

Abstract
The polarization of views on how best to exploit new information from the Human Genome Project for medicine reflects our ignorance of the genetic architecture underlying common diseases: are susceptibility alleles common or rare, neutral or deleterious, few or many? Single-nucleotide polymorphism (SNP) technology is almost in place to dissect such diseases and to create a personalized medicine, but success is critically dependent on the biology and "Nature to be commanded must be obeyed" (Francis Bacon, 1620, Novum Organum).
The genome as text
Shakespeare is thought to have had a working vocabulary of some 35,000 words, about twice that of an educated person today. A play is therefore an apposite metaphor for the genome, in which the principal interest lies not in the words or letters but in the way it is performed and brought to life on the biological stage. The sequencing of the human genome provides a sense that human biology has reached a new beginning in which the genomic text can be read but it is far from clear how it is played out. Despite this, new horizons are being proclaimed, in which complex diseases are explained, new drugs delivered and new models created. But science and theology are never far apart and nowhere more so than in unravelling the genetics of complex disease - the Croesus code that will bring wealth to some and health to others. The theological differences lie in how to apply new information from the Human Genome Project and whether or not it is an illusion that whole genome association studies using dense maps of single nucleotide polymorphisms (SNPs) in large patient populations will lead to new insights into common disease and to a safe, personalized medicine.
The case in favor
The key issues concern the genetic architecture of common diseases. On the one hand, Eric Lander (speaking at the Human Genome Organization's annual meeting, HGM2001, in Edinburgh, UK), along with David Reich and colleagues from MIT, has argued that the allelic diversity underlying disease is predictable and that it favors SNP mapping of large patient populations [1]. Four main factors account for this predictability. First, in human founder populations some 100,000 years ago, the effect of an allelic variant on reproductive fitness to a large extent determined its equilibrium frequency. An allele with a deleterious phenotype would reduce fitness and achieve a low equilibrium frequency, determined by a mutation-drift-selection balance [2]. An allele with little or no effect on fitness, as perhaps expected for genes influencing late-onset disorders such as type 2 diabetes mellitus or heart disease, could achieve a high equilibrium frequency. The second factor is the relatively rapid expansion of anatomically modern humans from a small founder pool of a few tens of thousands to the present 6 billion [3]. The third factor is the expectation that the predicted allelic diversity for neutral or selectively equivalent alleles in such a small founder pool is remarkably low, in fact close to one or a few alleles per locus - given by (1 + 4Neμ), where Ne is the effective population size and μ the average mutation rate per locus per generation [4]. As the human population expanded, however, the allelic diversity increased enormously, at the rate of about 175 new mutations per genome per generation. In principle, it is possible for every base of the genome to be mutated in at least one person alive today. Most of this variation is destined to disappear rapidly, but many young alleles reached significant frequencies within human subpopulations, prior to recent urbanization [5].
The fourth and key insight provided by Reich and Lander [1] is that if a disease-risk allele was common in the founder population, it takes a very long time before it is diluted out by the new alleles generated during population growth. Eventually, a new equilibrium will be reached, with a very high allelic diversity - in the tens of thousands of alleles per locus - but in some cases this may take over a million years. At present, this degree of diversity is only expected for alleles that were at low frequency in the founder population and so are more rapidly diluted out and equilibrated with younger alleles. Such a situation is already evident in the high allelic diversity of deleterious monogenic disorders. In short, if an allele was common in the distant past, it may well be common today and allelic diversity will be low. For example, the APOE*E4 allele that is associated with susceptibility to Alzheimer's disease was such a founder allele and remains common, with an allele frequency of 0.04-0.49, in all human populations today. On the other hand, if an allele was rare in the founder population, it will now be a substantially rarer member of a diverse allelic set. In the case of susceptibility alleles for common diseases, which Reich and Lander [1] assume to be more-or-less invisible to natural selection, these may have reached high allele frequencies in the past, in which case they will remain at high frequency today. The common disease/common variant hypothesis [6] is thus both predictable from population genetic theory and supported by empirical data (Table 1a). From this perspective, it is a short step to whole-genome association mapping of such variants using dense SNP maps in large samples of patients and controls.
