

Abstract
Recent evidence argues that the oncogenesis and growth of CNS tumors occurs through dysregulated molecular and cellular mechanisms of neural development. New insights have emerged that have had a significant impact on both research and treatment of these cancers.
Keywords: embryonal tumors, medulloblastoma, astrocytoma, stem cells, cerebellum, cerebellar granule cells
Recent discoveries have linked the molecular and cellular origins of CNS tumors to mechanisms of neural development. The strongest case can be made for CNS embryonal tumors, which are the most common malignant brain tumors of childhood. In this review, I focus on clinical and biological aspects of CNS tumors, with emphasis on their relationship to cellular and molecular development.
Clinical presentation of brain tumors and conventional therapeutics
Primary tumors of the CNS are the most common solid tumors of childhood and a major source of mortality and neurological disability for people of all ages. Their incidence is 5–15/100 000 person-years. Currently, there are ~360 000 people living with a brain tumor in the United States, including 26 000 children (CBTRUS, 2002).
Children with a malignant brain tumor have an estimated 5-year-survival rate of 60%; survival rates decline progressively with age to reach a level of >5% for those older than 65 years. For each individual, survival probability is linked closely with histological type. Although there are many histological types of CNS neoplasms recognized by the World Health Organization (WHO), this review will focus primarily on astrocytomas and medulloblastomas, which account for ~75% of tumors in children, and astrocytomas/oligodendrogliomas, which comprise >50% of all tumors in adults (Kleihues and Cavenee, 2000). Brain tumors are further classified into histological grades that generally correlate with biological behavior. Low-grade tumors (WHO grades I and II) are comprised of neoplastic cells with relatively compact and homogenous nuclei and cytological differentiation reminiscent of their normal cells of origin. High-grade tumors diverge from the appearance of their cells of origin. For example, anaplastic (less differentiated) astrocytomas (WHO grade III) have atypical pleomorphic nuclei, poorly differentiated cytoplasm and high rates of proliferation as evident by the presence of mitoses. The most malignant tumors (WHO grade IV) have extremely atypical nuclei and very high mitotic rates, often associated with striking microvascular proliferation and regions of necrosis. The overall probability of survival is substantially lower for patients with high-grade tumors when compared to lower-grade tumors of the same histological type.
Medulloblastomas occur exclusively in the cerebellum. Their peak incidence is in the latter half of the first decade of life, and they are rare beyond 20 years of age. When first diagnosed, children with medulloblastoma typically have signs and symptoms of a mass in the posterior fossa, including headache and vomiting from obstructive hydrocephalus, ataxia and cranial-nerve deficits. Because medulloblastomas tend to spread throughout the neuraxis, treatment consists of surgical resection, radiation to the entire brain and spine, and multi-drug chemotherapy for ~1 year. The overall 5-year-survival rate is 65–80%. Unfortunately, survivors almost invariably have significant learning disabilities and neuroendocrine hormonal deficiencies, largely because of the effects of aggressive therapy on the developing nervous system (Packer et al., 1994; Packer et al., 1999).
Traditionally, medulloblastomas have been divided into two principal histological subtypes, but all classes are malignant and therefore are considered to be WHO grade IV tumors. The majority are classical medulloblastomas or ‘small blue cell tumors’ that are highly cellular with compact nuclei and minimal cellular differentiation. They typically express neuron-specific enolase, synaptophysin and other markers that indicate a neuronal lineage. Approximately 25% of medulloblastomas are of a desmoplastic subclass, defined by the presence of dense, extracellular, stromal elements, that has foci with neuronal differentiation amidst densely cellular and highly proliferative zones. Favorable prognosis is associated with complete resection, age >3 years, and absence of metastasis at the time of initial diagnosis.
Astrocytomas and oligodendrogliomas arise throughout the CNS and can occur throughout life. Their incidence increases with age, peaking at 65–75 years (CBTRUS, 2002). The signs and symptoms of glial tumors vary with their location in the CNS. Children and adults with tumors of the cerebral hemispheres typically have seizures, headaches and focal neurological deficits as their presenting symptoms (DeAngelis, 2001). Tumors in the optic system induce loss of vision, whereas those that arise in the brainstem present with ataxia and cranial-nerve deficits. In nearly all cases, surgical biopsy is necessary to establish the diagnosis, but because the tumors tend to invade vital brain structures complete surgical removal often is not feasible. Patients with residual disease after surgery typically require further therapy with radiation and/or chemotherapy, especially if the tumors meet the histological criteria for high-grade malignancies.
Astrocytomas have a wide spectrum of histological subclasses, ranging from low-grade, relatively indolent (WHO grades I and II) tumors to highly malignant, invasive, WHO grades III (anaplastic) and IV (glioblastoma multiforme) tumors. Oligodendrogliomas have a similar range of histological phenotypes. Prognosis can be linked to multiple factors, with young age, low grade, oligodendroglial histology, complete resection and response to therapy correlated with more favorable probability of survival. Patients with favorable prognostic factors might live 20 years or more. In contrast, elderly patients with glioblastoma multiforme have a life expectancy measured in months, even after receiving maximally aggressive therapy.
