

Abstract
Many cells, including neuronal and glial progenitor cells, stem cells and microglial cells, have the capacity to move through the extracellular spaces of the developing and mature brain. This is particularly pronounced in astrocyte-derived tumors, gliomas, which diffusely infiltrate the normal brain. Although a significant body of literature exists regarding signals that are involved in the guidance of cells and their processes, little attention has been paid to cell-shape and cell-volume changes of migratory cells. However, extracellular spaces in the brain are very narrow and represent a major obstacle that requires cells to dynamically regulate their volume. Recent studies in glioma cells show that this involves the secretion of Cl− and K+ with water. Pharmacological inhibition of Cl− channels impairs their ability to migrate and limits tumor progression in experimental tumor models. One Cl−-channel inhibitor, chlorotoxin, is currently in Phase II clinical trials to treat malignant glioma. This article reviews our current knowledge of cell-volume changes and the role of ion channels during the migration of glioma cells. It also discusses evidence that supports the importance of channel-mediated cell-volume changes in the migration of immature neurons and progenitor cells during development. New unpublished data is presented, which demonstrates that Cl− and K+ channels involved in cell shrinkage localize to lipid-raft domains on the invadipodia of glioma cells and that their presence might be regulated by trafficking of these proteins in and out of lipid rafts.
Keywords: Cell migration, chloride, potassium, cell volume, chlorotoxin
INTRODUCTION
Astrocyte-derived tumors, which are referred to commonly as gliomas, are among the most deadly cancers. They display an unusual ability to disseminate in the brain and appear to do so by active migration of cells rather than passive, hematogenous spread as is typical for metastasizing cancers of the body. The migration of glioma cells is reminiscent of that in immature brain cells during development and, for this reason, glioma cells are used frequently as model to study cell migration in general. From a clinical point-of-view, the diffuse infiltration of these cancer cells into the healthy brain parenchyma makes complete surgical resection nearly impossible and focal radiation therapy difficult. In a previous series of studies (Soroceanu et al., 1999; Ransom et al., 2001; Ransom and Sontheimer, 2001) we examined glioma-cell migration, with particular attention to the changes in cell shape and volume that these cells undergo as they navigate through the narrow, tortuous extracellular spaces in brain. These studies reveal that glioma cells acquire an elongated, wedge-like shape (Soroceanu et al., 1999) and appear to shrink their cell volume to fit into the narrow spaces available. To accomplish a reduction in cell volume, glioma cells must reduce some of their cytoplasm, which appears to require the coordinated secretion of K+ and Cl− ions, and water. This process is illustrated in Fig. 1. Typically, movement of water occurs through water channels or aquaporins, whereas the movement of ions can occur either via ion channels or ion transporters. In this paper we review current understanding of the main pathways that contribute to glioma-cell shrinkage as they invade, and discuss similarities with other migratory cells. Moreover, we present new, unpublished data that indicates that ion channels involved in glioma migration are localized to specialized lipid domains on invadipodia, and that the number of functional channels in these invasive domains is regulated by protein trafficking in and out of these lipid domains.
Fig. 1. K+ and Cl− efflux aids glioma-cell shrinkage.
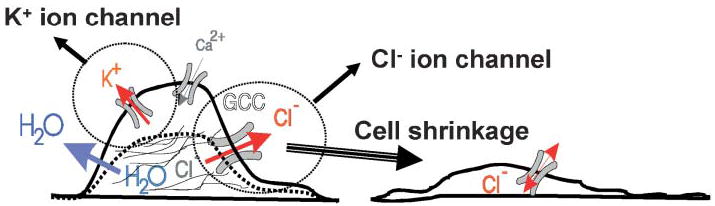
It is thought that cells must shrink as they invade the brain. To reduce their cytosplasmic content, cells release Cl− and K+ through ion channels, and water follows passively through water channels or aquaporins. For this mechanism to function, cells must accumulate Cl− and K+ above their respective electrochemical gradients. This is accomplished by the combined activity of the NKCC Cl− transporter and the Na+–K+ ATPase for K+ ions. Modified, with permission, from Soroceanu et al. (1999)
OBJECTIVE
This article, by mingling new data with previously published work, attempts to develop the hypothesis that glioma cells utilize a defined set of Cl− and K+ ion channels to adjust their cell volume as they invade the normal brain. Changes in volume are essential to allow cell migration and, hence, these ion channels represent an opportune pharmacological target.