Table 1.
Summary of allelic heterogeneity in support of the common disease/common variant or multiallele/multilocus hypotheses
| Disease type | Locus | Allele | Trait | Frequency | Effect | Comments |
| (a) Common disease/common variant hypothesis | ||||||
| Cardiovascular | APOE | *E4 | Alzheimer | 0.10-0.15 | Early onset | Allele present in primates and all world |
| disease | (Caucasian) | populations; possible interaction with | ||||
| dietary fats; may account for 20% of | ||||||
| Alzheimer disease | ||||||
| Age-related | 0.10-0.15 | Decreased risk | Well-established protective effect on | |||
| macular | age-related macular degeneration | |||||
| degeneration | ||||||
| Cardiovascular | 0.10-0.15 | Increased risk | Accounts for 10-16% of plasma | |||
| disease | cholesterol variance (western | |||||
| populations); increases risk of | ||||||
| cardiovascular disease (odds ratio | ||||||
| approximately 1.5) | ||||||
| F5 | R506Q | Venous | 0.02-0.08 | Increased risk | Carriers have around 10% lifetime risk | |
| thrombosis | for significant venous thrombosis | |||||
| Metabolic/ | PPARG | P12A | Type 2 diabetes | 0.85 | Increased risk | Relative risk 1.25 |
| nutritional | mellitus | (Caucasian) | ||||
| CAPN10 | Haplotypes | Type 2 diabetes | 0.03-0.29 (low | Increased risk in | Complex risk haplotypes that may | |
| 112 and 121 | mellitus | to high risk | 121/112 haplotype | include several SNPs, including | ||
| populations) | heterozygotes | CAPN10-g.4852G/A (UCSNP-43) | ||||
| HFE | C282Y | Haemochromatosis | 0.05 | Around 40% risk | High frequency in Caucasians, low in | |
| (Caucasian) | for homozygotes | Asiatics (suggesting admixture), so it may | ||||
| be a recent mutation (less than 50,000 | ||||||
| years ago) | ||||||
| Cancer | ELAC2 | S217L | Prostate cancer | 0.30 and 0.04 | Increased risk | Odds ratio 2.4-3.1 |
| and A541T | (Caucasian) | |||||
| BRCA2 | N372H | Breast cancer | 0.22-0.29 | Increased risk | Relative risk = 1.31 for HH compared to | |
| (Caucasian) | NN genotypes | |||||
| Infectious/ | MHC class I | HLA-B*2702, | Ankylosing | 0.09 | Increased risk | Odds ratio approximately 170, mechanism |
| inflammatory | 04, 05 | spondylitis | (Caucasian) | unclear; also associated with reactive | ||
| arthritis and uveitis; about 2% of B27- | ||||||
| positive carriers develop ankylosing | ||||||
| spondylitis | ||||||
| MHC class II | DQB1*0302- | Type 1 diabetes | 0.05 | Increased risk | Around 10% of heterozygotes for these | |
| DRB1*0401/ | mellitus | (European) | high risk haplotypes develop type 1 | |||
| DQB1*0201- | diabetes mellitus; relative risk | |||||
| approximately 20 | ||||||
| DRB1*03 | ||||||
| IL12B | 3' UTR | Type 1 diabetes | 0.79 | Increased risk | Interaction with HLA; increased | |
| allele 1 | mellitus | (Caucasian) | expression of IL12B in vitro | |||
| G6PD | A- | G6PD deficiency | Approximately | Decreased risk of | High allele frequency proposed to be | |
| (V68M/N126D) | 0.20 (West | severe malaria | due to balancing selection | |||
| African) | ||||||
| HBB | HbC (E6K) | Anaemia | 0.09 (West | Decreased risk of | High allele frequency proposed to be | |
| (homozygotes) | African) | severe malaria | due to balancing selection | |||
| CCR5 | Δ32-CCR5 | HIV-1 | 0.09 | Decreased HIV-1 | Recent origin - estimated approximately | |
| transmission | (Caucasian) | transmission | 700 years ago [13] | |||
| Developmental | PDGFRA | Promoter | Neural tube | 0.23 | Increased risk for | At least six polymorphic sites within |
| H1/H2α | defect | (Caucasian) | sporadic neural | each haplotype | ||
| haplotypes | tube defect | |||||
| (b) Multilocus/multiallele hypothesis | ||||||
| Cardiovascular | LDLR | > 735 alleles | Coronary artery | All rare, except in | Increased risk of | |
| disease | isolate or founder | coronary artery | ||||
| populations | disease | |||||
| APOB | > 24 alleles | Coronary artery | R3500Q 0.002, | Increased risk of | Single common R3500Q allele | |
| disease | remainder rare | coronary artery | ||||