Developmental mechanisms promote the growth of medulloblastomas and gliomas
Recent discoveries have established that brain tumors and other forms of cancer arise from either genetic mutations or epigenetic mechanisms that alter the expression of genes that are responsible for the normal growth and development of cells (Bishop, 1991; Hahn et al., 1999). From the perspective of cancer growth, growth-regulating genes might assume the role of genetically dominant proto-oncogenes or they might act as recessive tumor-suppressor genes when normal molecular mechanisms become dysregulated to promote uncontrolled cell proliferation (Weinberg, 1989). Typically, cancers do not arise de novo as malignancies, instead they evolve from more indolent growths into malignant tumors through the emergence and expansion of malignant subclones that gain competitive advantage over the remaining cells of the cancer by acquiring new genetic mutations (Nowell, 1976; Farber, 1984; Nowell, 1986). For example, clonal expansion of tumor cells with mutations of TP53, a tumor suppressor gene, has been linked to astrocytoma progression from low-grade to high-grade tumors (Sidransky et al., 1992). The evolution to malignancy might occur through multiple steps, as malignant subclones arise through the accumulation of an increasing number of genetic lesions (Nowell, 1986; Weinberg, 1989; Bishop, 1991). Low-grade tumors in early stages of cancer might appear very similar to their cells of origin, whereas highly malignant tumors in advanced stages of cancer progression are histologically very dissimilar to normal cells.
Early efforts to identify genetic mutations characteristic of medulloblastomas focused on chromosomal losses and gains detected by tumor karyotyping. Loss of chromosome 17p distal to the TP53 locus is the most prevalent chromosomal deletion, and is frequently associated with gain of chromosome 17q (isochromosome 17q) (Biegel et al., 1999; Kleihues and Cavenee, 2000). Despite years of investigation, the specific genes associated with this genetic abnormality have not been identified. A highly informative locus (9q22) with high medulloblastoma incidence has been linked to the basal cell carcinoma (Gorlin) syndrome (Gailani et al., 1992), an autosomal dominant disorder caused by germline mutation of the Shh receptor PTCH (Hahn et al., 1996; Johnson et al., 1996). Further investigations of a variant of Gorlin syndrome identified mutations of suppressor of fused (SUFU), a tumor suppressor that is downstream in the Shh/PTCH pathway (Rubin and Rowitch, 2002; Taylor et al., 2002). SUFU is part of a molecular complex that normally regulates GLI transcription factors that serve as effectors of Shh pathway activation. Approximately 15–20% of sporadic medulloblastomas have mutations of either PTCH or SUFU. Moreover, a small percentage of sporadic medulloblastomas have mutations of Smoothened (SMO), a transmembrane protein that interacts with PTCH and is derepressed when Shh binds to PTCH (Dong et al., 2000; Zurawel et al., 2000; Taylor et al., 2002). Both familial and sporadic medulloblastomas with mutations of molecules in the Shh/PTCH pathway are typically of the desmoplastic histological variant (Hynes et al., 1995; Pietsch et al., 1997; Pomeroy et al., 2002).
A second, informative, inherited disorder linked to medulloblastomas, Turcot syndrome, has been found to be associated with mutations of adenomatous polyposis coli (APC) (Hamilton et al., 1995). APC is part of a molecular complex that regulates the activity of β-catenin, a transcription regulatory element of the Wnt signaling pathway. Mutations of APC and β-catenin, and of axin, another member of the complex regulating β-catenin, have been found in a small percentage of sporadic medulloblastomas with both classic and desmoplastic histology (Zurawel et al., 1998; Dahmen et al., 2001; Koch et al., 2001).
Importantly, Shh also has been found to have a role in normal development of cerebellar granule cells. Cerebellar granule cell progenitors express PTCH and are induced to proliferate by Shh which is synthesized by Purkinje cells in the developing cerebellum (Wechsler-Reya and Scott, 1999). The Wnt pathway has been linked to later stages of granule cell development, promoting synaptogenesis and axonal maturation (Lucas and Salinas, 1997). Thus, mutations discovered from inherited syndromes have identified two molecular developmental pathways, Shh/PTCH and Wnt, that have an essential role in granule cell development and are also highly significant in the oncogenic transformation of granule cells to medulloblastomas.
The final established molecular lesion of medulloblastomas, amplification of cMyc, occurs in a highly malignant variant of medulloblastoma with characteristic large cell anaplastic histology (Eberhart et al., 2002). Interestingly, increased expression of cMyc has been linked to poor prognosis, independent of gene amplification (Herms et al., 2000; Grotzer et al., 2001). Because cMyc expression is known to be induced by activation of the Wnt pathway and nMyc by Shh/PTCH (He et al., 1998; Kenney et al., 2003; Oliver et al., 2003), these data collectively implicate Myc as a common regulatory element in the control of medulloblastoma growth.
