

Abstract
An efficient method for synthesis of oligonucleotide 2′-conjugates via amide bond formation on solid phase is described. Protected oligonucleotides containing a 2′-O-carboxymethyl group were obtained by use of a novel uridine 3′-phosphoramidite, where the carboxylic acid moiety was introduced as its allyl ester. This protecting group is stable to the conditions used in solid-phase oligonucleotide assembly, but easily removed by Pd(0) and morpholine treatment. 2′-O-Carboxymethylated oligonucleotides were then efficiently conjugated on a solid support under normal peptide coupling conditions to various amines or to the N-termini of small peptides to give products of high purity in good yield. The method is well suited in principle for the preparation of peptide–oligonucleotide conjugates containing an amide linkage between the 2′-position of an oligonucleotide and the N-terminus of a peptide.
Introduction
There are many methodologies available for preparing natural oligonucleotides, as well as their analogues containing modified phosphate groups, nucleobases and sugars.1 The advances in solid-phase synthesis inspired various strategies for preparation of oligonucleotide conjugates.2,3 Oligonucleotide conjugation is predominantly carried out by use of a nucleophilic group on an oligonucleotide to react with an electrophilic group on a tag or a solid support. This strategy still predominates because the common oligonucleotide deprotection is performed by base treatment e.g. ammonia or methylamine which are inherently nucleophilic. Indeed, there are many methods of preparation of oligonucleotides modified with amino or thiol groups at a variety of positions.1 On the other hand, there are many situations when researchers wish to introduce an electrophilic group into oligonucleotides. 4 In this vein, we developed a phosphoramidite incorporating a carboxylic acid function protected by a 2-chlorotrityl group.5a The reagent is suitable for solid-phase synthesis of 5′-carboxylated oligonucleotides that may be conjugated to various amines on solid supports after activation by a suitable peptide coupling reagent.5 The monomer is now commercially available.5b Similarly, 5′-carboxy-modifier C10 is sold by Glen Research that contains a preformed carboxylic acid N-hydroxysuccinimide ester and can be used directly in solid-phase conjugation.6
In previous work in this field we7 and others8,9 described syntheses of 2′-O-7,8 or 2′-S-carboxymethyloligonucleotides,9 where an alkyl ester was chosen as a carboxylic acid protecting group. After completion of solid-phase oligonucleotide assembly, support-bound oligonucleotides containing methyl7,8b or ethyl8a ester were hydrolysed by treating with aq. NaOH or treated by an appropriate amine to afford either the carboxymethyl group or the corresponding amides, respectively, after final deprotection. As a result, these methods are frequently plagued by low yields, long reaction times, the need for large excesses of reactant(s), and the formation of by-products that are difficult to separate.
Recently, we reported our preliminary studies10,11 that show that oligonucleotides containing a 2′-carboxymethyl group can be conjugated to amines, amino acid derivatives and small peptides by amide bond formation on solid phase under peptide coupling conditions or in aqueous media by a water-soluble carbodiimide-mediated reaction.
We would like to report here the synthesis of the 2′-O-carboxymethylated oligonucleotides by use of a uridine 3′-phosphoramidite, where the carboxylic acid function carries an allyl protecting group, which is readily removed under mild and specific conditions of Pd(0)-catalysis. Subsequent conjugation of the 2′-carboxylic acid to a range of primary aliphatic and aromatic amines and peptides is achieved by peptide coupling reagent-promoted amide bond formation on a solid support. We demonstrate examples of the synthesis of 2′-O-methyl oligoribonucleotides containing a 2′-O-carboxymethyl group and their conjugation to a range of amines and peptides.