| disease | ||||||
| Cancer | BRCA1 | > 483 alleles | Familial breast- | All rare, except in | Increased risk | |
| ovarian cancer | isolate or founder | |||||
| populations | ||||||
| BRCA2 | > 404 alleles | Familial breast | All rare, except in | Increased risk | Common N372H allele (frequency | |
| cancer | isolate or founder | approximately 0.25) with relative | ||||
| populations | risk 1.31 | |||||
| MLH1 | > 143 alleles | Hereditary non- | All rare | Increased risk | ||
| polyposis colorectal | ||||||
| cancer (HNPCC) | ||||||
| MSH2 | > 108 alleles | Hereditary non- | All rare | Increased risk | ||
| polyposis colorectal | ||||||
| cancer (HNPCC) | ||||||
| P53 | > 144 alleles | Multiple cancers | All rare | Increased risk | ||
| Neurosensory | ABCA4 | > 350 alleles | Stargardt disease, | Most rare, G863A | Increased risk | |
| retinitis pigmentosa | allele approximately | |||||
| 0.014 (Europeans) | ||||||
| RHO | > 88 alleles | Retinitis pigmentosa, | All rare | Increased risk | ||
| congenital stationary | ||||||
| night blindness | ||||||
| GJB2 | > 45 alleles | Non-syndromic | Most rare, 30delG | Increased risk | 30delG absent from non-European | |
| deafness | allele around 0.015 | populations | ||||
| (Europeans) | ||||||
| Metabolic/ | CFTR | > 963 alleles | Cystic fibrosis | Most rare, | ||
| nutritional | ΔF508 accounts for | Increased risk ΔF508 allele recent | ||||
| approximately 70% | -estimated to have arisen 3,000 | |||||
| of cystic fibrosis | years ago [14] | |||||
| alleles in Caucasians | ||||||
Data are from the Online Mendelian inheritance in Man database [30].
The case against
The common disease/common variant model is elegant, appealing and politically correct, but there are objections. The essential one is that it fundamentally misrepresents the nature of common disease. By definition, complex traits have what Kenneth Weiss and Joseph Terwilliger [7] call low "detectance" - a low probability of carrying any particular susceptibility genotype given that the individual has a particular disease or trait phenotype. This is because, unlike Mendelian disorders, common diseases clearly result from the interaction of many genetic and environmental influences, so that the correlation with any one factor is weak. There may be uncertainty about the extent of oligogenic versus polygenic influences on a trait, but few doubt that non-genetic factors play a major role in the common late-onset disorders of western societies, many of which have fluctuated in prevalence within the last 50-100 years (type 2 diabetes, obesity, auto-immune diseases, asthma, hypertension and coronary artery disease) [8]. Several of these disorders show a steep decline in heritability as age-of-onset rises, implicating generalized ageing processes that are not strongly influenced by genetic differences [8,9]. These diseases are common because of highly prevalent non-genetic influences, not because of common 'disease alleles' in the population. The majority of cases are not genetically determined to any meaningful extent. Such weakly disease-associated alleles as do exist can undoubtedly reach high frequencies if they are truly invisible to selection, but a key issue is the proportion of them that exert non-trivial influences on late-onset phenotypes. An inverse relationship between the magnitude of genetic effect and allele frequency was postulated many years ago [10,11], suggesting that few variants of clinical consequence will be common (Figure 1b). More recently, modeling of complex diseases by Jonathan Pritchard [12] predicts that neutral susceptibility alleles contribute little to the genetic variance underlying disease, since they tend to be either lost or close to fixation in the population. By contrast, alleles under weak selection may constitute the bulk of the genetic variance, especially at loci showing high mutation rates. This predicts extensive allelic heterogeneity underlying disease, although the collective frequency of these alleles may be quite high.