Astrocytomas have long been known to progress spontaneously from low-grade to high-grade tumors (Kleihues and Cavenee, 2000). Early molecular events that promote the growth of low-grade tumors include inactivating mutations of the TP53 tumor suppressor gene and expression of both the α and β isoforms of platelet-derived growth factor (PDGF) and the PDGF receptor-α (PDGFRA) (Fleming et al., 1992; Hermanson et al., 1992). Co-expression of PDGF and its receptor is functionally significant because tumor growth can be inhibited by either inactivating PDGFR or blocking its expression (Shamah et al., 1993). Mutations of the p16INK4a/ARF locus (chromosome 9p) inactivating the p16–cdk4–pRb pathway might combine with PDGF/PDGFR expression to promote progression to high-grade (grades III and IV) astrocytomas (Dai et al., 2001). Ultimately, progression to glioblastoma multiforme might occur with loss of chromosome 10 and associated loss of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) which is known to be inactivated in a variety of advanced cancers (Steck et al., 1997).
Glioblastomas might also arise de novo as primary tumors, typically with amplification, rearrangement and/or overexpression of the epidermal growth factor receptor (EGFR) gene, often with associated loss of the p53 binding protein mouse double minute 2 (MDM2). These tumors appear to arise quite suddenly, most often in older adult patients, and have a distinctive small cell histology (Burger et al., 2001). Typically, high-grade astrocytomas with EGFR amplification are highly aggressive tumors that are less responsive to therapy than secondary glioblastomas that arise through progression of lower grade forms (Barker et al., 2001; Kunwar et al., 2001).
Oligodendrogliomas frequently have allelic loss of chromosomes 1p and 19q and, similar to astrocytomas, grade III anaplastic tumors might have inactivation of the p16–cdk4–pRb pathway (Cairncross et al., 1998; Smith et al., 2000; Ino et al., 2001). Anaplastic oligodendrogliomas with loss of 1p and 19q have a high rate of response to treatment with the chemotherapy drug combination procarbazine, CCNU and vincristine (Cairncross et al., 1998), whereas tumors lacking these mutations do not respond. It is not known if the genes deleted by these chromosomal losses are related functionally to chemotherapy response or whether the chromosomal losses are only markers of association.
Cellular origin of medulloblastomas and astrocytomas
Traditionally, the classification and presumed origin of brain tumors is based on the histological similarity of tumors with normal cells in the developing CNS. An early classification scheme, proposed in 1926 by Bailey and Cushing, focused on states of differentiation within a cell lineage, proposing that brain cancers might arise from cells that essentially are arrested at specific stages in their development (Bailey and Cushing, 1926). Although many aspects of the cell-lineage relationships they proposed have proved incorrect, the basic idea that brain cancers can be classified by resemblance to normal cells at different stages of development has had sustained acceptance. It provided a conceptual understanding for the context within which brain tumors arise, which gave some insights into their clinical behavior and prognosis. Thus, the Bailey and Cushing schema became the prototype for modern taxonomies, including the brain tumor classification system currently in use (Kleihues and Cavenee, 2000).
As details of the molecular basis of brain tumors have emerged, however, it has become clear that classification on morphologic criteria alone is inadequate (Gould, 1986). Tumors with identical histology and presumed state of differentiation might have very different growth characteristics and prognosis, and they apparently arise from different developmental lineages (Pomeroy et al., 2002). This is in agreement with the conclusion that cancer is a primarily a genetic disease. Although the cellular/developmental context is important, ultimately, it is the molecular mechanism that drives cancer growth, renders a tumor either susceptible or resistant to therapies and, ultimately, determines whether a cancer can be cured if it is not possible to physically remove it from the body through surgical excision.
The cellular origin of medulloblastomas has been debated for decades. One perspective, based largely on their histological appearance, is that they are a cerebellar subclass of primitive neuroectodermal tumors (PNET) that arise by oncogenic transformation of cells from subventricular germinal centers throughout the CNS (Rorke, 1983). An alternative view is that they are derived from the cerebellar granule cell lineage (Kadin et al., 1970). Although classifying medulloblastomas as a type of PNET provides an appealing conceptual schema, the idea that medulloblastomas are tumors of granule cell origin is attractive because granule cells are by far the most abundant neuronal class in the cerebellum (Williams and Herrup, 1988). Their abundance and prolonged proliferative phase during development make granule cells a probable target for transformation and oncogenesis.
The identification of granule cell-specific molecular markers and analysis of gene-expression profiles provide evidence that medulloblastomas are molecularly different to PNETs and that frequently they are derived from cerebellar granule cells (Kozmik et al., 1995; Yokota et al., 1996; Pomeroy et al., 2002). The case for granule cell origin is strongest for desmoplastic medulloblastomas whose gene-expression patterns indicate dysregulated Shh signaling (Pomeroy et al., 2002). Classic medulloblastomas might arise from granule cells through mutations involving other molecular pathways; alternatively, in some cases their gene-expression patterns might reflect a different cell of origin (Katsetos et al., 1995; Buhren et al., 2000; Pomeroy et al., 2002). Mice heterozygous for mutation of the murine homologue of the Shh receptor Ptc develop medulloblastomas that clearly arise within the granule cell lineage (Goodrich et al., 1997; Wechsler-Reya and Scott, 2001).
