

Abstract
Taking each coding sequence from the human genome in turn and identifying the subcellular localization of the corresponding protein would be a significant contribution to understanding the function of each of these genes and to deciphering functional networks. This article highlights current approaches aimed at achieving this goal.
The spatial and temporal regulation of biochemical reactions in eukaryotic cells is achieved by a high degree of compartmentalization. Each protein is part of a functional biochemical network and all proteins within a particular network are at least once in their lifetime localized close to each other, within (or at) a particular organelle or compartment. This facilitates interactions and yet allows the segregation of different networks. Exchange of information between different organelles, and of proteins between networks, is essential for the proper function of the cell as an entity and is achieved by the active transport of material.
One of the best examples of such an assembly of networks is the secretory pathway. Secretory proteins move sequentially through the distinct membrane-bounded organelles of this pathway, receiving at each step specific enzymatic modifications necessary for their quality control and proper function. The communication and specific transfer of material between membrane organelles is mediated by distinct small membrane-bounded transport carrier vesicles containing a myriad of regulatory proteins. A key feature of any protein functionally involved in the secretory pathway is its permanent or transient localization to one of the appropriate transport carriers or organelles. Extending this concept to the whole cell, the determination of the subcellular localization of a novel protein is one of the essential steps in resolving its function. This includes imaging not only the protein's steady-state distribution but also the changes in localization that can occur in response to environmental conditions, during specific stages of the cell cycle or of cell differentiation. Indeed, changes in localization can also be caused by the breakdown of remote but functionally related organelles and/or cellular structures, such as Golgi fragmentation resulting from microtubule reorganization (see for example Figure 1c,d).
Figure 1.
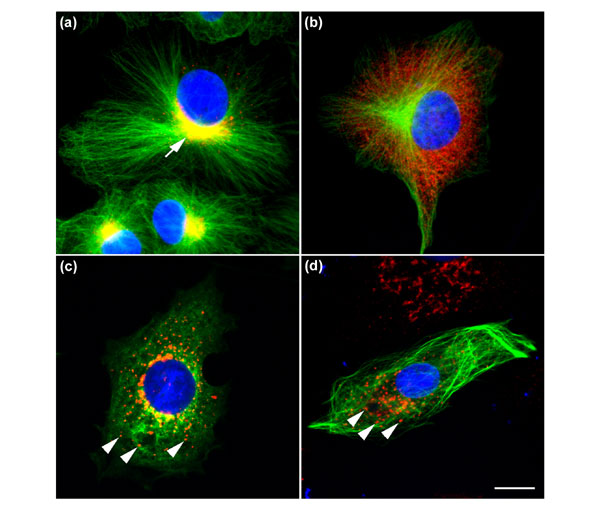
Highly dynamic and interdependent organization of distinct subcellular structures. The Vero cells in (a) show the normal arrangement of microtubules (green) radiating from the microtubule-organizing center. The Golgi complex (indicated by the arrow), a membrane-bounded organelle through which all secretory proteins pass en route to the cell surface, is stained with antibodies against the coat protein complex COPI (red; where red and green staining coincide they appear yellow). The Golgi complex resides as a tight structure at a central perinuclear location. The cell in (b) has been treated with the drug brefeldin A, which causes rapid removal of the COPI coat from Golgi membranes into a cytoplasmic pool, followed by disassembly of the Golgi apparatus. The microtubule network remains unaffected by this treatment, however. (c) Treatment of a cell with the drug nocodazole causes disassembly of the microtubules into their respective cytoplasmic tubulin monomers. This breakdown of the microtubule network, a key component of cell architecture, also results in the breakdown of the Golgi complex into distinct fragments spread throughout the cell (as indicated by the arrowheads). The cell in (d) has been transfected with a GFP-tagged novel cDNA, which when expressed localizes along the entire microtubule network (green). But as the expression level of this protein increases, it interferes with the microtubule network with the concomitant result that the Golgi is fragmented in a similar manner to that observed in (c) (as indicated by the arrowheads). This phenotypic effect illustrates the dynamic interdependency of organelles exemplified by Golgi morphology and the microtubule network. The nuclei of all the cells have also been stained with the DNA-chelating agent diamino phenylindole (DAPI; blue), showing that this organelle appears not to be affected by the various treatments. The bar indicates 10 μm.