Results and discussion
The route to the 2′-O-(allyloxycarbonyl)methyluridine building block 6 is illustrated in Scheme 1. The starting 3′,5′-O-(tetraisopropyldisiloxan-1,3-diyl)uridine 1 was obtained from uridine as reported previously12,13 and purified by column chromatography on silica gel in CHCl3–EtOAc (9:1, then 2:1 v/v). Protection of the N3 of uracil is required, since the subsequent alkylation of 3′,5′-O-protected uridine preferentially occurs at the base moiety. To protect the imido function from this side reaction, we chose to block the N3-position with an ammonia-labile pivaloyloxymethyl (Pom) group. The group also prevents undesirable side reactions during oligonucleotide assembly. Furthermore, the greater lipophilicity of the Pom derivative is helpful for chromatographic separation of the target compound. Initially, Pom was reported as a protecting group for the lactam function of a uracil moiety.14 This group was introduced by the transient protection procedure, which involved 2′-O-trimethylsilylation followed by N3-protection.14b On the other hand, Sekine15 showed earlier that various N3-protected uridine derivatives may be obtained selectively in the presence of an unprotected 2′-OH group under conditions of phase-transfer catalysis. These results led us to consider a phase-transfer reaction to convert 1 into 2. Indeed, when 1 was allowed to react with 10 equiv. of chloromethyl pivalate in a biphasic system 0.2 M Na2CO3–CH2Cl2 (2:1 v/v) in the presence of 0.2 equiv. of tetra-n-butylammonium hydrogen sulfate (TBAHS) as a phase-transfer catalyst at room temperature for 48 h under vigorous stirring, N3-pivaloyloxymethyl-3′,5′-O-(tetraisopropyldisiloxan-1,3-diyl)uridine 2 was isolated in 70% yield after column chromatography. General methods of 2′-O-alkylation of ribonucleosides have been reviewed by Zatsepin et al.16 We previously adopted a convenient procedure7 for 2′-O-alkylation by use of a strong sterically hindered organic base 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP) described in a number of publications.17,18 Later we replaced BEMP by another phosphazene base P1-tert-butyltris(tetramethylene) (BTPP), which is cheaper and even more basic, though less sterically hindered than BEMP.19 Thus, we found that alkylation of compound 2 with 2.5 equiv. of allyl chloroacetate and 2.8 equiv. of BTPP gave 3 in 90% yield after 3–5 h. Compound 3 was then desilylated smoothly with TBAF7 or, better, triethylamine trihydrofluoride13 in THF and then converted into the 5′-O-dimethoxytrityl (DMTr) derivative 5 by the known procedure.20 Subsequent phosphitylation of the 3′-hydroxy group in an inert atmosphere using bis(N,N-diisopropylamino)-2-cyanoethoxyphosphine in CH2Cl2 in the presence of diisopropylammonium tetrazolide21 afforded the phosphoramidite 6. This was used successfully in machine-assisted solid-phase oligonucleotide synthesis. The average coupling efficiency of 6 at a 0.2 M concentration in dry MeCN and 30 min reaction time was found to be greater than 97%. The modified phosphoramidite was utilised in the synthesis of two 2′-O-methyloligoribonucleotides I and II (Table 1). Decamer I was synthesised as a model to select and optimise the specific conditions of deblocking, purification and conjugation. Oligonucleotide II is complementary to the HIV-1 TAR RNA apical stem-loop, the binding site for the HIV-1 trans-activator protein Tat.22
Scheme 1.
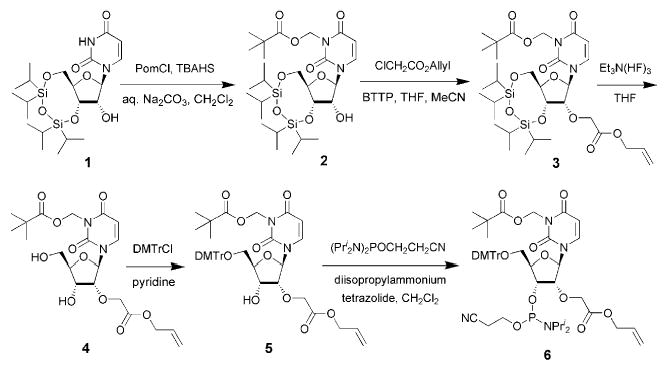
Preparation of 2′-O-(allyloxycarbonyl)methyluridine 3′-phosphoramidite (7). Abbreviations: TIPS - 1,1,3,3-tetraisopropyldisiloxan- 1,3-diyl, Pom - pivaloyloxymethyl, TBAHS - tetra-n-butylammonium hydrogen sulfate, BTPP - phosphazene base P1-tert-butyltris(tetramethylene), DMTr - 4,4′-dimethoxytrityl.
Table 1.
