

Abstract
What is the minimum number of genes or functions necessary to support cellular life? The concept of a 'minimal genome' has become popular, but is it a useful concept, and if so, what might a minimal genome encode? We argue that the concept may be useful, even though the goal of defining a general minimal genome may never be attained.
Attempts to define life have touched nearly every scientific discipline and have been a long-standing subject in philosophy. Even when limited to the realm of biology, the definitions of life fall short of universal agreement and remain a widely debated subject. The practice of reductionism, whereby life is considered solely from the perspective of the genetic material, also generates controversy and complications. Many of these complications have been appreciated recently as a result of the large-scale application of whole-genome sequencing to microbes. The specific issue we discuss here is an attempt to define the minimum number of genes or functions necessary to support cellular life. The appeal of this subject lies largely in the fact that it represents a fundamental question in biology.
We have approached the concept of a 'minimal genome' in a manner not unlike that taken by physicists interested in understanding the nature of the universe: we have relied on an underlying assumption or guiding force that states that a simple set of rules must exist. The difficulty lies in finding the proper way to strip away the outer layers of complexity in order to uncover what is truly general and therefore satisfactorily describes all things. It is our belief that some set of basic parameters and principles are common to all cellular life on this planet. On the surface, especially in the age of genomics, it would appear that we will indeed be able to draw inferences and define commonalities. The advent of whole-genome shotgun sequencing [1] has changed biology in ways that we will not fully appreciate for many years to come. Advances in functional genomics, structural genomics and computational biology have also contributed greatly to our ability to make 'sense' of the huge quantities of DNA sequence data being generated. As we begin to apply these methodologies to the question of the minimal genome, we are faced with new perspectives which emphasize that, like any good scientific endeavor, the more knowledge we acquire, the less equipped we feel to answer our original question.
A model organism with a minimal genome?
Over the years, a number of experiments have been undertaken to approximate the number of essential genes within various model organisms [2,3]. Most often this was done through the generation of a set of mutations that were subsequently classified with respect to lethality. Through extrapolation, one derives an estimate of the number of genes or the amount of DNA required to support life. One of the difficulties in these early studies was that the data generated were analyzed and interpreted in the absence of complete genomic sequence, and therefore without our current awareness of the relatively large amount of potential functional redundancy in most microbial genomes examined to date. Paralogs arise through gene duplication events, not by vertical descent. In some cases, paralogous gene families within a genome are represented by simple gene pairs, whereas other families have many individual members with varied chromosomal arrangements. It is assumed that maintenance of most paralogous genes in a microbial genome is due to the increased adaptive potential the organism derives from their presence. But the existence of paralogous genes in genomes greatly hampers the interpretation of genome-scale mutagenesis experiments. On the basis of sequence analysis alone, we are not currently able to distinguish whether two or more members of a paralogous gene family have identical or non-redundant functions. This ambiguity forces investigators to take care to discriminate between dispensable genes and dispensable functions.
Mycoplasmas have been viewed as minimal organisms for many years [4]. This perception was initially based on their extreme fastidiousness: growth of these microbes in the laboratory requires the addition of many extracts and complex nutrient sources. The validity of the minimalist viewpoint would have to await advances in DNA technology, such as pulsed-field gel electrophoresis (PFGE). Among the mycoplasmas, the one that appears to have the smallest genome is Mycoplasma genitalium. Initial estimates, based on PFGE of chromosomal restriction fragments, indicated that the M. genitalium genome was approximately 600 kilobases (kb) in size [5]. This result was quite startling and was sufficient to attract the collective attention of Clyde Hutchison and one of us (S.N.P.) at The University of North Carolina, Chapel Hill, and Craig Venter and the other of us (C.M.F.) at The Institute for Genomic Research. In both instances, our instincts were to sequence the genomic DNA of this curious microbe, to satisfy an urge to know what genes are present within its freakishly small genome [6]. Ultimately, this curiosity provided the motivation for determining the complete sequence of the 580 kb M. genitalium genome [7]; it encodes a mere 480 proteins. The completion of the M. genitalium sequence was followed by the determination of the complete sequence of M. genitalium's closest relative, M. pneumoniae, just one year later [8]. The M. pneumoniae genome has approximately 685 identified open reading frames (ORFs). With the recent report of the Ureaplasma urealyticum genomic sequence [9], and the expected completion of three other mycoplasma species in the next year or two, the mycoplasmas have become a rich resource for comparative genomics. The high density of sequences from the mycoplasma lineage will provide great potential for understanding the reductive evolution of genomes.