Figure 1.
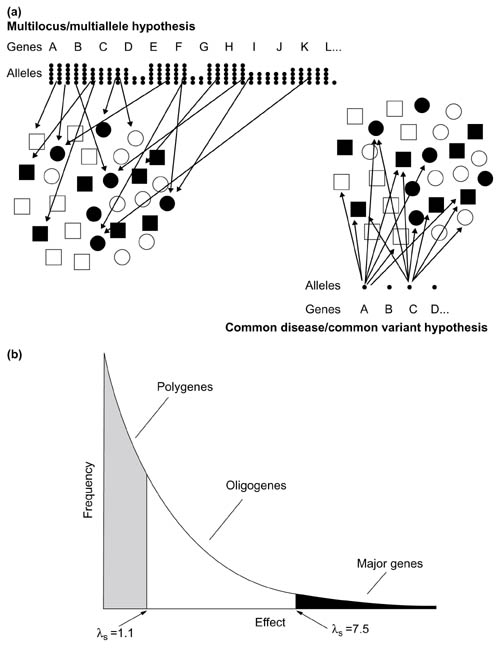
(a) Illustration of the common variant/common disease and multilocus/multiallele hypotheses (see text for details). Shaded symbols indicate carriers of a disease or trait; open symbols are non-carriers. (b) Inverse relationship between allele frequency and phenotypic effect, as postulated by Sewall Wright [10]. The arbitrary division between alleles with small (polygene), intermediate (oligogene) or large (major) effects is based on Morton [11]; λS, relative risk to sibs.
The empirical observation that late-onset Mendelian-inheritance disorders, in which causal genes should also have failed to influence reproductive fitness, show broad allelic diversity contradicts the common disease/common variant hypothesis (Table 1b). An example is the diversity of rare disease-causing BRCA2 alleles (of which there are more than 400) compared with only one out of six common alleles that shows any effect on breast cancer risk (see Table 1; N372H relative risk 1.3) [13]. Similarly, premature coronary artery disease due to familial hypercholesterolaemia is caused by over 735 different alleles of the low-density lipoprotein (LDL) receptor, but there are no common LDL receptor variants with significant clinical effects. Conversely, some deleterious alleles, such as the ΔF508 CFTR allele associated with cystic fibrosis, are at high population frequency (approximately 1.5% of Caucasian chromosomes). This allele appears to be of relatively recent origin (estimated at 3,000 years ago), explaining why it is found largely in northern Europeans [14].
These and similar observations (Table 1) suggest, firstly, that genetic effects do not conveniently parcel themselves into early-onset versus late-onset, with corresponding effects that are either visible or invisible to selection: high allelic diversity is evident in many late-onset disorders, suggesting significant adverse selection and low allele frequencies in founder populations. It is commonplace for a gene to show both early and late patterns of expression, which may or may not overlap in time or space, some of which represent a trade-off between early advantageous and late deleterious effects (antagonistic pleiotropy), others early deleterious and later neutral or advantageous effects [15]. Variants with deleterious phenotypic consequences can also survive and reach high population frequencies if they confer a selective advantage at reduced dosage or during times of high mortality. Secondly, chance and population history are major determinants of extant patterns of variation.
