chromosome 1 with a proximal recombination breakpoint between D1Mit215 (47 cM) and D1Mit334 (49 cM) and a distal limit between D1Mit187 (64 cM) and D1Mit200 (75 cM) on the C3HeB/FeJ genetic background using 10 backcrosses. Importantly, the congenic interval transferred from the B6 resistant background did not include the Slc11a1 gene (formerly known as Nramp1), which is located at 39.2 cM. Thus, the sst1 resistant congenic mouse strain C3H.B6-sst1 carries the same allele of the Nramp1 as the parental sst1 susceptible C3HeB/FeJ mice. The B6.C3H-sst1 mice were obtained by transferring the sst1 susceptible allele on the B6 genetic background using 10 backcrosses. The transgenic C3H-TgN(SRA-Ipr1) mice were established by expressing the C57BL/6J-derived Ipr1 gene under the control of a macrophage specific Scavenger Receptor A (SRA) promoter 29 on the C3HeB/FeJ genetic background (see Supplementary Methods for details).
Infection of mice with MTB.
For i.v. infection 1 x 105 of live MTB were injected via tail vein in 100 μl of PBS. Aerosol infections were performed using aerosol apparatus manufactured by the College of Engineering Shops at the University of Wisconsin (Madison, WI). Mice were exposed to aerosol for 20 min, which resulted in the deposition of 15 to 30 CFU per mouse. Mice were sacrificed using halothane anesthesia. Organs were homogenized in PBS containing 0.05% Tween 80 and serial 10-fold dilutions were cultured on 7H10 agar enriched with 10% OADC (Difco, MI) for 3 weeks at 37°C.
Isolation and infection of murine bone marrow-derived macrophages (BMDM) with MTB and L. monocytogenes (LM) in vitro.
BMDM were isolated from femurs and tibias of male C3H, C3H.B6-sst1 and C3H-TgN(SRA-Ipr1) mice (6 to 8 weeks old) and infected with LM strain 10403S as previously described30. Macrophage monolayers were infected at multiplicity of infection 1 MTB Erdman per 10 macrophages (MOI 1:10). After 6h the cells were washed with PBS containing 1% FCS (PBS-1% FCS). The cells were incubated in complete medium containing 10% FCS and three coverslips were removed from the culture at indicated time points and separately lysed with 0.1% Triton X-100. Serial 10-fold dilutions of cell lysates were plated on 7H10 agar containing OADC and incubated for 3 weeks at 37°C.
Differentiation of apoptotic and necrotic pathway of macrophage cell death.
Macrophages were infected at MOI 1:10. At indicated time points, cells were stained with 10 nM DiOC6 (Molecular Probes) and 0.8 mM Ethidium Bromide (Sigma) for 20 min at 37°C, washed three times with PBS, fixed with 1% paraformaldehyde 20 min and washed once again with PBS. Cells were analyzed using BD FACScan flow cytometer (BD Biosciences) to differentiate between live (DiOC6highEB−); apoptotic (DiOC6lowEB−) and necrotic(EB+) cells. For Annexin V staining cells were incubated in Annexin binding buffer (10 mM Hepes, 140 mM NaCl and 2.5 mM CaCl2) and stained with 10 μl Annexin V-Alexa 488 (Molecular Probes) for 20 min, then counterstained with propidium iodide (PI, 1 μg/ml), washed with cold PBS twice, fixed with 1% paraformaldehyde for 30 min, and washed once with PBS. FACS analysis was used to differentiate between early apoptotic (Annexin V+ PI), late apoptotic (Annexin V+ PI+) and necrotic (Annexin V− PI+) cells.
Isolation and analysis of the Ipr1 cDNA.
The Ifi75-specific oligonucleotide primers (F1-2 and R1-3) are presented in Supplementary Methods and in Fig.2c. RACE-PCR was performed using the SMART RACE cDNA Amplification Kit (BD Clontech). The cDNAs were synthesized from the lungs of M. tuberculosis–infected C3HeB/FeJ and C3H.B6-sst1 mice. The RACE amplification products were purified using PCR purification columns (Qiagen), cloned into the plasmid vector pGEM-T (Promega) and sequenced using T7 and SP6 primers. A full length sequence of Ipr1 was confirmed by sequencing the “end-to-end” PCR product obtained using the F1 and R3 primers.
Statistical analysis.