The evolution of Mycoplasma genomes is thought to have occurred through a series of DNA deletions from larger Gram-positive ancestors. The fact that mycoplasmas are predominantly found in association with humans, plants or animals suggests that their diminutive genome size is a direct reflection of their parasitic lifestyle. Given our current level of sampling of microbes, the smallest genomes among free-living organisms appear to be those of pathogens. It is not known precisely what selective pressures generate minimal genomes, but it is conceivable that an organism begins the process through the acquisition of new functions that enable it to parasitize a host cell. This newly acquired ability has a dramatic impact on the selective value of many genes present in the genome. Under relaxed selection, gene loss may become prevalent. This is demonstrated most clearly in the genomes of M. genitalium and M. pneumoniae. The numerous genes required to synthesize all 20 amino acids, and to synthesize cell wall components, as well as genes encoding enzymes of the citric acid cycle and the majority of all other biosynthetic genes have been lost in these two organisms, presumably because they have evolved in such a way as to acquire these products from their host in vivo.
Can the minimal genome be defined?
Ultimately, a definition of a minimal set of genes required to sustain cellular life requires an agreed-upon definition of what is living and what is not. Gene loss in M. genitalium has come at some expense in terms of fitness in the laboratory setting. (M. genitalium is able to grow in culture in a cell-free environment and therefore meets the definition of free-living.) This is evident in terms of doubling time when comparing M. pneumoniae (680 ORFs) to M. genitalium (480 ORFs). The doubling time of wild-type M. pneumoniae in culture is approximately 6 hours compared to about 12 hours for M. genitalium. Experiments currently being performed in our laboratory aimed at introducing a saturating number of mutations into the M. genitalium chromosome indicate that the fitness of the cultures is gradually reduced upon successive rounds of mutagenesis (unpublished observations). It is anticipated that at some point the ability to maintain active cultures will become severely challenged. In many ways it seems plausible, if not likely, that the fitness reductions observed through loss of gene function could be compensated for through changes in the growth media (provided by an omniscient microbial physiologist). This point emphasizes the intimate relationship of environment and a minimal genome definition, and the difficulty of ever defining a minimal genome.
A minimal genome is, in reality, only a theoretical construction. It is not something one could find in nature. A minimal cell would require an 'ideal' environment, free of any selective pressure. It may be instructive to consider all genomes as being comprised of two types of genes. The first set includes those that confer the core set of functions required for basic cellular processes. By definition, these genes would be essential in any conceivable environment. For example, it may be safely assumed that all cells require an ability to replicate their DNA, produce message through some basic transcription machinery and translate that message to produce protein. In addition, all cells will have some requirements for energy production and transport of raw materials across the cell membrane and maintenance of cellular homeostasis.
It is well established that some features of a genome are selectively neutral and others maladaptive. If we ignore these cases, the remaining genes in any genome are presumably those that confer a specific selective advantage to an organism within the environmental niche it occupies. These functions may be dedicated to immune-response avoidance, expanded metabolic and biosynthetic potential, or stress-management skills, to name a few. It is assumed that the number of core functions required by any cell is relatively similar. We know that eubacterial genomes range in size from less than 0.6 Mb to greater than 9 Mb [10]. The number of accessory or adaptive proteins maintained in various genomes is therefore highly variable and in fact indicates a lot about the environment and selective pressures each organism faces. According to current evolutionary thinking, the environmental niche occupied by a microbe affects the set of genes retained in its genome. This idea gives rise to the possibility that one day we will be able to derive information about the microenvironment that an organism lives in through an examination of its gene complement. Likewise, the specific environment also determines whether a gene is essential or dispensable. This statement is true in nature and also in the laboratory. It is meaningless to refer to a gene as being essential or dispensable without an accompanying statement that defines the environment or growth conditions in which the categorization was made. For all of these reasons, discussion of minimal genomes often becomes a matter of semantics and definitions.
How does a minimal genome arise?