As has been found for astrocytomas and other cancers, medulloblastomas arise in multiple steps. In normal mice, proliferation of granule cell progenitors is driven by Shh expressed by Purkinje cells (Wechsler-Reya and Scott, 1999). Granule cell proliferation occurs in the outer half of the external granule cell layer (EGLa, Fig. 1) and is completed within the first 2–3 weeks postnatally. The committed progenitors then migrate inward from the inner EGLb layer past the molecular and Purkinje cell layers to form the internal granule cell layer (IGL). By the end of the first postnatal month in mice, the EGL has largely disappeared. Although generally, granule cell development proceeds normally in heterozygous Ptc mutant mice, ~50% of Ptc +/− mice have foci of persistently proliferating granule cell progenitors (Corcoran and Scott, 2001; Kim et al., 2003). Approximately 10–15% of these mice then evolve frank tumors that aberrantly express postmitotic molecular markers while actively proliferating (Kim et al., 2003). Tumor incidence peaks at ~5–6 months in these mice and then rapidly falls so that the tumors rarely occur after 12 months of age. This indicates a model of tumorigenesis in which granule cells are susceptible to oncogenic transformation during their proliferative phase of development. Although it is formally possible that granule cells remain susceptible after they exit the cell cycle as early post-mitotic progenitors, the potential to form tumors declines to low levels once they are terminally differentiated into neurons of the IGL (Fig. 2A). The time lag of several months between the disappearance of the EGL and the symptomatic growth of tumors might be explained by either the time needed for multiple steps of tumor progression or, possibly, the retention of a small number of progenitor cells after the EGL has regressed. This model presumably also applies to human medulloblastomas, which peak in incidence during the first decade of life and then decrease to a very low incidence thereafter (CBTRUS, 2002).
Fig. 1. Photomicrograph of human cerebellar cortex from a 2-month-old female.
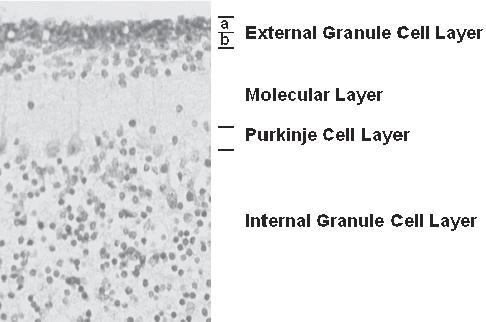
Cerebellar granule cell progenitors proliferate in the outermost layer of the external granule cell layer (EGLa). Once they leave the cell cycle, granule cell progenitors migrate from the deeper external layer (EGLb) through the molecular and Purkinje layers to form the internal granule cell layer.
Fig. 2. Models of oncogenic transformation.
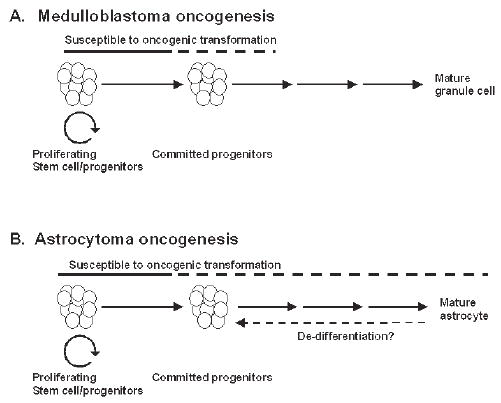
(A) Experimental and clinical evidence indicates that granule cells are susceptible to oncogenic transformation into medulloblastomas as proliferating progenitors (solid line) and, possibly, early post-mitotic progenitors (dotted line), but the potential for tumorigenesis declines when they become mature neurons. (B) By contrast, for astrocytomas the period of susceptibility for oncogenesis might extend to mature forms of astrocytes within the CNS.
Human astrocytomas have a different pattern of occurrence, with tumor incidence increasing beyond childhood and throughout adult years to peak at 65–75 years (CBTRUS, 2002). It is not known whether this represents a fundamental difference in susceptibility for oncogenic transformation of astrocytes compared with cerebellar granule cells. Terminally differentiated astrocytes might undergo malignant transformation or, possibly, dedifferentiate into a susceptible cell type (Fig. 2B). Alternatively, either astrocyte progenitors or stem cells might be present throughout adult life, unlike granule cell progenitors, which largely disappear by 1–2 years of age in humans.