Statgraphics Plus, release 4, 1999 (Statgraphics Corp., Rockville, MD) and GraphPad Prizm 3.0 (GraphPad, CA) software were used for the analysis. Comparison of bacterial loads was performed using Student’s t-test. Results are presented as the mean ± SD. A threshold for statistical significance was p < 0.05. Kaplan Meier Survival curves were generated and compared using the log-rank test (GraphPad Prizm). Intracellular bacterial growth and cell death were analysed by two factors ANOVA (time, genetic backgrounds and experiment). The statistical significance was tested at p<0.05 as critical value using the Student-Newman-Keuls post-test to compare means between both genetic backgrounds. Data are presented as the mean ± 95% confidence interval for mean (95% CI).
Supplementary Material
Acknowledgments
The authors are grateful to Ilona Breiterene, Kristine Vasquez, Kirsten Sigrist and Christina Mottley for expert technical assistance, to Drs. Raju Kucherlapati, Peter Demant, William F. Dietrich, Sergei Agoulnik and Donald Bloch for helpful discussions and support throughout the project. This work was supported by the National Institutes of Health (USA).
Footnotes
This work was supported by National Institutes of Health
The Ipr1 sequence GenBank accession number is AY845948.
Competing interests statement The authors declare that they have no competing financial interests.
Abbreviations: MTB – Mycobacterium tuberculosis; AFB – acid-fast bacilli
Supplementary Information accompanies the paper on www.nature.com/nature.
References
- 1.Raviglione MC. The TB epidemic from 1992 to 2002. Tuberculosis (Edinb) 2003;83:4–14. doi: 10.1016/s1472-9792(02)00071-9. [DOI] [PubMed] [Google Scholar]
- 2.Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- 3.Bellamy R, et al. Genetic susceptibility to tuberculosis in Africans: a genome-wide scan. Proc Natl Acad Sci U S A. 2000;97:8005–9. doi: 10.1073/pnas.140201897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kramnik I, Dietrich WF, Demant P, Bloom BR. Genetic control of resistance to experimental infection with virulent Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2000;97:8560–5. doi: 10.1073/pnas.150227197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bleed D, Dye C, Raviglione MC. Dynamics and control of the global tuberculosis epidemic. Curr Opin Pulm Med. 2000;6:174–9. doi: 10.1097/00063198-200005000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Corbett EL, et al. The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch Intern Med. 2003;163:1009–21. doi: 10.1001/archinte.163.9.1009. [DOI] [PubMed] [Google Scholar]
- 7.Bloom BR. Tuberculosis--the global view. N Engl J Med. 2002;346:1434–5. doi: 10.1056/NEJM200205093461902. [DOI] [PubMed] [Google Scholar]
- 8.Cosma CL, Sherman DR, Ramakrishnan L. The secret lives of the pathogenic mycobacteria. Annu Rev Microbiol. 2003;57:641–76. doi: 10.1146/annurev.micro.57.030502.091033. [DOI] [PubMed] [Google Scholar]
- 9.Taylor JL, et al. Pulmonary necrosis resulting from DNA vaccination against tuberculosis. Infect Immun. 2003;71:2192–8. doi: 10.1128/IAI.71.4.2192-2198.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamath AB, Alt J, Debbabi H, Behar SM. Toll-like receptor 4-defective C3H/HeJ mice are not more susceptible than other C3H substrains to infection with Mycobacterium tuberculosis. Infect Immun. 2003;71:4112–8. doi: 10.1128/IAI.71.7.4112-4118.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kramnik I, Demant P, Bloom BB. Susceptibility to tuberculosis as a complex genetic trait: analysis using recombinant congenic strains of mice. Novartis Found Symp. 1998;217:120–31. doi: 10.1002/0470846526.ch9. discussion 132–7. [DOI] [PubMed] [Google Scholar]
- 12.Hsu T, et al. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci U S A. 2003;100:12420–5. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guinn KM, et al. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol. 2004;51:359–70. doi: 10.1046/j.1365-2958.2003.03844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agulnik S, Plass C, Traut W, Winking H. Evolution of a long-range repeat family in chromosome 1 of the genus Mus. Mamm Genome. 1993;4:704–10. doi: 10.1007/BF00357793. [DOI] [PubMed] [Google Scholar]