Misconceived mapping
The common disease/common variant hypothesis is not just an interesting idea to be discussed in ivory towers. The real opposition to it stems from its wide acceptance as justification for SNP mapping of complex disease and pharmacogenomic traits in large population samples [16,17,18]. It is acknowledged that these methods will rarely work unless the theory is substantially correct. Association mapping requires enrichment for a common ancestral predisposing (or protective) allele within groups sharing a common disease or drug-response trait [19]. Traditionally, trait mapping is achieved by the simple but powerful strategy of studying families with more than one member either exhibiting or correlated for the trait. The greater the familial correlation, the more likely that a large genetic effect is involved. This strategy, the mainstay of Mendelian mapping, increases the signal-to-noise ratio by reducing the proportion of those studied whose trait results from non-genetic factors. Other strategies for increasing the detectance of a disease locus include ascertaining by extreme age-of-onset (for example, early-onset adult cancers or coronary artery disease); by studying those showing extreme values of a sub-clinical phenotype, which is genetically simpler than the disease itself (for example, plasma lipid profiles); by disease severity (for example, recurrent or bipolar depression); by studying a high prevalence ethnic group despite similar environmental exposure (for example, type 2 diabetes in Mexican Americans); by low environmental risk (for example obstructive lung disease in non-smokers, or coronary artery disease in rural Mediterraneans); by studying an isolated subpopulation (so that a small founder size minimizes the number of risk alleles entering the population); or by studying a clinical-aetiological subgroup (for example, HLA-matched type 1 diabetics) [20]. Generally, a combination of such strategies is required to minimize the background noise of non-genetic cases and to enrich for those with a common genetic susceptibility [7,20]. Weiss and Terwilliger [7] argue that such methods of ascertainment are crucial, providing the major buffer against low detectance and loading the dice in favor of the investigator. The odds are heavily stacked against gene mapping in complex disease. If locus or allelic heterogeneity is high, association studies of single affected individuals, especially if they show late onset, or studies of parent-child trios, are all inherently flawed.
Pervasive diversity
There is formidable diversity within complex traits, not only in their environmental determinants but also in the genetic components of risk. The low success rate of complex trait mapping stems from a combination of poor study design and extreme locus and allelic heterogeneity [7,20,21,22]. Locus heterogeneity - where more than one locus contributes to disease risk - is perhaps the biggest problem for complex traits and will undoubtedly make association mapping extremely difficult. No geneticist correctly predicted the extent of locus heterogeneity in 'simple' Mendelian disorders (for example, see Figure 2). It is correspondingly both difficult and painful to consider that such heterogeneity may be orders of magnitude greater in more physiologically complex disorders such as coronary artery disease or asthma. We can easily conceive of a few tens of disease loci but not many hundreds, which interact in different combinations in different individuals to influence the trait (Figure 1a). Some researchers, like HGM2001 speaker John Todd (University of Cambridge, UK), openly admit that this scenario is too depressing to contemplate, so we tend to proceed as if "our disease" will be the exception. Scientific reductionists are trained to minimize complexity. Modern medicine may find it hard to accept that our current state of knowledge in hypertension, coronary artery disease or asthma is similar to that in 1900 for conditions such as mental handicap, anaemia, heart failure or blindness, for each of which hundreds of distinct causes are now routinely delineated. The common disease/common variant hypothesis is ringingly silent on the problem of locus heterogeneity. Similarly, if allelic heterogeneity is as extensive in common disorders as it evidently is in their Mendelian subgroups (Table 1), most association studies will be paralyzed [20,21].
Figure 2.