Properties of 2′-carboxy-modified 2′-O-methyloligonucleotides and their conjugates
| # | Oligonucleotide sequence, 5′-3′/conjugated molecule | MALDI-TOF MS calc./found m/z) | Retention timea/min | Yield b (%) |
|---|---|---|---|---|
| I | UUUUUU*UUUU | 3183.9/3182.6 | 15.5 | – |
| II | CUCCCAGGCU*CA | 3942.6/3942.7 | 15.7 | – |
| I.7 | N, N, N-Tris(aminoethyl)amine (7) | 3311.6/3310.2 | 14.5 | >90 |
| I.8 | Cyclohexylamine (8) | 3264.6/3264.1 | 16.0 | >90 |
| I.9 | 3-Amino-1,2-propanediol (9) | 3256.5/3254.8 | 15.0 | >90 |
| I,10 | Histamine (10) | 3276.5/3277.8 | 14.8 | 75 |
| I.11 | Spermine (11) | 3367.7/3366.6 | 14.7 | 85 |
| I.12 | N, N, N′, N′-Tetrakis(3-aminopropyl)-1,4-butanediamine (12) | 3481.8/3482.6 | 14.9 | 80 |
| II.13 | 1-Aminopyrene (13) | 4141.8/4138.4 | 27.1 | 65 |
| II.14 | 1-Pyrenemethylamine (14) | 4155.9/4153.4 | 27.5 | 85 |
| II.15 | H-Phe-NH215) | 4088.8/4084.3 | 17.2 | >90 |
| II.16 | H-Leu-NH2 (16) | 4054.8/4051.0 | 16.2 | >90 |
| II.17 | H-Phe-Phe-NH2 (17) | 4235.9/4230.4 | 21.6 | >90 |
| II.18 | H-Leu-Phe-NH2 (18) | 4201.9/4199.4 | 16.9 | >90 |
| II.19 | H-Gly-Leu-Met-NH2 (19) | 4243.0/4245.8 | 17.1 | >90 |
| II.20 | H-Pro-Leu-Gly-NH2 (20) | 4208.9/4206.3 | 21.7 | 75 |
| II.21 | H-Asn-Arg-Asn-Phe-Leu-Arg-Phe-NH2 (21) | 488.7/4894.0 | 21.5 | 80 |
For conditions, see Experimental section.
Conversion of oligonucleotide peak to the conjugate peak, calculated from RP-HPLC traces.
To remove the 2′-allyl protecting group on the solid phase, we used a mixture of tetrakis(triphenylphosphine)palladium(0), triphenylphosphine and morpholine7 in dry CH2Cl2 for 50 min (Scheme 2). During the procedure, other protecting groups remain intact and the oligonucleotide is still linked to a polymer support.23
Scheme 2.
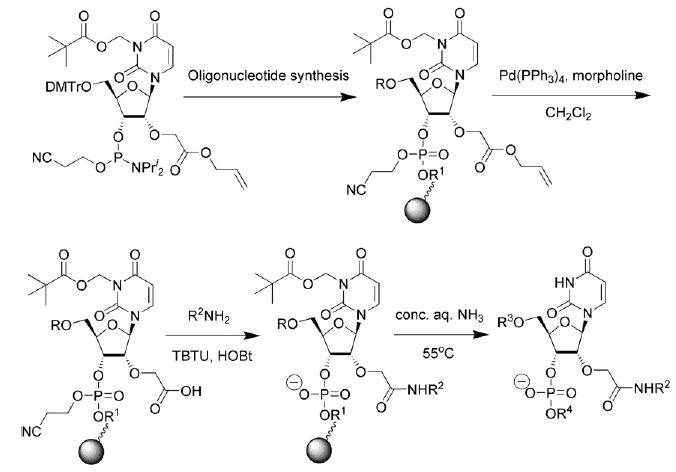
Solid-phase synthesis, selective deprotection and conjugation of 2′-O-carboxymethylated oligonucleotides. Abbreviations: R, R1 = protected oligonucleotide chain, R2 = amine or peptide residue, R3, R4 = unprotected oligonucleotide chain; TBTU - O-benzotriazol-1-yl-N,N,N′,N′- tetramethyluronium tetrafluoroborate, HOBt - 1-hydroxybenzotriazole.
Further conjugations of 2′-O-carboxymethylated oligonucleotides I and II were carried out on solid phase in organic solvent (Scheme 2). The choice of amine was influenced by the expected application for the conjugate e.g. introduction of positive charge(s) or nucleophilic amino groups (N,N,N-tris( aminoethyl)amine 7, histamine 10, spermine 11 and N,N,N′,N′-tetrakis(3-aminopropyl)-1,4-butanediamine 12), chemoselective ligation (3-amino-1,2-propanediol 9)22 or fluorescent labeling (1-aminopyrene 13 and 1-pyrenemethylamine 1413). In the case of spermine that contains both primary and secondary amino groups, we expected selective reaction with a primary amino group facilitated by the excess of amine.5a For further conjugation experiments, we have chosen several amino acid derivatives 15 and 16 and short peptides 17–20 and FMRF amide-related peptide 21 demonstrated to be cardioactive neuropeptide. 24 To prevent the formation of byproducts both amino acids and peptides were utilised as C-terminal amides. To obtain a high yield of structurally diverse types of conjugates (Table 1) we used pre-activation of polymer-bound 2′-O-carboxymethyloligonucleotide with TBTU–HOBT (1:1) at 37 °C in dry DMF for 40 min, followed by addition of the amine, amino acid or peptide and further incubation.25 Reaction times for the amines differed significantly from those for the amino acids and peptides. Whilst all amines reacted within 3 h, amino acids and peptides required overnight reaction. After completion of the reaction, polymer-bound conjugates were cleaved from their solid supports and deprotected by concentrated aqueous ammonia treatment at 55 °C overnight. Reaction mixtures obtained were analysed by reversed-phase HPLC and MALDI-TOF MS. Only a single product was observed in the case of spermine conjugate I.11. Examples of typical RP-HPLC traces are shown in Fig. 1. Noteworthy, the two unprotected arginine residues in peptide 21 did not interfere with its successful conjugation at the N-terminus. We ascribe this to the presence of an excess of HOBt that may protonate the guanidino group of arginine and thus protect it from acylation.