Genome evolution does not occur with any preconceived internal logic. Microbial genomes are no longer seen as static entities evolving through the accumulation of point mutations and infrequent chromosomal rearrangements. Gene acquisition and gene loss may be far more prevalent in microbes than was previously appreciated. Gene acquisition may allow a microbe to occupy a new environmental niche. Once resident in this new environment, the selective value of each gene in the genome is necessarily redefined and may allow the loss of previously useful genes. Gene loss is much more likely to be a one-way street in the course of genome evolution. Despite the presumed prevalence of horizontal gene transfer in nature, the number and types of functions that have evolved in the history of cellular life is far greater than the number of functions 'sampled' by any single organism. Mycoplasmas have dispensed with many metabolic functions and have retained very few genes required for transcription, repair and recombination, for example.
The loss of any given gene from a genome may limit the number and types of losses that are tolerated in the future: gene loss has consequences for the future evolution of the remaining genes in the genome. For example, it is known that the repertoire of transporters in mycoplasmas is fewer than in most microbes [11]. It has been speculated that the reduction of transporters has been compensated for by a broadened substrate specificity of the retained transporters [12]. It is also possible that M. genitalium and M. pneumoniae have been able to shed many of the genes involved in transcriptional regulation by shifting the burden of regulation toward translational processes ([13] and S.N.P., unpublished observations). An examination of the genes present in the M. genitalium genome can fool one into thinking that the organism has found the minimal solutions for basic cellular needs. This is of course very unlikely to be true for every pathway and every functional category within the cell. It is anticipated that future genome-sequencing efforts and functional characterization of genes will identify novel and more economical solutions for various cellular functions compared to those present in the genome of M. genitalium. On the basis of these arguments, it is evident that an experimentally defined minimal genome is lineage-specific, stating little, if anything, about what a minimal gene set might be for another phylogenetic lineage.
The theoretical minimal genome
A theoretical approach has been taken towards defining a minimal gene set [14]. This study derived a minimal genome by comparing what at the time were the only two fully sequenced microbial genomes (Haemophilus influenzae and M. genitalium). An all-against-all whole-genome comparison was performed in order to identify genes held in common by both organisms. The reasoning behind this comparison was simple but powerful and starts with a straightforward assumption: genes conserved across large phylogenetic distances are likely to be essential. Thus, if one compares two genomes that have diverged from a common ancestor a very long time ago (1,500 million years ago in this example), the genes in common will be highly enriched for those that carry out the most basic cellular functions common to all organisms. The application of this idea resulted in the definition of a minimal gene set consisting of about 250 genes. While the starting assumptions of this analysis appear reasonable, the two organisms in question are both human parasites and so may have too much in common, with regard to environmental selective pressures since the time of divergence, to be representative of genomes as a whole. It was the speculation of at least these authors [14] that the results would also be improved if one were to repeat the analysis at a later time when more microbial genomes could be sampled, representing a more diverse phytogeny (see below for further discussion of this issue).
Soon after the report of the M. pneumoniae genome sequence, a comparison of the M. pneumoniae and M. genitalium genomes was performed [15]. This analysis revealed some interesting features of the two genomes and their respective evolution. A comparison of amino-acid sequences among orthologs in M. pneumoniae and M. genitalium reveals an average of 65% identity, suggesting a much larger distance between the two genomes than was previously thought. The M. genitalium genome is essentially a proper subset of the M. pneumoniae genome. Approximately six major chromosomal rearrangements together with many tens of chromosomal deletions account for the large-scale differences between the two genomes. These facts also support the idea that there is substantial evolutionary distance between the two genomes. The overall conservation of gene order (synteny) is remarkably high over very large distances in many cases, however; this observation is consistent with the notion that the two species are very close relatives.
An attempt to explain this apparent dichotomy - divergence of individual sequences but close relationship of gene order - has been provided by the possibility that mycoplasmas have a significantly increased mutation rate relative to most bacterial species [16]; this is presumably due to the loss of several DNA-mismatch repair systems and mutator loci [17]. Virtually every gene in the M. genitalium genome has an ortholog in the M. pneumoniae genome. Given that these genomes were formed through reductive evolution from ancestors with larger genomes, it may be reasonable to assume that some, if not most, of the differences between the two genomes reflect gene loss in M. genitalium that occurred after divergence from their common ancestor. It is therefore notable, if not downright bizarre, that the net gene loss that has taken place in these genomes - after speciation - is unidirectional. This is counter to expectations. Given that the two organisms reside in different host sites (the lung versus the urogenital tract), and are presumably under different selective pressures, one would expect gene loss to have occurred differentially in the two genomes: some genes, lost from M. genitalium, would be present in M. pneumoniae, and vice versa. Perhaps the asymmetry of gene loss indicates that the environments these two human parasites occupy are substantially different. It is thought that both are extracellular pathogens. It is conceivable that M. genitalium has adapted to a new environment with a much reduced selective pressure compared to M. pneumoniae. One way to explain the gene-content differences of these genomes is to speculate that M. genitalium has acquired the status of an intracellular pathogen. Direct experiments may be warranted to examine this possibility.