Although studies to follow these dynamic events have been a difficult task in the past, the availability of green fluorescent protein (GFP) and its spectral variants has now facilitated localization experiments particularly aimed at observing protein dynamics in living cells [1,2,3,4]. The cDNA encoding GFP was cloned several years ago and encodes a 27 kDa protein that emits green fluorescence when excited with blue light, without the need for any co-factors. Thus, any cDNA can be fused with the coding sequence of GFP, and the localization of the expressed GFP fusion can be followed in living cells. This unique feature of GFP has led to the development of a number of 'localization screening assays', which can be performed in a systematic 'high-throughput' manner as typically required for large-scale post-genome projects.
GFP-based techniques
Most GFP-based techniques fuse either fragments of genomic libraries or individual clones from cDNA libraries to the coding sequence of GFP, then express the fusions in cells or tissues and determine their subcellular localizations by microscopic inspection. Subsequently, the respective cDNAs or genes are rescued from the cells or tissues, cloned and sequenced. Such strategies have already been conducted on a genome-wide scale in yeast [5,6] and have identified the localization of so-far uncharacterized proteins, or fragments thereof. The GFP-tagged proteins can be immediately followed in living cells by time-lapse microscopy to determine their cellular dynamics, which adds a further level of information to such screens. At least 50% of the cDNAs isolated in this way are already known and well characterized, however [6,7,8,9]. Furthermore, the same cDNA clones are isolated several-fold in one screen, as the primary criterion for selection is simply localization [5]. These aspects are major disadvantages of such morphological screens and make them inefficient. For example, in an attempt to isolate novel nuclear-envelope proteins, 550,000 starting cDNA clones were required to identify 27 clones localizing to this compartment, of which only two proved to be novel [9].
When tagging cDNA libraries with GFP, consideration must also be given to the effect of the reporter on masking targeting signals contained within the expressed proteins. Amino-terminal fusions of GFP to target proteins potentially block signal sequences associated with import into mitochondria or the endoplasmic reticulum, for example. Conversely, when using either random DNA fragments or even non-full-length cDNAs (of which there are significant numbers in cDNA libraries), the expressed proteins may appear to clearly localize, but the recorded localization may be aberrant, resulting simply from exposing a peptide sequence normally hidden in the full-length protein. This was clearly demonstrated in the 'motif-trap method' by which a large number of cryptic mitochondrial targeting signals were isolated - many corresponding to sequences derived from non-coding genomic DNA [10]. In an attempt to circumvent the problem of hidden amino-terminal targeting sequences, in one study [11] cDNAs were cloned from a library containing cDNA fragments upstream of GFP, and a retrovirus-mediated expression system was used to determine the cellular localizations of the encoded fusion products. Although this expression system is highly effective, the authors themselves concede that none of their cDNAs was full-length, and that the interpretation of the localization results is dependent upon the targeting sequences being present in the partial cDNA [11]. Thus, strategies using GFP tagging of whole cDNA or genomic libraries generate significant amounts of redundant or inaccurate data, all of which are time-consuming, and therefore expensive, to eliminate.
Methods are therefore now being devised to focus more rapidly specifically on those localizations of interest. For example, one possibility is first to isolate GFP-positive cells from the non-fluorescent cells using fluorescence-activated cell sorting (FACS), which is able to sort thousands of GFP-expressing cells within minutes into individual wells of multiwell plates, and subsequently to clone them. In this way only GFP-expressing cells have to be examined microscopically, which increases the speed of analysis. An improved variant of such an approach was described recently [7] with the aim of identifying proteins localizing to the nucleus. Pichon and co-workers first mildly permeabilized intact cells with detergent, in order to remove cytosolic but not nuclear GFP-fusion proteins, and then sorted the remaining GFP-positive cells using FACS. This resulted in a 70-fold enrichment of cells expressing GFP-fusion proteins in the nucleus compared to cultures that had not been treated and sorted.