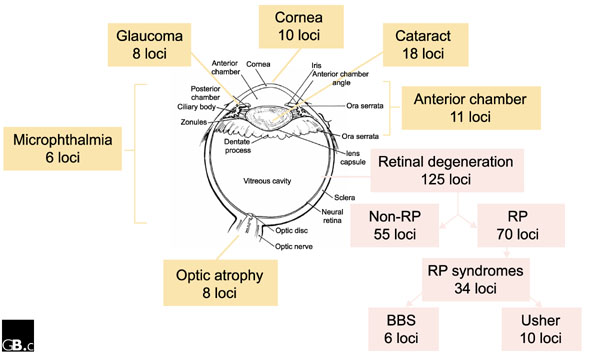
Locus heterogeneity in Mendelian disorders. The diagram shows the diversity of disease loci in Mendelian forms of blindness. A rough relationship between tissue and physiological complexity and the number of identified disease loci can be discerned. Data are from the Online Mendelian Inheritance in Man [30] and RetNet [31] databases. RP, retinitis pigmentosa; Usher, Usher syndrome; BBS, Bardet-Biedl syndrome.
Mapping made easy
The common variant is certainly a player in some common disorders (Table 1), but how many, and how significant are their effects for medicine or biology? There are an estimated 2-3 million common SNP variants [22] but it is often subtle combinations and permutations that influence disease, as proposed for predisposing and protective haplotypes in type 2 diabetes and in several autoimmune disorders (Table 1). It now seems that SNP haplotypes are, in general, less diverse than expected. David Cox (Perlegen, Inc. and Stanford University, USA) outlined at HGM2001 some of the first fruits of large-scale SNP haplotyping of human chromosomes, in which somatic cell hybrids are being used to separate chromosomal homologs so as to give unambiguous haplotypes. The results for chromosome 21 show that the potential haplotype diversity is not nearly as great as expected, facilitating the identification of common disease-associated haplotypes. For example, only two or three of the 64 possible six-marker SNP haplotypes are detectable at many loci, so that the ancestral chromosome 21 genome can be summarized in some 3,000 haplotypes, each representing small (on average 12-15 kilobase) regions within which alleles are in strong linkage disequilibrium. These can be typed using 'wafers' containing 96 chips, each with 400,000 arrayed oligonucleotides. Extrapolating from chromosome 21, some 300 wafers could provide coverage of the whole genome. Lander and colleagues [23] have recently suggested that the northern European genome can be summarized in some 30,000 ancestral haplotypes, with conserved regions of linkage disequilibrium that average 60 kilobases in length. The observation that sub-Saharan Africans show seven-to eight-fold smaller regions of linkage disequilibrium is consistent with a tight population 'bottleneck' somewhere between 27,000 and 53,000 years ago that dramatically reduced European diversity relative to that of sub-Saharan Africans.
Resolution?
Where does this leave those who are in a position to capitalize, in every sense, on the new technologies? Regardless of one's theology, there are certainly awkward questions to be asked of the common disease/common variant hypothesis. Its success as a model will depend on the survival of common alleles that are today capable of significantly influencing health or drug response. These alleles may either have been more-or-less invisible to selection (and most of our ancestors died before the age of 30-40 years), weakly selected against [12], or even positively selected, as is proposed for major histocompatibility complex (MHC) class II haplotypes associated with autoimmune disorders, which can be seen as the flip side of a strong immune response. Selectively neutral alleles are a random selection of variants arising throughout evolutionary history, the sum total of which reflects chance, past demographic events and mutation rates. It seems unlikely that large numbers of these random, functionless events will significantly influence common disease traits. Disease modeling also suggests that they contribute little to the genetic variance underlying common disease [12]. Previous selection is a more promising argument, since there is a case that several of the common variants underlying disease today have increased within the last 5,000 years as a result of selection (Table 1). These variants may have exerted significant phenotypic effects in the past and so are more likely to do so again today under changed environmental circumstances. Against this is set what should perhaps be seen as the 'default' multilocus/multiallele hypothesis. Historically, we have always underestimated the number of loci and alleles in the population that influence disease traits. Medical genetics has largely confined itself to the clinical extremes (Figure 1b), where the number of loci capable of exerting such large effects is small - those regulating key, rate-limiting pathways, for example. Even here, Mendelian diversity was grossly underestimated. What of those phenotypes nearer the centre of the distribution? How many genes will contribute to these traits?