Fig. 1.
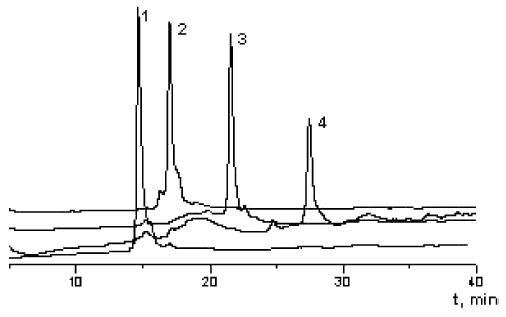
Reversed-phase HPLC traces of crude oligonucleotide 2′-conjugates: (1) oligonucleotide II; (2) conjugate II.20; (3) conjugate II.17; (4) conjugate II.14. For HPLC conditions, see Experimental section.
In conclusion, we have described an efficient and reliable method for preparation of 2′-O-carboxymethyloligonucleotides using protected uridine 3′-phosphoramidite containing a 2′-carboxymethyl group protected as an allyl ester. After deprotection under conditions of Pd(0) catalysis, such modified oligonucleotides may be conjugated, while still attached to a solid support, to a range of primary or secondary aliphatic or aromatic amines or to the N-termini of short peptides. The method described could prove useful in synthesis of small molecule libraries of oligonucleotide conjugates for diagnostic or therapeutic evaluation. In addition, this conjugation method may be a useful route for attachment of membrane-penetrating peptides, particularly those containing multiple arginine residues, to the 2′-position of oligonucleotides and their analogues for cellular uptake studies.
Experimental
General
Chemicals were obtained from commercial suppliers and used without further purification unless otherwise noted. Chloromethyl pivalate, tetra-n-butylammonium hydrogen sulfate, allyl chloroacetate, triethylamine trihydrofluoride, tetrakis(triphenylphosphine) palladium(0), N,N,N-tris(aminoethyl)amine, 3-amino-1,2-propanediol, histamine, spermine, N,N,N′,N′-tetrakis(3-aminopropyl)-1,4-butanediamine, 1-aminopyrene and 1-pyrenemethylamine hydrochloride were purchased from Aldrich. Phosphazene base P1-tert-butyltris(tetramethylene), bis(N,N-diisopropylamino)-2-cyanoethoxyphosphine, triphenylphosphine, morpholine, 1-hydroxybenzotriazole, O-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) and cyclohexylamine were from Fluka. 4,4′-Dimethoxytrityl chloride (DMTrCl) was from Avocado. Amino acids and peptides were bought from Bachem. Diisopropylammonium tetrazolide was prepared from diisopropylamine (BDH) and 1H-tetrazole solution in acetonitrile (Glen Research).21 Dichloromethane (BDH) was used after refluxing over and distillation from CaH2. DMF (Fisher) was distilled in vacuo and used fresh. Other solvents: benzene, chloroform, ethyl acetate, acetone, acetonitrile, THF, hexane, absolute ethanol and methanol were used as received.
1H NMR spectra were recorded on a Bruker DRX 500 spectrometer (500.13 MHz for 1H and 202.4 MHz for 31P) and 2–5 mM solutions. Chemical shifts (δ, ppm) for 1H and 31P are referenced to internal solvent resonances and reported relative to SiMe4 and 85% aq. H3PO4, respectively. 2D spectra involved use of adapted COSY and HMQC techniques. Chemical shifts are accurate to within 0.01 ppm for 1H. CSSI are accurate to within 0.25 Hz. MALDI-TOF spectra were recorded on a Voyager DE system (Applied Biosystems) or Reflex IV (Bruker) in positive ion mode using either a 1:1 (v/v) mixture of 2,6-dihydroxyacetophenone (2,6-DHAP) (40 mg cm−3 in MeOH) and aq. diammonium hydrogen citrate (80 mg cm−3) for all oligonucleotides, and 2,5-dihydroxybenzoic acid (2,5-DHBA) (10 mg cm−3 in 50% aq. MeOH) or 2,4,6-trihydroxyacetophenone (2,4,6-THAP) (10 mg cm−3 in 50% aq. MeOH) for low molecular weight compounds as a matrix. TLC was carried out on Merck DC Kieselgel 60 F254 aluminium sheets. Compounds were visualised under short-wavelength UV and stained by trifluoroacetic acid vapours for DMTr-containing species. Column chromatography was carried out on Kieselgel 60 0.040–0.063 mm (Merck).