The recent report of a third complete mycoplasma genome sequence, that of U. urealyticum (0.75 Mb) [9], has allowed further comparative genomics of minimalism. The unusual genomic relationship of M. genitalium and M. pneumoniae does not apply to the 613-ORF genome of U. urealyticum. In this case, the findings are more as expected: unequal gene loss has occurred between U. urealyticum and the other two mycoplasma genomes. Phylogenetically, U. urealyticum is more distantly related to M. genitalium and M. pneumoniae than the latter two are to each other. A comparison of the more distantly related genomes has allowed greater insights into mycoplasma evolution to be made. Only 324 genes are shared between U. urealyticum and M. genitalium; and M. genitalium has some genes (74) that are absent from U. urealyticum. On the surface, it might be tempting to assume that these 74 genes are dispensable in M. genitalium. But as the authors point out [9], 10 of these 74 encode functions involved in energy production, in this case glycolysis. Since U. urealyticum has evolved a unique means of generating ATP from urea hydrolysis, there has been a wholesale substitution of glycolytic genes for this system. Another notable feature of the U. urealyticum genome sequence is the apparent lack of the chaperone proteins GroES and GroEL, as well as the cytokinesis protein ftsZ. It is unclear whether these functions are truly absent from this mycoplasma, or alternatively whether the functions are being carried out by proteins with sequences unlike previously annotated proteins of the same function.
Irrespective of which possibility is more likely, this finding emphasizes a strong predisposition held by almost all molecular biologists, namely to define expectations on the basis of what is clear and evident in the most well-studied model microorganisms, Escherichia coli, Bacillus subtilis and Saccharomyces cerevisiae. The wealth of knowledge that these model systems have brought to our understanding of cellular mechanisms and general principles is undisputed. Perhaps for the first time, since the advances in genome sequencing we are perceiving that although these models are very useful, they are not in every case representative. This statement should not be thought of as blasphemous, but rather as an obvious point, considering the large number of specialized functions that have evolved in microbes. Whole-genome analysis and gene annotation is intimately linked to those precious few genes for which actual functional experiments have been conducted in the laboratory. It is still true that most functional assignments that have a foundation in laboratory experiments are limited to the great model systems.
An experimental approach
Our experimental approach for defining a minimal genome identified, on a rather large-scale, transposon-insertion events in M. genitalium and M. pneumoniae that, due to their location on the chromosome and presence in viable cells, are likely to have disrupted the activity of a gene function that could be considered dispensable to the cell [18]. Despite the technical difficulties associated with working with these organisms, and the lack of genetic and reverse-genetic tools, the fact that natural selection had already been working on them in a natural 'minimal genome project' for millions of years made them a very attractive model system. It is fair to say that before initiating this study we probably did not have a strong set of expectations as to the number of genes that would be dispensable in M. genitalium. It would not have been completely surprising if the genome of M. genitalium harbored only a few dispensable functions or several dozen. The fact that we were approaching a question for which little insight or predictive power was available made all the more important the use of good control experiments: we needed to gain confidence that the transposon insertions were capable of reliably identifying dispensable genes. We made use of the fact that the M. genitalium genome is a proper subset of the M. pneumoniae genome as a rationale for the assumption that the additional 210 genes present in M. pneumoniae should be dispensable in the laboratory setting. We considered the M. genitalium genome as containing two types of genes, essential and dispensable, whereas the M. pneumoniae genome contains three types of genes: shared (in M. genitalium and M. pneumoniae) essential genes, shared dispensable genes and dispensable pneumoniae-specific genes. We predicted that the proportion of recovered gene-disruption events should be greater in the pneumoniae-specific portion of the genome than in the shared portion of the genome. We did in fact recover approximately six times the frequency of insertions from the pneumoniae-specific portion of the M. pneumoniae genome [18]. Altogether, we identified 129 genes present in M. genitalium that were apparently dispensable. We used statistical analyses based on the Poisson distribution and extrapolation of our data set to estimate that between 180 and 215 dispensable genes exist in the M. genitalium genome.