Experimental organisms provide an unreliable measure of locus diversity since they generally contain only a fraction of the total locus variability found in their wild populations. Simple, selectively neutral traits, such as bristle number in Drosophila melanogaster, tend to be influenced by a large number of loci (estimated at 22-26), a few of which exert large (oligogenic) effects while the majority exert small (polygenic) effects [24]. The current debate relates less to the relative frequency of large versus small effects but more to the absolute numbers of variant loci and the diversity and frequency of their alleles. Compared with simple traits like bristle number, the greater physiological and genetic complexity of coronary artery disease or diabetes in a more diverse (at least for young alleles) and outbred human population would seem to be self-evident. Where does this leave progress towards a personalized medicine?
The prospects for identifying and predicting individualized pharmacogenetic responses would seem to be more favorable, since the response to a small molecule and its metabolites may be more predictable, either on the basis of expression profiles or a knowledge of structure-function relationships. SNP variability within target genes can be readily sought and the haplotypes used to screen even for low-frequency variants in non-responders or other subgroups. Whether this will lead to lower drug-development costs and significantly increased efficacy is less clear, since greater knowledge of disease mechanisms is also required. Peter Goodfellow (GlaxoSmithKline, UK) pointed out at HGM2001 the less than two-fold increase in new drug targets identified in the past 20 years. New therapeutic possibilities now reflect the more than 10-fold increase in drug targets arising from the Human Genome Project: from around 400 in 1995 to between 4,000 and 40,000 today.
The need to reduce adverse effects is no longer perceived as a local problem for the pharmaceutical industry, since in 1994 they were found to be the fourth major cause of death in USA [25]. In the pursuit of personalized medicine, Goodfellow cited the example of genetic unresponsiveness to HMG CoA reductase inhibitors, the statins, which have proven efficacy in heart disease and stroke. One person in seven with coronary artery disease will not respond to statins because of a readily identifiable cholesteryl ester transfer protein (CETP) genotype [26]. Discovering the interaction with CETP was the easy part, however: the hard part was finding an effective therapy in the first place. It is a salutary thought that the statins were largely developed on the back of studies that elucidated key steps and pathways in cholesterol metabolism in familial hypercholesterolaemia, an early-onset and familial form of premature coronary artery disease. Are the major insights therefore going to come not from whole-genome association studies but from affected sib-pair and family linkage studies, which are robust to allelic heterogeneity and can exploit association methods for fine mapping. This strategy had sufficient power to detect the role of the NOD2 locus (IBD1) in Crohn's disease, despite multiple disease alleles, the most common of which has a prevalence of only 4% and a penetrance as low as 0.1-1.4% [27,28,29].
The excitement is now tangible amongst researchers, and the race is on to identify new regulatory steps and disease pathways. The prizes may come fastest to those who apply the central dogma of human gene mapping - ascertain the study population so as to maximize detectance and familial correlation. The technology is impressive, but it is the underlying biology that will determine who succeeds or fails.
References
- Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001. [DOI] [PubMed]
- Hartl DL, Clark AG. Principles of Population Genetics Sunderland: Sinauer; 1997.
- Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992;9:552–569. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- Hartl DL, Campbell RB. Allelic multiplicity in simple Mendelian disorders. Am J Hum Genet. 1982;34:866–873. [PMC free article] [PubMed] [Google Scholar]
- Thompson EA, Neel JV. Private polymorphisms: how many? How old? How useful for genetic taxonomies? Mol Phylogenet Evol. 1996;5:220–231. doi: 10.1006/mpev.1996.0015. [DOI] [PubMed] [Google Scholar]
- Lander ES. The new genomics: global views of biology. Science. 1996;274:536–539. doi: 10.1126/science.274.5287.536. [DOI] [PubMed] [Google Scholar]
- Weiss KM, Terwilliger JD. How many diseases does it take to map a gene with SNPs? Nat Genet. 2000;26:151–157. doi: 10.1038/79866. [DOI] [PubMed] [Google Scholar]
- King RA, Rotter JI, Motulsky AG, (eds.) The Genetic Basis of Common Diseases New York: Oxford University Press; 1992.
- Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994;330:1041–1046. doi: 10.1056/NEJM199404143301503. [DOI] [PubMed] [Google Scholar]
- Wright S. Evolution and the Genetics of Populations Volume 1, Genetic and Biometric Foundations Chicago: University of Chicago Press: 1968.
- Morton NE. Significance levels in complex inheritance. Am J Hum Genet. 1998;62:690–697. doi: 10.1086/301741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healey CS, Dunning AM, Teare MD, Chase D, Parker L, Burn J, Chang-Claude J, Mannermaa A, Kataja V, Huntsman DG, et al. A common variant in BRCA2 is associated with both breast cancer risk and prenatal viability. Nat Genet. 2000;26:362–364. doi: 10.1038/81691. [DOI] [PubMed] [Google Scholar]
- Slatkin M. Allele age and a test for selection on rare alleles. Philos Trans R Soc Lond B Biol Sci. 2000;355:1663–1668. doi: 10.1098/rstb.2000.0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC. Pleiotropy, natural selection and the evolution of senescence. Evolution. 1957;11:398–411. [Google Scholar]
- Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Chakravarti A. Population genetics - making sense out of sequence. Nat Genet. 1999;21:56–60. doi: 10.1038/4482. [DOI] [PubMed] [Google Scholar]
- Collins FS, Guyer MS, Chakravarti A. Variations on a theme: cataloging human DNA sequence variation. Science. 1997;278:1580–1581. doi: 10.1126/science.278.5343.1580. [DOI] [PubMed] [Google Scholar]
- Risch NJ. Searching for genetic determinants in the new millennium. Nature. 2000;405:847–856. doi: 10.1038/35015718. [DOI] [PubMed] [Google Scholar]
- Wright AF, Carothers AD, Pirastu M. Population choice in mapping genes for complex diseases. Nat Genet. 1999;23:397–404. doi: 10.1038/70501. [DOI] [PubMed] [Google Scholar]
- Terwilliger JD, Weiss KM. Linkage disequilibrium mapping of complex disease: fantasy or reality? Curr Opin Biotechnol. 1998;9:578–594. doi: 10.1016/s0958-1669(98)80135-3. [DOI] [PubMed] [Google Scholar]
- The International SNP Map Working Group. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, Lander ES. Linkage disequilibrium in the human genome. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- Mackay TF. Quantitative trait loci in Drosophila. Nat Rev Genet. 2001;2:11–20. doi: 10.1038/35047544. [DOI] [PubMed] [Google Scholar]
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998;279:1200–1205. doi: 10.1001/jama.279.15.1200. [DOI] [PubMed] [Google Scholar]
- Kuivenhoven JA, Jukema JW, Zwinderman AH, de Knijff P, McPherson R, Bruschke AV, Lie KI, Kastelein JJ. The role of a common variant of the cholesteryl ester transfer protein gene in the progression of coronary atherosclerosis. The Regression Growth Evaluation Statin Study Group. N Engl J Med. 1998;338:86–93. doi: 10.1056/NEJM199801083380203. [DOI] [PubMed] [Google Scholar]
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard J-P, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease and chromosome 16. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- Hampe J, Cuthbert A, Croucher PJP, Mirza MM, Mascheretti S, Fisher S, Frenzel H, King K, Hasselmeyer A, MacPherson AJS, et al. Association between insertion mutation in NOD2 gene and Crohn's disease in German and British populations. Lancet. 2001;357:1925–28. doi: 10.1016/S0140-6736(00)05063-7. [DOI] [PubMed] [Google Scholar]
- Online Mendelian Inheritance in Man http://www.ncbi.nlm.nih.gov/Omim/
- RetNet - Retinal Information Network http://www.sph.uth.tmc.edu/Retnet/