Mouse models indicate that even mature astrocytes might serve as the cell of origin for astrocytomas. Expression of a constitutively active form of EGFR that is targeted to either an early, nestin-expressing glial lineage or mature cells that express glial fibrillary acidic protein in Ink4a/ArfA−/− mice induces glioma growth, but the efficiency of glioma induction is higher in the immature, nestin-expressing cells (Holland et al., 1998). The differential increased sensitivity of immature cells was not seen in Ink4a/ArfA−/− cells transduced with EGFR in vitro prior to orthotopic transplantation. Mature astrocytes have the same permissiveness for tumor formation as neural stem cells (Bachoo et al., 2002). These data indicate that astrocytes might be fundamentally different to cerebellar neurons in that they can undergo malignant transformation even when they are mature. Thus, although it is possible that astrocyte progenitors are present in the adult brain and are susceptible to genetic mutations that promote tumor growth, further research is needed to determine whether terminally differentiated astrocytes can also undergo malignant transformation or whether they dedifferentiate to a susceptible cell population.
While a basic model of tumorigenesis is beginning to emerge, there are many questions that remain. Does the normal developmental stage at which tumorigenesis occurs define the growth characteristics of the tumor? Developmental stage appears to be permissive, defining the timing of susceptibility. For example, medulloblastomas would not arise from PTCH mutations without expression of SMO and other downstream effector molecules of the Shh pathway, which are only expressed in later stages of granule cell-development. Is the biological phenotype defined by the molecular lesions that promote oncogenesis independent of the developmental stage at oncogenesis? Some mutations, such as amplification of cMyc, promote rapid medulloblastoma growth and are associated with poor clinical outcome (Reardon et al., 2000; Eberhart et al., 2002). Although this might indicate that specific mutations have a dominant effect in defining the biological phenotype, it cannot be excluded that these highly malignant tumors emerge only when cMyc amplification occurs at a specific, normal, developmental stage. It also is clear that the biological effects of a molecular mechanism in a tumor might not be predicted by the normal response in wild type cells. For example, TrkC activation promotes the survival and differentiation of cerebellar granule cells, but it promotes apoptosis of medulloblastomas (Kim et al., 1999). Clearly, more research is needed before we understand how genetic mutations interact with developmental stage to define tumor phenotype.
For human disease, medulloblastoma tumorigenesis has been placed within the context of developmental stage by comparing gene-expression profiles of human medulloblastomas to a gene-expression map of the cerebellar molecular developmental sequence in mice (Kho et al., 2004). Tumor expression profiles were most consistent with the patterns of gene expression of P1–P10 mouse cerebellum; metastatic tumors mapped to the profiles of P5 cerebellum whereas less invasive tumors mapped to P7. Murine medulloblastomas derived from Ptc+/− mice show a similar pattern of gene expression (Lee et al., 2003; Kho et al., 2004). The interval of P5–P7 corresponds to the period of maximal proliferation and migration of granule cell precursor in mice, indicating that medulloblastomas retain properties of granule cell progenitors.
When viewed at a cellular level, medulloblastomas harbor a minority tumor cell population that retains stem cell properties. These brain tumor stem cells (BTSC) are positive for the neural stem cell surface marker CD133+, and they retain the capacity for proliferation, self-renewal and differentiation in vitro into cells that resemble the tumor from which the BTSC fraction was derived (Singh et al., 2003). Although the presence of BTSC within medulloblastomas might argue that these tumors arise from stem cells from an earlier stage of development than committed granule cell progenitors, it also is possible that the molecular events that lead to tumorigenesis promote the acquisition or maintenance of stem cell properties within tumor cell populations. The extent to which medulloblastoma growth is dependent on BTSC is not known. Moreover, it is uncertain whether stem cells will have selective vulnerability to therapy. Although this exciting discovery has raised questions, it underscores the conclusion that molecular and cellular developmental mechanisms are central to the biology of medulloblastomas. Understanding and exploiting vulnerabilities inherent to these developmental processes may provide a mechanism to develop specific, targeted, biologically based therapies that destroy the tumor while leaving the nervous system intact.
Therapeutic implications
As details of the molecular mechanisms of medulloblastomas and astrocytomas emerge, it is anticipated that selective, targeted therapies will become available to augment and, in time, replace conventional radiation and chemotherapy. Development of new therapies designed to block signal transduction mechanisms or otherwise inhibit effector molecules is well underway. For medulloblastomas, specific, potent blockers of Shh signaling that are derived from the teratogen cyclopamine appear promising in preclinical testing in vitro and in mouse models (Taipale et al., 2000). Phase II trials are planned for small molecule inhibitors of PDGFRA, the ras/MAPK signaling pathway and Neuregulin, which have been linked with invasive and metastatic tumor growth (Gilbertson et al., 1997; MacDonald et al., 2001). Developmental molecular markers, including TrkC, HER2 and cMyc, which are predictive of medulloblastoma outcome, are being validated currently for risk stratification in national therapy trials (Segal et al., 1994; Gilbertson et al., 1997; Herms et al., 2000; Pomeroy et al., 2002).
Targeted therapies that inhibit tyrosine kinases, farnesyl-transferases and other molecules in signal transduction pathways are also in Phase I–II clinical trials for malignant astrocytomas. Other therapeutic approaches focus on the role of matrix metalloproteinases in tumor invasion and on the mechanisms of tumor angiogenesis.