Clearly, tagging sequenced full-length cDNAs on an individual basis retains the advantages but overcomes many drawbacks of the approaches described above [12,13]. One advantage is the availability of a large clone resource from genome projects, the cDNA sequences of which can be prescreened for already-known genes or species variants, so that only novel cDNAs need to be GFP-tagged and screened. In addition, different versions of full-length GFP fusions - tagged at either the amino or the carboxyl terminus - can be generated and compared, helping to circumvent the risk of masking targeting sequences. Indeed, as expected, often only one version of a GFP-tagged protein shows proper subcellular localization [13]. Although the tagging of full-length cDNAs is a relatively low-throughput process and is reliant upon the identification of novel cDNAs by other means such as systematic sequencing [14], it has a further clear advantage that no additional cloning is required once an interesting localization has been identified. Tagging of full-length cDNAs suffered until recently from the problem that conventional restriction-enzyme-based cloning had to be used, which is tedious and virtually impossible to do for any large set of molecules [12]. To overcome this problem, we have recently devised a method that uses a recombination-based cloning system to systematically tag with GFP open reading frames of full-length cDNAs that have been identified and sequenced by large-scale genome projects [13,14]. The whole procedure is amenable to automation, and other characterization studies (for example, mutagenesis, protein dynamics and identification of interacting partners) can follow the localization screen immediately without further generation of new reagents or lengthy cloning procedures to identify the full-length cDNAs.
In silico methods
Several bioinformatic tools have been developed with the aim of predicting protein localization on the basis of sequence features within the respective gene or cDNA. One of the early methods, PSORT [15,16], detects in sequences the signals required for sorting proteins to particular subcellular compartments. Although PSORT is a well-accessed program and is widely applicable to different organisms, its overall accuracy - at best, for yeast - is still in the region of 50%. Others have used phylogenetic profiles [17], more careful use of annotated databases such as the Meta-A evaluation of SWISS-PROT entries [18], or expression levels [19] as means to tap into the knowledge that can be gained from determining localization. More profitable, perhaps, is to concentrate on specific organelles and the sequence motifs that direct proteins to them. For example, defined signals for directing proteins to mitochondria, the secretory pathway or chloroplasts are now well characterized, and the success rate of prediction can be as high as 90%. Even the correct prediction of cleavage sites for the signal sequences is possible with more than 50% success rate [20]. Certainly the speed and cost of these methods is currently unsurpassed. As a result of more genome sequencing projects being completed, more data for comparisons are available, and so the quality of results using screening algorithms based on sequence homologies rises steadily. More databases, which integrate all this information, are therefore being implemented [21,22]. Experimental data gathered for individual genes, and ideally proteins, also funnels into such databases information that is then accessible to in silico tools. For many novel proteins, however, these tools remain at present suggestive at best, and for these molecules there is still no alternative to actual experimental verification.
In summary, a protein's localization and its subcellular dynamics are important parameters to know when trying to determine its function. With the availability of GFP and its variants, new in vivo approaches have been made possible, and these have already identified novel proteins in various desired locations. In due course, these techniques will undoubtedly be applied and perfected on a genome-wide scale. Furthermore, the reagents generated during the course of such projects (such as GFP-tagged proteins) are extremely useful for subsequent microscope-based functional studies with different foci - for example, the analysis of a protein's posttranslational modifications or the dynamics of interactions with binding partners in living cells [4]. This will ultimately allow us to identify functional networks of proteins in a morphological context and will greatly contribute to our understanding of whole-cell function.
Acknowledgments
Acknowledgements
We thank Jan Ellenberg for useful comments on the manuscript. J.C.S. was in part supported by an EMBO Long Term fellowship. The Wiemann and Pepperkok laboratories are supported by grants from the BMBF numbers 01KW9987 (German cDNA Consortium), 01KW0012 (to S.W.) and 01KW0013 (to R.P).