3′,5′-O-(Tetraisopropyldisiloxane-1,3-diyl)uridine (1)
This was prepared as described previously.13
N3-Pivaloyloxymethyl-3′,5′-O-(tetraisopropyldisiloxan-1,3-diyl)uridine (2)
To a two-phase solution of compound 1 (4.86 g, 10 mmol) in CH2Cl2 (200 cm3)–0.2 M aq. Na2CO3 (400 cm3) were added chloromethyl pivalate (14.3 cm3, 99.2 mmol) and tetra-n-butylammonium hydrogen sulfate (0.74 g, 2.1 mmol). The resulting mixture was vigorously stirred at ambient temperature for 48 h. Then the organic phase was collected and washed with 5% NaHCO3 (2 × 200 cm3). The organic layers were combined, dried (Na2SO4) and filtered. The filtrate was evaporated, co-evaporated with benzene (3 × 25 cm3) and the residue was chromatographed on silica gel (stepwise gradient of 0→2→4→6→8→10% EtOAc in benzene, v/v) to give compound 2 which was obtained as a white foam (4.2 g, 70%). Rf 0.85 (CHCl3–EtOH, 9:1 v/v). MALDI-TOF (2,5-DHBA): [M + Na]+ calc. m/z 623.85, found 624.05. 1H NMR (CDCl3): δ 7.81 (d, 1H, H-6, J5,6 = 7.5), 5.92 (AB system, 2H, NCH2OCOBut), 5.81 (s, 1H, H-1′), 5.76 (d, 1H, H-5, J5,6 = 7.5), 4.67 (br d, 2H, H-3′, H-4′), 4.22 (br s, 2H, H-5′), 4.0 (d, 2H, H-2′), 1.19 (s, 9H, But), 1.1–0.9 (m, 28H, Pri).
2′-O-Allyloxycarbonylmethyl-N3-pivaloyloxymethyl-3′,5′-O-(tetraisopropyldisiloxan-1,3-diyl)uridine (3)
Compound 2 (4.2 g, 7 mmol) was dried by co-evaporation with dry MeCN (3 × 20 cm3) and was then dissolved in dry MeCN–THF (100 cm3, 1:1 v/v). Phosphazene base P1-tert-butyltris(tetramethylene) (6 cm3, 19.6 mmol) followed immediately by allyl chloroacetate (2 cm3, 17.5 mmol) were added to the stirred mixture at ambient temperature. TLC (CHCl3–EtOH, 97:3 v/v) showed complete reaction after 3 h. The reaction mixture was evaporated to dryness in vacuo. The yellow oil was dissolved in CHCl3 (100 cm3) and washed with brine (1 × 100 cm3) and water (2 × 100 cm3). The combined organic layers were dried (Na2SO4), filtered, evaporated and co-evaporated with benzene (3 × 25 cm3). The crude product was purified by column chromatography on silica gel (stepwise gradient of 0→2→4→6→8% EtOAc in benzene, v/v). Compound 3 was obtained as a pale yellow oil (4.4 g, 90%). Rf 0.58 (CHCl3–EtOH, 97:3 v/v). MALDI-TOF (2,5-DHBA): [M + H]+ calc. m/z 699.97, found 699.32. 1H NMR (CDCl3): δ 7.81 (d, 1H, H-6, J5,6 = 7.5), 5.92 (AB system, 2H, NCH2OCOBut), 5.89 (ddt, 1H, CH═CH2), 5.81 (s, 1H, H-1′), 5.76 (d, 1H, H-5, J5,6 = 7.5), 5.53 (d, 1H, ═CH2-(Z)), 5.24 (d, 1H, ═CH2-(E)), 4.67 (br d, 2H, H-3′, H-4′), 4.58, 4.43 (AB system, 2H, CO2CH2CH═), 4.25 (A part of AB system, 2H, CO2CH2CH═), 4.22 (br s, 2H, H-5′), 4.0 (overlap, 2H, H-2′, B part of AB system, CO2CH2CH═), 1.19 (s, 9H, But), 1.1–0.9 (m, 28H, Pri).