The development of targeted therapies has also created a need to develop novel methods of drug delivery. Agents have been developed that can disrupt the blood–brain barrier, and there are now methods to infuse solutions of large, complex molecules that specifically target tumor cells directly into the brain (Bobo et al., 1994). These and other new methods of drug delivery might be necessary as targeted therapies become available for brain tumors.
The growing understanding of the cellular and molecular basis of brain tumors has enabled the beginning of a new era of personalized treatment of brain tumors in which therapies are tailored to the individual patient, based on the molecular and cellular content of their tumors. The era of brain-tumor therapy in which diagnosis was based largely on histological appearance and therapies were destructive is evolving rapidly to a stage where diagnosis relies on the identification of molecular signatures that indicate which growth mechanisms provide therapeutic targets.
Acknowledgments
Thanks to Chuck Stiles and David Rowitch for comments on the manuscript. The author’s work described in this review has been supported by the National Institutes of Health, the Children’s Brain Tumor Foundation and the Kyle Mullarkey Medulloblastoma Research Fund.
References
- Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, Tang Y, DeFrances J, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- Bailey P. and Cushing H. (1926) A classification of the tumors of the glioma group on histogenetic basis with a correlated study of prognosis. Philadelphia: J.D. Lippincott.
- Barker FG, 2nd, Simmons ML, Chang SM, Prados MD, Larson DA, Sneed PK, Wara WM, Berger MS, et al. EGFR overexpression and radiation response in glioblastoma multiforme. International Journal of Radiation Oncology, Biology, Physics. 2001;51:410–418. doi: 10.1016/s0360-3016(01)01609-1. [DOI] [PubMed] [Google Scholar]
- Biegel JA, Zhou JY, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germ–line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Research. 1999;59:74–79. [PubMed] [Google Scholar]
- Bishop JM. Molecular themes in oncogenesis. Cell. 1991;64:235–248. doi: 10.1016/0092-8674(91)90636-d. [DOI] [PubMed] [Google Scholar]
- Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection–enhanced delivery of macromolecules in the brain. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:2076–2080. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhren J, Christoph AH, Buslei R, Albrecht S, Wiestler OD, Pietsch T. Expression of the neurotrophin receptor p75NTR in medulloblastomas is correlated with distinct histological and clinical features: evidence for a medulloblastoma subtype derived from the external granule cell layer. Journal of Neuropathology and Experimental Neurology. 2000;59:229–240. doi: 10.1093/jnen/59.3.229. [DOI] [PubMed] [Google Scholar]
- Burger PC, Pearl DK, Aldape K, Yates AJ, Scheithauer BW, Passe SM, Jenkins RB, James CD. Small cell architecture—a histological equivalent of EGFR amplification in glioblastoma multiforme? Journal of Neuropathology and Experimental Neurology. 2001;60:1099–1104. doi: 10.1093/jnen/60.11.1099. [DOI] [PubMed] [Google Scholar]
- Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, Silver JS, Stark PC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. Journal of the National Cancer Institute. 1998;90:1473–1479. doi: 10.1093/jnci/90.19.1473. [DOI] [PubMed] [Google Scholar]
- CBTRUS (2002) Statistical report: Primary brain tumors in the United States, 1955–1999.
- Corcoran RB, Scott MP. A mouse model for medulloblastoma and basal cell nevus syndrome. Journal of Neurooncology. 2001;53:307–318. doi: 10.1023/a:1012260318979. [DOI] [PubMed] [Google Scholar]
- Dahmen RP, Koch A, Denkhaus D, Tonn JC, Sorensen N, Berthold F, Behrens J, Birchmeier W, et al. Deletions of AXIN1, a component of the WNT/wingless pathway, in sporadic medulloblastomas. Cancer Research. 2001;61:7039–7043. [PubMed] [Google Scholar]
- Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes and Development. 2001;15:1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeAngelis LM. Brain tumors. New England Journal of Medicine. 2001;344:114–123. doi: 10.1056/NEJM200101113440207. [DOI] [PubMed] [Google Scholar]