References
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- Ellenberg J, Lippincott-Schwartz J, Presley JF. Dual-color imaging with GFP variants. Trends Cell Biol. 1999;9:52–56. doi: 10.1016/s0962-8924(98)01420-2. [DOI] [PubMed] [Google Scholar]
- Bastiaens PI, Pepperkok R. Observing proteins in their natural habitat: the living cell. Trends Biochem Sci. 2000;25:631–637. doi: 10.1016/s0968-0004(00)01714-x. [DOI] [PubMed] [Google Scholar]
- Ding DQ, Tomita Y, Yamamoto A, Chikashige Y, Haraguchi T, Hiraoka Y. Large-scale screening of intracellular protein localization in living fission yeast cells by the use of a GFP-fusion genomic DNA library. Genes Cells. 2000;5:169–190. doi: 10.1046/j.1365-2443.2000.00317.x. [DOI] [PubMed] [Google Scholar]
- Sawin KE, Nurse P. Identification of fission yeast nuclear markers using random polypeptide fusions with green fluorescent protein. Proc Natl Acad Sci USA. 1996;93:15146–15151. doi: 10.1073/pnas.93.26.15146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichon B, Mercan D, Pouillon V, Christophe-Hobertus C, Christophe D. A method for the large-scale cloning of nuclear proteins and nuclear targeting sequences on a functional basis. Analyt Biochem. 2000;284:231–239. doi: 10.1006/abio.2000.4674. [DOI] [PubMed] [Google Scholar]
- Merkulov GV, Boeke JD. Libraries of green fluorescent protein fusions generated by transposition in vitro. Gene. 1998;222:213–222. doi: 10.1016/S0378-1119(98)00503-4. [DOI] [PubMed] [Google Scholar]
- Rolls MM, Stein PA, Taylor SS, Ha E, McKeon F, Rapoport TA. A visual screen of a GFP-fusion library identifies a new type of nuclear envelope membrane protein. J Cell Biol. 1999;146:29–44. doi: 10.1083/jcb.146.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejarano LA, Gonzalez C. Motif trap: a rapid method to clone motifs that can target proteins to defined subcellular localizations. J Cell Sci. 1999;112:4207–4211. doi: 10.1242/jcs.112.23.4207. [DOI] [PubMed] [Google Scholar]
- Misawa K, Nosaka T, Morita S, Kaneko A, Nakahata T, Asano S, Kitamura T. A method to identify cDNAs based on localization of green fluorescent protein fusion products. Proc Natl Acad Sci USA. 2000;97:3062–3066. doi: 10.1073/pnas.060489597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoja MR, Wahlestedt C, Hoog C. A visual intracellular classification strategy for uncharacterized human proteins. Exp Cell Res. 2000;259:239–246. doi: 10.1006/excr.2000.4948. [DOI] [PubMed] [Google Scholar]
- Simpson JC, Wellenreuther R, Poustka A, Pepperkok R, Wiemann S. Systematic subcellular localization of novel proteins identified by large-scale cDNA sequencing. EMBO Rep. 2000;1:287–292. doi: 10.1093/embo-reports/kvd058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann S, Weil B, Wellenreuther R, Gassenhuber J, Glassl S, Ansorge W, Bocher M, Blocker H, Bauersachs S, Blum H, et al. Toward a catalog of human genes and proteins: sequencing and analysis of 500 novel complete protein coding human cDNAs. Genome Res. 2001;11:422–435. doi: 10.1101/gr.154701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai K, Horton P. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci. 1999;24:34–36. doi: 10.1016/s0968-0004(98)01336-x. [DOI] [PubMed] [Google Scholar]
- PSORT http://psort.ims.u-tokyo.ac.jp
- Marcotte EM, Xenarios I, van Der Bliek AM, Eisenberg D. Localizing proteins in the cell from their phylogenetic profiles. Proc Natl Acad Sci USA. 2000;97:12115–11220. doi: 10.1073/pnas.220399497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber F, Bork P. Wanted: subcellular localization of proteins based on sequence. Trends Cell Biol. 1998;8:169–170. doi: 10.1016/s0962-8924(98)01226-4. [DOI] [PubMed] [Google Scholar]
- Drawid A, Jansen R, Gerstein M. Genome-wide analysis relating expression level with protein subcellular localization. Trends Genet. 2000;16:426–430. doi: 10.1016/s0168-9525(00)02108-9. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- Yudate HT, Suwa M, Irie R, Matsui H, Nishikawa T, Nakamura Y, Yamaguchi D, Peng ZZ, Yamamoto T, Nagai K, et al. HUNT: launch of a full-length cDNA database from the Helix Research Institute. Nucleic Acids Res. 2001;29:185–188. doi: 10.1093/nar/29.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUNT: Human full length cDNA database http://www.hri.co.jp/HUNT/