2′-O-Allyloxycarbonylmethyl-N3-pivaloyloxymethyluridine (4)
To a solution of 3 (4.4 g, 6.3 mmol) in THF (15 cm3) in a 30 cm3 screw-capped Teflon vial (Nalgene) was added triethylamine trihydrofluoride (2.6 cm3, 15.7 mmol) and the mixture was left for 1.5 h at room temperature, the completion of deprotection was checked by TLC (CHCl3–EtOH, 9:1 v/v), then diluted with EtOAc (50 cm3), washed with 5% NaHCO3 (2 × 50 cm3), water (50 cm3), 5% citric acid (2 × 50 cm3), and brine (50 cm3), then dried (Na2SO4), evaporated to dryness and co-evaporated with CHCl3 (3 × 25 cm3). The residue was chromatographed on a silica gel column (0→1→2→3→4% MeOH in CHCl3, v/v). Yield (2.3 g, 80%). Rf 0.15 (CHCl3–EtOH, 9:1 v/v). MALDI-TOF (2,5-DHBA): M+ calc. m/z 456.45, found 456.26, [M + Na]+ calc. m/z 479.44, found 479.11, [M + K]+ calc. m/z 495.56, found 497.04. 1H NMR (CDCl3): δ7.81 (d, 1H, H-6, J5,6 = 7.5), 5.92 (AB system, 2H, NCH2OCOBut), 5.89 (ddt, 1H, CH═CH2), 5.78 (d, 1H, H-1′, J1′,2′ = 10), 5.76 (d, 1H, H-5, J5,6 = 7.5), 5.53 (d, 1H,═CH2-(Z)), 5.24 (d, 1H, ═CH2-(E)), 4.67 (br d, 2H, H-3′, H-4′), 4.61 (AB system, 2H, OCH2CO), 4.58, 4.43 (AB system, 2H, CO2CH2CH═), 4.03, 4.0 (dd, 2H, H-3′, H-4′), 3.88 (dd, 1H, H-2′), 3.66 (dt, 1H, H-5′), 3.58 (dt, 1H, H-5′), 3.0 (s, 1H, OH), 1.19 (s, 9H, But).
2′-O-Allyloxycarbonylmethyl-5′-O-(4,4′-dimethoxytrityl)- N3-pivaloyloxymethyluridine (5)
Compound 4 (2.3 g, 5 mmol) was co-evaporated with dry pyridine (3 × 20 cm3), dissolved in dry pyridine (50 cm3), cooled in an ice-bath, and DMTrCl (2.05 g, 6.05 mmol) was added in one portion. The reaction was monitored by TLC until the starting nucleoside disappears completely. Then the excess of DMTrCl was quenched with MeOH (2 cm3), and after 10 min the mixture was partially evaporated, diluted with CHCl3 (100 cm3), washed with 5% NaHCO3 (2 × 100 cm3), and brine (100 cm3), then dried (Na2SO4), evaporated, co-evaporated with benzene (3 × 25 cm3) and the residue was chromatographed on a silica gel column (0→20→25→30→50→100% CHCl3 in benzene and further 1→2→5% MeOH in CHCl3 + 1% pyridine v/v/v). Yield (3.4 g, 90%). Rf 0.4 (CHCl3–EtOH, 95:5 v/v). MALDI-TOF (2,4,6-THAP): [M + Na]+ calc. m/z 780.81, found 780.57. 1H NMR (CDCl3): δ7.8 (d, 1H, H-6, J5,6 = 7.5), 7.38–7.17 (m, 10H, Ar), 6.85 (d, 4H, o-Ar), 5.92 (AB system, 2H, NCH2OCOBut), 5.89 (m, 1H, CH═CH2), 5.82 (d, 1H, H-1′), 5.58 (d, 1H, H-5, J5,6 = 7.5), 5.22 (d, 2H, CH2CH═CH2), 4.69 (s, 2H, OCH2CO), 4.64 (d, 2H, CH2CH═CH2), 4.39 (t, 1H, H-3′), 4.32 (t, 1H, H-2′), 4.22 (br s, 1H, H-4′), 3.8 (s, 6H, OCH3), 3,52, 3.43 (dd, 2H, H-5′), 1.19 (s, 9H, But).