- Dong J, Gailani MR, Pomeroy SL, Reardon D, Bale AE. Identification of PATCHED mutations in medulloblastomas by direct sequencing. Human Mutation. 2000;16:89–90. doi: 10.1002/1098-1004(200007)16:1<89::AID-HUMU18>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Eberhart CG, Kratz JE, Schuster A, Goldthwaite P, Cohen KJ, Perlman EJ, Burger PC. Comparative genomic hybridization detects an increased number of chromosomal alterations in large cell/anaplastic medulloblastomas. Brain Pathology. 2002;12:36–44. doi: 10.1111/j.1750-3639.2002.tb00420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber E. The multistep nature of cancer development. Cancer Research. 1984;44:4217–4223. [PubMed] [Google Scholar]
- Fleming TP, Matsui T, Heidaran MA, Molloy CJ, Artrip J, Aaronson SA. Demonstration of an activated platelet-derived growth factor autocrine pathway and its role in human tumor cell proliferation in vitro. Oncogene. 1992;7:1355–1359. [PubMed] [Google Scholar]
- Gailani MR, Bale SJ, Leffell DJ, DiGiovanna JJ, Peck GL, Poliak S, Drum MA, Pastakia B, et al. Developmental defects in Gorlin syndrome related to a putative tumor suppressor gene on chromosome 9. Cell. 1992;69:111–117. doi: 10.1016/0092-8674(92)90122-s. [DOI] [PubMed] [Google Scholar]
- Gilbertson RJ, Perry RH, Kelly PJ, Pearson AD, Lunec J. Prognostic significance of HER2 and HER4 coexpression in childhood medulloblastoma. Cancer Research. 1997;57:3272–3280. [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Gould VE. Histogenesis and differentiation: a re-evaluation of these concepts as criteria for the classification of tumors. Human Pathology. 1986;17:212–215. doi: 10.1016/s0046-8177(83)80213-5. [DOI] [PubMed] [Google Scholar]
- Grotzer MA, Hogarty MD, Janss AJ, Liu X, Zhao H, Eggert A, Sutton LN, Rorke LB, et al. MYC messenger RNA expression predicts survival outcome in childhood primitive neuroectodermal tumor/medulloblastoma. Clinical Cancer Research. 2001;7:2425–2433. [PubMed] [Google Scholar]
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, et al. The molecular basis of Turcot’s syndrome. New England Journal of Medicine. 1995;332:839–847. doi: 10.1056/NEJM199503303321302. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, et al. Identification of c–MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B, Nister M. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Research. 1992;52:3213–3219. [PubMed] [Google Scholar]
- Herms J, Neidt I, Luscher B, Sommer A, Schurmann P, Schroder T, Bergmann M, Wilken B, et al. C-MYC expression in medulloblastoma and its prognostic value. International Journal of Cancer. 2000;89:395–402. [PubMed] [Google Scholar]
- Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes and Development. 1998;12:3675–3685. doi: 10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes M, Porter JA, Chiang C, Chang D, Tessier-Lavigne M, Beachy PA, Rosenthal A. Induction of midbrain dopaminergic neurons by Sonic hedgehog. Neuron. 1995;15:35–44. doi: 10.1016/0896-6273(95)90062-4. [DOI] [PubMed] [Google Scholar]
- Ino Y, Betensky RA, Zlatescu MC, Sasaki H, Macdonald DR, Stemmer-Rachamimov AO, Ramsay DA, Cairncross JG, et al. Molecular subtypes of anaplastic oligodendroglioma: implications for patient management at diagnosis. Clinical Cancer Research. 2001;7:839–845. [PubMed] [Google Scholar]
- Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- Kadin ME, Rubinstein LJ, Nelson JS. Neonatal cerebellar medulloblastoma originating from the fetal external granular layer. Journal of Neuropathology and Experimental Neurology. 1970;29:583–600. doi: 10.1097/00005072-197010000-00005. [DOI] [PubMed] [Google Scholar]
- Katsetos CD, Herman MM, Krishna L, Vender JR, Vinoresearch SA, Agamanolis DP, Schiffer D, Burger PC, et al. Calbindin–D28k in subsets of medulloblastomas and in the human medulloblastoma cell line D283 Med. Archives of Pathology and Laboratory Medicine. 1995;119:734–743. [PubMed] [Google Scholar]
- Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- Kho AT, Zhao Q, Cai Z, Butte AJ, Kim JY, Pomeroy SL, Rowitch DH, Kohane IS. Conserved mechanisms across development and tumorigenesis revealed by a mouse development perspective of human cancers. Genes and Development. 2004;18:629–640. doi: 10.1101/gad.1182504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Nelson AL, Algon SA, Graves O, Sturla LM, Goumnerova LC, Rowitch DH, Segal RA, et al. Medulloblastoma tumorigenesis diverges from cerebellar granule cell differentiation in patched heterozygous mice. Developmental Biology. 2003;263:50–66. doi: 10.1016/s0012-1606(03)00434-2. [DOI] [PubMed] [Google Scholar]
- Kim JY, Sutton ME, Lu DJ, Cho TA, Goumnerova LC, Goritchenko L, Kaufman JR, Lam KK, et al. Activation of neurotrophin-3 receptor TrkC induces apoptosis in medulloblastomas. Cancer Research. 1999;59:711–719. [PubMed] [Google Scholar]
- Kleihues P. and Cavenee W.K. (2000) Tumours of the Nervous System Lyon: IARC Press.