This somewhat startling result raises several interesting questions. How are we to interpret the findings? It is important to mention first that an examination of the M. genitalium genome reveals a lack of any recently evolved paralogous gene families. Unfortunately, as stated before, it is not possible to be certain whether existing genes with paralogous relationships within the M. genitalium genome carry out identical functions. The accuracy of the assumption that there are no redundant functions in the M. genitalium genome determines the certainty with which we are able to interpret our disruption data. The natural next question to ask is whether the catalog of dispensable functions identified in 'a one gene at a time' study could be tolerated in combination, and if so, to what extent? As mentioned before, we have initiated a saturation mutagenesis of the M. genitalium genome. While mutagenesis is still ongoing, it appears unlikely that any cell in the population will be recovered harboring more than 100 functionally null genes (S.N.P., unpublished observations). There are two reasonable explanations for this apparent discrepancy between the 'gene by gene' approach and the combinatorial approach. First, some genes in the M. genitalium genome do carry out redundant or partially redundant functions, so while single-gene disruptions are tolerated, the combination of two disruptions is not. The second, and probably more important, explanation is that dispensable genes, especially in M. genitalium, are not likely to be lost without some discrete reduction in the overall fitness level of the organism. In this regard, it maybe appropriate to consider dispensable genes to be associated with a quantifiable fitness contribution to the cell in any given environment. If one considers the combination of genes in the wild-type M. genitalium genome as providing a global fitness value above some minimal threshold for survival, then ultimately the minimal genome will be obtained by sequential disruption of genes that individually contribute the least to the fitness of the cell. Viewed in this manner, it seems that minimal genomes are more an interesting mathematical problem or, worse yet, a matter for an accountant's spreadsheet, than an issue that can be defined experimentally. We would argue that it is perhaps more important to attempt to grasp the quantitative selective value that any gene contributes to the fitness of the organism than to attempt to define a minimal gene set.
What do the transposon results tell us about the overall architecture of gene functions in a genome? It is assumed that the activity of individual enzymes within the cell has an impact on other pathways and processes, giving rise to a view that the cell is an intricate sensing system that is responding not only to the outside environment but also to its own actions. This ability to modulate is especially important to bacteria, where waste is not considered a virtue. But the fact that we were able to disrupt so many gene functions in a minimal cell suggests something subtle about the nature of gene functions within genomes: it suggests that there is in fact a lack of interconnectivity among gene functions. It appears that pathways evolve which in many cases show interdependency, and epistasis is a well-defined genetic idea that speaks directly to the functional interdependency of genes. But among dispensable functions, our ability to recover a living organism bearing disruptions of nearly one-third of its gene complement lends support to the view that interdependency beyond epistatic relationships may be uncommon.
If we examine gene disruption data in terms of the functional roles of the genes in the cell, we are able to gain some additional insights about minimal genomes. The group of genes for which we recovered by far the largest number of disruptive insertions is the group of unknown function (Figure 1). This is in contrast to the number of insertions tolerated in genes involved in translation, for example. The functional group for which the highest proportion of putative disruptions were identified was the genes encoding cell-envelope proteins. A precise interpretation of this result is not possible without extensive experimentation, but it may serve to highlight the previously mentioned idea that the definition of the dispensability of a gene is dependent on the environment. It is conceivable that the group of cell-envelope proteins plays a significant role in the context of a host infection but is of relatively little value to the cell in laboratory growth media. By examining the data shown in Figure 1, we can see that not all functional roles in M. genitalium are equally dispensable; some functional systems are closer to minimal than others. One can obtain a visual sense of the functional distribution of genes in a minimal cell by examining the relative proportions of genes in each functional group; it is interesting that nearly two-thirds of the cell is dedicated to the translation of message into protein and genes for which we do not yet know the function or cellular role.
Figure 1.