2′-O-Allyloxycarbonylmethyl-3′-O-(N,N-diisopropylamino- 2-cyanoethoxyphosphinyl)-5′-O-(4,4′-dimethoxytrityl)-N3- pivaloyloxymethyluridine (6)
Compound 5 (3.4 g, 4.5 mmol) was co-evaporated with dry CH2Cl2 (3 × 20 cm3), dissolved in dry CH2Cl2, diisopropylammonium tetrazolide (1.1 g, 6.7 mmol) and bis(N,N-diisopropylamino)-2-cyanoethoxyphosphine (2.3 cm3, 7.3 mmol) were added, and the reaction mixture was stirred at 25 °C overnight. After TLC (CH2Cl2–Et3N, 98:2 v/v) showed the completion of the reaction, the mixture was diluted with CH2Cl2 (50 cm3), washed with brine (2 × 50 cm3), dried (Na2SO4), and evaporated to dryness. The residue was dried in vacuo to afford 7 as a white foam (4.1 g, 95%). Rf 0.45 (CH2Cl2–Et3N, 98:2 v/v). MALDI-TOF (2,6-DHAP–diammonium hydrogen citrate): [M + H]+ calc. m/z 960.06, found 960.13. 31P NMR (CD3CN): δ 153.0, 153.5 (two diastereomers).
Oligonucleotide synthesis
2′-O-Methyloligoribonucleotides were assembled on an ABI 394B DNA Synthesizer by the phosphoramidite method according to the manufacturer’s recommendations. Protected 2′-O-methylribonucleoside phosphoramidites and S-ethylthiotetrazole were purchased from Glen Research (via Cambio). Prepacked 0.4 μmol functionalised columns of controlled pore glass (Glen Research) were used throughout. For couplings with modified phosphoramidite 6, 0.2 M concentration in dry MeCN was used, and the coupling time was increased to 30 min.
Deprotection of carboxylic acid function of oligonucleotides I, II
The 2′-allyl protecting group was removed from the support-bound modified oligonucleotides by treatment with a solution of morpholine (0.03 cm3), tetrakis(triphenylphosphine)-palladium(0) (5 mg) and triphenylphosphine (5 mg) in dry CH2Cl2 (0.2 cm3) for 40 min at ambient temperature. The supernatant was then decanted and the support was rinsed with CH2Cl2 (2 × 0.15 cm3), EtOH (0.15 cm3), water (0.15 cm3) and CH2Cl2 (0.15 cm3). The support was then dried on air.
Coupling of amines, amino acids and peptides to 2′-O-carboxymethyloligonucleotides I, II
After the completion of the 2′-carboxylic acid group deprotection, a solution of TBTU (5 mg) and HOBt (2 mg) in dry DMF (0.2 cm3) was added. The slurry was incubated at 37 °C for 40 min with occasional swirling, then an amine, amino acid or peptide was added (100 equiv.). The reaction was carried out at 37 °C for 3 h in the case of amines or overnight for amino acid derivatives or peptides and shaking, then the supernatant was discarded, and glass beads washed successively with DMF (2 × 0.2 cm3), water (2 × 0.2 cm3) and EtOH (2 × 0.2 cm3). Cleavage from the support and deprotection of phosphate and nucleobase residues were performed using conc. aq. ammonia overnight at 55 °C. Reaction mixtures were analysed by RPHPLC, conjugates were purified by RP-HPLC on a Gilson HPLC system using a Beckman Ultrasphere ODS column (4.6 × 250 mm) and dual wavelength detection (215 and 254 nm); buffer A: 5% of MeCN (v/v) in 0.1 M triethylammonium acetate, buffer B: MeCN; flow rate 1 cm3 min−1, gradient of B in A: 0–5%, 5 min, 5–15%, 10 min, 15–40, 30 min, 40–80%, 10 min, 80–0%, 10 min. Combined fractions containing the conjugate were evaporated, redissolved and precipitated by 4 M sodium acetate solution (0.2 cm3) and ethanol (1.5 cm3) or 2 M LiClO4 (0.2 cm3) and acetone (1.5 cm3). Molecular masses of purified conjugates were then checked by MALDI-TOF MS.
Acknowledgments
This work was supported by the Wellcome Trust CRIG 069419 and AstraZeneca UK, Ltd. The authors thank Dr A. D. Malakhov and Mrs A. N. Muravieva for help with HPLC purification.
Footnotes
Electronic supplementary information (ESI) available: MALDI-TOF spectra. See http://www.rsc.org/suppdata/ob/b4/b409496d/
References
- 1.Current Protocols in Nucleic Acid Chemistry, ed. S. L. Beaucage, D. E. Bergstrom, G. D. Glick and R. A. Jones, John Wiley & Sons, Inc., New York, 2000.
- 2.Zubin EM, Romanova EA, Oretskaya TS. Russ Chem Rev. 2002;71:239–264. [Google Scholar]
- 3.Virta P, Katajisto J, Niittymäki T, Lönnberg H. Tetrahedron. 2003;59:5137–5174. [Google Scholar]
- 4.Kachalova AV, Zubin EM, Oretskaya TS. Russ Chem Rev. 2002;71:1041–1059. [Google Scholar]
- 5.a Kachalova AV, Stetsenko DA, Romanova EA, Tashlitsky VN, Gait MJ, Oretskaya TS. Helv Chem Acta. 2002;85:2409–2416. [Google Scholar]; (b) further information on use of our reagent is available from Link Technologies, Ltd. on www.linktech.co.uk/Downloads/TIS-BC-02v1.3.pdf
- 6.R. I. Hogrefe and M. M. Vaghefi, US Pat., 6,320,041, 2001.