- 15.Traut W, Rahn IM, Winking H, Kunze B, Weichehan D. Evolution of a 6-200 Mb long-range repeat cluster in the genus Mus. Chromosoma. 2001;110:247–52. doi: 10.1007/s004120100152. [DOI] [PubMed] [Google Scholar]
- 16.Weichenhan D, et al. Source and component genes of a 6-200 Mb gene cluster in the house mouse. Mamm Genome. 2001;12:590–4. doi: 10.1007/s00335-001-3015-9. [DOI] [PubMed] [Google Scholar]
- 17.Krog HH, Moutier R. Identification of inbred strains of mice: II. Characterization of different substrains of the C3H strain. J Hered. 1978;69:66–70. doi: 10.1093/oxfordjournals.jhered.a108905. [DOI] [PubMed] [Google Scholar]
- 18.Bloch DB, et al. Sp110 localizes to the PML-Sp100 nuclear body and may function as a nuclear hormone receptor transcriptional coactivator. Mol Cell Biol. 2000;20:6138–46. doi: 10.1128/mcb.20.16.6138-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wasylyk C, Schlumberger SE, Criqui-Filipe P, Wasylyk B. Sp100 interacts with ETS-1 and stimulates its transcriptional activity. Mol Cell Biol. 2002;22:2687–702. doi: 10.1128/MCB.22.8.2687-2702.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sternsdorf T, Jensen K, Reich B, Will H. The nuclear dot protein sp100, characterization of domains necessary for dimerization, subcellular localization, and modification by small ubiquitin-like modifiers. J Biol Chem. 1999;274:12555–66. doi: 10.1074/jbc.274.18.12555. [DOI] [PubMed] [Google Scholar]
- 21.Surdo PL, Bottomley MJ, Sattler M, Scheffzek K. Crystal structure and nuclear magnetic resonance analyses of the SAND domain from glucocorticoid modulatory element binding protein-1 reveals deoxyribonucleic acid and zinc binding regions. Mol Endocrinol. 2003;17:1283–95. doi: 10.1210/me.2002-0409. [DOI] [PubMed] [Google Scholar]
- 22.Bottomley MJ, et al. The SAND domain structure defines a novel DNA-binding fold in transcriptional regulation. Nat Struct Biol. 2001;8:626–33. doi: 10.1038/89675. [DOI] [PubMed] [Google Scholar]
- 23.Watashi K, et al. Modulation of retinoid signaling by a cytoplasmic viral protein via sequestration of Sp110b, a potent transcriptional corepressor of retinoic acid receptor, from the nucleus. Mol Cell Biol. 2003;23:7498–509. doi: 10.1128/MCB.23.21.7498-7509.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castrillo, A. & Tontonoz, P. Nuclear Receptors in Macrophage Biology: At the Crossroads of Lipid Metabolism and Inflammation. Annu Rev Cell Dev Biol (2004). [DOI] [PubMed]
- 25.Kadereit S, Gewert DR, Galabru J, Hovanessian AG, Meurs EF. Molecular cloning of two new interferon-induced, highly related nuclear phosphoproteins. J Biol Chem. 1993;268:24432–41. [PubMed] [Google Scholar]
- 26.Saito T, et al. Genetic variations in humans associated with differences in the course of hepatitis C. Biochem Biophys Res Commun. 2004;317:335–41. doi: 10.1016/j.bbrc.2004.03.056. [DOI] [PubMed] [Google Scholar]
- 27.Nicewonger J, Suck G, Bloch D, Swaminathan S. Epstein-Barr virus (EBV) SM protein induces and recruits cellular Sp110b to stabilize mRNAs and enhance EBV lytic gene expression. J Virol. 2004;78:9412–22. doi: 10.1128/JVI.78.17.9412-9422.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hofmann TG, Will H. Body language: the function of PML nuclear bodies in apoptosis regulation. Cell Death Differ. 2003;10:1290–9. doi: 10.1038/sj.cdd.4401313. [DOI] [PubMed] [Google Scholar]
- 29.Horvai A, et al. Scavenger receptor A gene regulatory elements target gene expression to macrophages and to foam cells of atherosclerotic lesions. Proc Natl Acad Sci U S A. 1995;92:5391–5. doi: 10.1073/pnas.92.12.5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyartchuk V, et al. The host resistance locus sst1 controls innate immunity to Listeria monocytogenes infection in immunodeficient mice. J Immunol. 2004;173:5112–20. doi: 10.4049/jimmunol.173.8.5112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.