- Koch A, Waha A, Tonn JC, Sorensen N, Berthold F, Wolter M, Reifenberger J, Hartmann W, et al. Somatic mutations of WNT/wingless signaling pathway components in primitive neuroectodermal tumors. International Journal of Cancer. 2001;93:445–449. doi: 10.1002/ijc.1342. [DOI] [PubMed] [Google Scholar]
- Kozmik Z, Sure U, Ruedi D, Busslinger M, Aguzzi A. Deregulated expression of PAX5 in medulloblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:5709–5713. doi: 10.1073/pnas.92.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunwar S, Mohapatra G, Bollen A, Lamborn KR, Prados M, Feuerstein BG. Genetic subgroups of anaplastic astrocytomas correlate with patient age and survival. Cancer Research. 2001;61:7683–7688. [PubMed] [Google Scholar]
- Lee Y, Miller HL, Jensen P, Hernan R, Connelly M, Wetmore C, Zindy F, Roussel MF, et al. A molecular fingerprint for medulloblastoma. Cancer Research. 2003;63:5428–5437. [PubMed] [Google Scholar]
- Lucas FR, Salinas PC. WNT–7a induces axonal remodeling and increases synapsin I levels in cerebellar neurons. Developmental Biology. 1997;192:31–44. doi: 10.1006/dbio.1997.8734. [DOI] [PubMed] [Google Scholar]
- MacDonald TJ, Brown KM, LaFleur B, Peterson K, Lawlor C, Chen Y, Packer RJ, Cogen P, et al. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nature Genetics. 2001;29:143–152. doi: 10.1038/ng731. [DOI] [PubMed] [Google Scholar]
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- Nowell PC. Mechanisms of tumor progression. Cancer Research. 1986;46:2203–2207. [PubMed] [Google Scholar]
- Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, Wickramasinghe R, Scott MP, et al. Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7331–7336. doi: 10.1073/pnas.0832317100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer RJ, Cogen P, Vezina G, Rorke LB. Medulloblastoma: clinical and biologic aspects. Neuro-oncology. 1999;1:232–250. doi: 10.1215/15228517-1-3-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer RJ, Sutton LN, Elterman R, Lange B, Goldwein J, Nicholson HS, Mulne L, Boyett J, et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. Journal of Neurosurgery. 1994;81:690–698. doi: 10.3171/jns.1994.81.5.0690. [DOI] [PubMed] [Google Scholar]
- Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, Sorensen N, Berthold F, et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Research. 1997;57:2085–2088. [PubMed] [Google Scholar]
- Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- Reardon DA, Jenkins JJ, Sublett JE, Burger PC, Kun LK. Multiple genomic alterations including N-myc amplification in a primary large cell medulloblastoma. Pediatric Neurosurgery. 2000;32:187–191. doi: 10.1159/000028932. [DOI] [PubMed] [Google Scholar]
- Rorke LB. The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. Journal of Neuropathology and Experimental Neurology. 1983;42:1–15. [PubMed] [Google Scholar]
- Rubin JB, Rowitch DH. Medulloblastoma: a problem of developmental biology. Cancer Cell. 2002;2:7–8. doi: 10.1016/s1535-6108(02)00090-9. [DOI] [PubMed] [Google Scholar]
- Segal RA, Goumnerova LC, Kwon YK, Stiles CD, Pomeroy SL. Expression of the neurotrophin receptor TrkC is linked to a favorable outcome in medulloblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:12867–12871. doi: 10.1073/pnas.91.26.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamah SM, Stiles CD, Guha A. Dominant-negative mutants of platelet-derived growth factor revert the transformed phenotype of human astrocytoma cells. Molecular and Cellular Biology. 1993;13:7203–7212. doi: 10.1128/mcb.13.12.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky D, Mikkelsen T, Schwechheimer K, Rosenblum ML, Cavanee W, Vogelstein B. Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature. 1992;355:846–847. doi: 10.1038/355846a0. [DOI] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Research. 2003;63:5821–5828. [PubMed] [Google Scholar]
- Smith JS, Perry A, Borell TJ, Lee HK, O’Fallon J, Hosek SM, Kimmel D, Yates A, et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. Journal of Clinical Oncology. 2000;18:636–645. doi: 10.1200/JCO.2000.18.3.636. [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nature Genetics. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, et al. Mutations in SUFU predispose to medulloblastoma. Nature Genetics. 2002;31:306–310. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya R, Scott MP. The developmental biology of brain tumors. Annual Review of Neuroscience. 2001;24:385–428. doi: 10.1146/annurev.neuro.24.1.385. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. Oncogenes, antioncogenes, and the molecular bases of multistep carcinogenesis. Cancer Research. 1989;49:3713–3721. [PubMed] [Google Scholar]
- Williams RW, Herrup K. The control of neuron number. Annual Review of Neuroscience. 1988;11:423–453. doi: 10.1146/annurev.ne.11.030188.002231. [DOI] [PubMed] [Google Scholar]
- Yokota N, Aruga J, Takai S, Yamada K, Hamazaki M, Iwase T, Sugimura H, Mikoshiba K. Predominant expression of human zic in cerebellar granule cell lineage and medulloblastoma. Cancer Research. 1996;56:377–383. [PubMed] [Google Scholar]
- Zurawel RH, Chiappa SA, Allen C, Raffel C. Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Research. 1998;58:896–899. [PubMed] [Google Scholar]
- Zurawel RH, Allen C, Chiappa S, Cato W, Biegel J, Cogen P, de Sauvage F, Raffel C. Analysis of PTCH/SMO/SHH pathway genes in medulloblastoma. Genes Chromosomes Cancer. 2000;27:44–51. doi: 10.1002/(sici)1098-2264(200001)27:1<44::aid-gcc6>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]