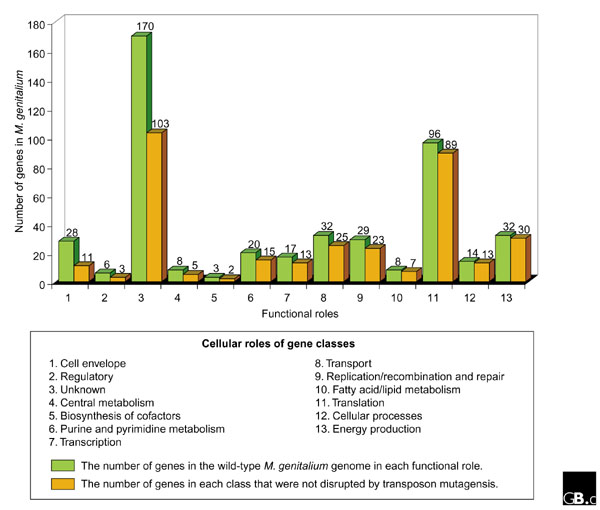
The genes of Mycoplasma genitalium categorized according to function and whether or not they were disrupted by transposon mutagenesis.
The fact that an estimated one third of the essential set of genes in this minimal genome are of undefined function is an important result that has at least two potential interpretations. First, it draws dramatically into question a basic assumption held by many biologists that the fundamental mechanisms and functions underlying cellular life have for the most part been identified and well characterized. If approximately 100 genes in the simplest functioning cell are of unknown function and are essential to basic cellular processes, this assumption becomes quite dubious. An alternative perspective is that many of the 100 genes of unknown function in the minimal gene set encode well-characterized functions in E. coli, but are simply not recognizable as orthologs in M. genitalium. It is likely that both explanations are at least partly valid, but the conclusion remains the same: we have much work to do before we can claim to have a clear understanding of even the simplest cell and its functions.
Comparing many more genomes
Since the time of the work on the comparison of the H. influenzae and M. genitalium genomes [14], many new microbial genomes have been sequenced, representing many lineages of eubacteria, archaebacteria and eukaryotes. In theory, this additional data set should allow the comparative approach to more precisely define a minimal gene set. Recently, 21 completed genomes were compared, this time employing the more sensitive gene-comparison tool Clusters of Orthologous Groups of proteins (COGs) to find orthologs common to all genomes [19]. COGs are defined by a grouping of at least three proteins from distantly related organisms that are more similar to each other than to any other gene in their respective genomes [20]. In this way, COGs define orthology groups and ancestral relationships. The added sensitivity derived from the COGs analysis arises because COGs are not dependent on BLAST cutoff scores and arbitrary sequence-relatedness criteria; genes with relatively unimpressive sequence identity can be placed into an orthology group. Some interesting facts arose from this analysis [19]. First, 55-83% of proteins encoded by bacterial and archaeal genomes can be placed into discrete COGs, suggesting that many genes present in bacteria and archaea are highly conserved. Very few genes appear to be universally, or nearly universally, conserved in all 21 organisms analyzed, however, and it was this criterion that was used in the original analysis for defining the essential gene set of a cell [14]. In this expanded study [19], only 80 genes were universally conserved across all organisms. Clearly, this number of genes is insufficient to serve as a satisfactory answer to the question 'what is the minimal number of functions required for cellular life?'
Was there something wrong with the starting set of assumptions made in the comparative approach? Among the possible interpretations of this troubling result, one is that there is something fundamentally faulty about COGs analysis, which is unlikely. A second possibility is that many genes and functions arose a very long time ago. These 'ancient' genes would therefore have diverged to such a great extent that they are unrecognizable by any sequence-relatedness algorithms. A third, and extremely interesting, possibility is that many gene functions have evolved independently more than once since the beginning of cellular life on the planet. The term that has been used to describe this occurrence is non-orthologous gene displacements (NODs) [21]. There is scattered evidence for this idea for various genes found in nature, but the COGs analysis with 21 genomes places a potential magnitude on the phenomenon. The implications of this idea, if correct, are significant but beyond the scope of this article.
Despite the arguments put forward here to highlight the complications and potential inappropriateness associated with attempts to define minimal genomes, it is clearly instructive to try. The analogy we would make is with the knowledge we gain from sequencing microbial genomes. We learn a lot by sequencing each one, but far more by comparing the similarities and differences of many genomes. Similarly, we have learned much about genome evolution and the gene functions required for cellular existence - and hopefully will continue to do so - and anticipate a future ability to compare the results of similar experiments from models other than 'the minimal genome'. The discussion here highlights the importance of generating functional data for genes in organisms from diverse lineages. Gene diversity in the microbial world is truly a resource. It is unlikely that we will exploit this resource efficiently until many more microbial model organisms are developed and studied at a similar level to E. coli or yeast. We are forging ahead in genomics at unprecedented speed, with too few frames of reference, and there is a risk of becoming lost in a sea of sequence data without a functional framework, unless we ensure that functional analysis does not lag too far behind. We believe that the concept of the minimal genome is a useful tool in attempting to organize our thoughts about gene function - even though we may never, in practice, be able to reach a definition of a minimal gene set that is applicable to all types of organism.