- 7.Kachalova AV, Zatsepin TS, Romanova EA, Stetsenko DA, Gait MJ, Oretskaya TS. Nucleosides, Nucleotides Nucleic Acids. 2000;19:1693–1707. doi: 10.1080/15257770008045453. [DOI] [PubMed] [Google Scholar]
- 8.(a) C. A. Buhr and M. D. Matteucci, Eur. Pat., EP 0942000A2, 1999; b Prakash TP, Kawasaki AM, Lesnik EA, Owens SR, Manoharan M. Org Lett. 2003;5:403–406. doi: 10.1021/ol027131k. [DOI] [PubMed] [Google Scholar]
- 9.Ozaki H, Moriyama S, Yokotsuka K, Sawai H. Tetrahedron Lett. 2001;42:677–680. [Google Scholar]
- 10.A. V. Kachalova, E. M. Zubin, T. S. Zatsepin, Yu. V. Agapkina, Yu. M. Ivanova, D. A. Stetsenko, M. J. Gait, T. S. Oretskaya, in Innovation & Perspectives in Solid Phase Synthesis & Combinatorial Libraries 2004, ed. R. Epton, Mayflower Worldwide, Kingswinford, in press.
- 11.Kachalova AV, Stetsenko DA, Gait MJ, Oretskaya TS. Bioorg Med Chem Lett. 2004;14:801–804. doi: 10.1016/j.bmcl.2003.10.069. [DOI] [PubMed] [Google Scholar]
- 12.Beijer B, Grøtli M, Douglas M, Sproat B. Nucleosides Nucleotides. 1994;13:1905–1928. [Google Scholar]
- 13.Korshun VA, Stetsenko DA, Gait MJ. J Chem Soc, Perkin Trans 1. 2002:1092–1104. [Google Scholar]
- 14.a Sekine M, Hata T. J Am Chem Soc. 1986;108:4581–4586. [Google Scholar]; b Grøtli M, Eritja R, Sproat B. Tetrahedron. 1997;53:11317–11346. [Google Scholar]
- 15.Sekine M. J Org Chem. 1989;54:2321–2326. [Google Scholar]
- 16.Zatsepin TS, Romanova EA, Oretskaya TS. Russ Chem Rev. 2002;71:513–534. [Google Scholar]
- 17.Schwesinger R. Chimia. 1985;39:269–273. [Google Scholar]
- 18.Grøtli M, Douglas M, Beijer B, Garsia RG, Eritja R, Sproat B. J Chem Soc, Perkin Trans 1. 1997:2779–2788. [Google Scholar]
- 19.For further information on phosphazene bases see, Chem Files (Fluka), 2003, 3, that can be downloaded from http://www.sigmaaldrich.com/Brands/Fluka__Riedel_Home/Literature/ChemFiles/Vol_3_No_1.html
- 20.Smith M, Rammler DH, Goldberg IH, Khorana HG. J Am Chem Soc. 1962;84:430–440. [Google Scholar]
- 21.Caruthers MH, Barone AD, Beaucage SL, Dodds DR, Fisher EF, McBride LJ, Matteucci M, Stabinsky Z, Tang JY. Methods Enzymol. 1987;154:287–313. doi: 10.1016/0076-6879(87)54081-2. [DOI] [PubMed] [Google Scholar]
- 22.Zatsepin TS, Stetsenko DA, Arzumanov AA, Romanova EA, Gait MJ, Oretskaya TS. Bioconjugate Chem. 2002;13:822–830. doi: 10.1021/bc020016+. [DOI] [PubMed] [Google Scholar]
- 23.Zubin EM, Romanova EA, Volkov EM, Tashlitsky VN, Korshunova GA, Shabarova ZA, Oretskaya TS. FEBS Lett. 1999;456:59–62. doi: 10.1016/s0014-5793(99)00921-7. [DOI] [PubMed] [Google Scholar]
- 24.Mercier AJ, Orchard I, Tebrugge V, Skerrett M. Peptides. 1993;14:137–143. doi: 10.1016/0196-9781(93)90021-8. [DOI] [PubMed] [Google Scholar]
- 25.Ribeiro PD, Alves EW, Machado OLT. Protein Peptide Lett. 1999;6:203–208. [Google Scholar]