References
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random shotgun sequencing and assembly of Haemophilus influenza Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Goebl MG, Petes TD. Most of the yeast genomic sequences are not essential for cell growth and division. Cell. 1986;46:983–992. doi: 10.1016/0092-8674(86)90697-5. [DOI] [PubMed] [Google Scholar]
- Itaya M. An estimation of minimal genome size required for life. FEBS Lett. 1995;362:257–260. doi: 10.1016/0014-5793(95)00233-y. [DOI] [PubMed] [Google Scholar]
- Morowitz HJ. The completeness of molecular biology. Isr J Med Sci. 1984;20:750–753. [PubMed] [Google Scholar]
- Colman SD, Hu Pc, Litaker W, Bott KF. A physical map of the Mycoplasma genitalium genome. Mol Microbiol. 1990;4:683–687. doi: 10.1111/j.1365-2958.1990.tb00638.x. [DOI] [PubMed] [Google Scholar]
- Peterson SN, Hu PC, Bott KF, Hutchison CA III. A survey of the Mycoplasma genitalium genome using random sequencing. J Bacteriol. 1993;175:7918–7930. doi: 10.1128/jb.175.24.7918-7930.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, Bult CJ, Kerlavage AR, Sutton G, Kelley JM, et al. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R. Complete sequence analysis of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24:4420–4449. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass JI, Lefkowitz EJ, Glass JS, Heiner CR, Chen EY, Cassell GH. The complete sequence of the mucosal pathogen Ureaplasma urealyticum. Nature. 2000;407:757–762. doi: 10.1038/35037619. [DOI] [PubMed] [Google Scholar]
- Kuspa A, Vollrath D, Cheng Y, Kaiser D. Physical mapping of the Myxococcus xanthus genome by random cloning in yeast artificial chromosomes. Proc Natl Acad Sci USA. 1989;86:8917–8921. doi: 10.1073/pnas.86.22.8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, Eisen J, Fleischmann RD, Ketchum KA, Peterson S. Comparative genomics and understanding of microbial biology. Emerg Infect Dis. 2000;6:505–512. doi: 10.3201/eid0605.000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saurin W, Dassa E. In the search of Mycoplasma genitalium lost substrate-binding proteins: sequence divergence could be the result of a broader substrate specificity. Mol Microbiol. 1996;22:389–390. [PubMed] [Google Scholar]
- Wasinger VC, Pollack JD, Humphery-Smith I. The proteome of Mycoplasma genitalium. Chaps-soluble component. Eur J Biochem. 2000;267:1571–1582. doi: 10.1046/j.1432-1327.2000.01183.x. [DOI] [PubMed] [Google Scholar]
- Mushegian AR, Koonin EV. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci USA. 1996;93:10268–10273. doi: 10.1073/pnas.93.19.10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelreich R, Plagens H, Hilbert H, Reiner B, Herrmann R. Comparative analysis of the genomes of the bacteria Mycoplasma pneumoniae and Mycoplasma genitalium. Nucleic Acids Res. 1997;25:701–712. doi: 10.1093/nar/25.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR, Maniloff J, Zablen LB. Phylogenetic analysis of the mycoplasmas. Proc Natl Acad Sci USA. 1980;77:494–498. doi: 10.1073/pnas.77.1.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA. A phylogenomic study of the MutS family of proteins. Nucleic Acids Res. 1998;18:4291–4300. doi: 10.1093/nar/26.18.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison CA III, Peterson SN, Gill SR, Cline RT, Richardson D, White O, Fraser CM, Smith HO, Venter JC. Global transposon mutagenesis and the minimal Mycoplasma genome. Science. 1999;286:2165–2169. doi: 10.1126/science.286.5447.2165. [DOI] [PubMed] [Google Scholar]
- Koonin EV. How many genes can make a cell - the minimal gene set concept. Annu Rev Genomic Hum Genet. 2000. [DOI] [PMC free article] [PubMed]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Mushegian AR, Bork P. Non-orthologous gene displacement. Trends Genet. 1996;12:334–336. [PubMed] [Google Scholar]