METHODS
Many of the methodologies used in this manuscript are covered extensively in the manuscripts from which data is reviewed and from which figures are taken. Therefore, only important additional methodological details are given here.
Cell lines
Human glioma cell lines D54-MG (obtained from Dr. D.D. Bigner, Duke University) and CCF-STTG-1 (ATCC) were maintained in DMEM-F12 supplemented with 2 mM L-glutamine (Life Technologies) and 7% heat-inactivated fetal bovine serum (FBS) (Hyclone). Cortical astrocytes were isolated from Sprague-Dawley rat pups at postnatal day 0 and cultured as described (Ye and Sontheimer, 1996).
Glioma invasion assays
The three assays used were matrigel invasion, spheroid confrontation and tissue-slice invasion. All three are described in detail in Soroceanu et al. (1999) and Ransom et al. (2001). For tissue-slice-invasion assays we generated a stably transfected daughter cell line from D54-MG cells that stably expresses EGFP. This allows us to visualize their migration into acute and organotypic brain slices by confocal microscopy. The microscope used was an Olympus Fluoview with a 40× water-immersion 0.8NA lens. Cells were maintained in a Leiden chamber (World Precision Instruments) to maintain temperature at 37°C and CO2:O2 at 95%:5% throughout the experiment. Images were acquired with the Argon 488 nm laser.
Antibodies and reagents
Polyclonal antibodies that recognize ClC-3 were purchased from Alpha Diagnostic International. Antibodies directed against the BK K+ channel were purchased from Chemicon International. Rabbit polyclonal antibodies directed to caveolin-1 were purchased from Santa Cruz Biotechnology. Alexa Fluor 546 goat anti-rabbit conjugated secondary antibodies (Molecular Probes) were used for immunofluorescence at the manufacturer’s recommended dilution. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibodies purchased from BioRad Laboratories were used for chemiluminescent Western blot analysis. The cholesterol-chelating agent filipin and FITC-labeled cholera toxin B subunit (FITC-CTxB), used to specifically label lipid rafts, were purchased from Sigma-Aldrich.
Synthesis and purification of recombinant chorotoxin
As described in Deshane et al. 2003.
Overlay assays (Far Westerns)
Briefly, proteins processed from membrane fractions, cytosolic fractions and total cell lysates were analyzed by Far Westerns using protocols described (Fenster et al., 2000). Membranes were probed with 300–500 nM His-Cltx followed by detection of the bound proteins using an anti-His mAb (Clonetech, 1:5000).
Biotinylation of cell-surface proteins
Cell-surface proteins were biotinylated following published procedures (Ye et al., 1999). Cells at 80% confluency in 100 mm dishes were incubated with 3.8 ml of a 1.5 mg ml−1 sulfo-LC-NHS biotin (Pierce) in PBS/Ca/Mg solution for 30 minutes at 4°C with gentle shaking. Following quenching of biotinylation in PBS/Ca/Mg plus 100 mM glycine, and further incubation for 30 minutes at 4°C with gentle shaking, the glycine solution was aspirated and cells rinsed twice with PBS/Ca/Mg. Cells were then lysed and biotinylated cell-surface fractions and intracellular fractions separated using agarose-immobilized streptavidin (Pierce) and analyzed by immunoblotting as described (Ye et al., 1999).
Coupling recombinant Cltx to Actigel-ALD
Recombinant chlorotoxin (Cltx) was coupled to Actigel-ALD (Sterogene) and stored following manufacturer’s recommendations. His-Cltx was added to Actigel-ALD (0.5 mg ml−1 of resin) followed by ALD-coupling solution (1 M NaCNBH3) to a final concentration of 0.1 M (0.2 ml ml−1 resin).
Affinity purification
Cell debris was removed by centrifugation at 2000×g for 5 minutes at 4°C and the supernatant was collected and recentrifuged at 100 000×g in Beckman Instruments T70.1 rotor for 60 minutes at 4°C. Pellets, representing the total cell membrane fraction, were resuspended in homogenization buffer (HB) supplemented with protease inhibitors containing 1% SDS followed by addition of 7-fold excess volume of 1% Triton X-100. The samples were then heated to 48°C for 5 minutes. This lysate was then pre-cleared with unconjugated Actigel-ALD beads with end-over-end rotation (100 μl bead slurry per 1 ml lysate) for 4 hours at 4°C. Following a brief centrifugation at 100×g, the supernatant was removed and incubated with the His-Cltxconjugated Actigel-ALD beads (250 μl per ml of pre-cleared lysate) for 4 hours at 4°C or overnight. The beads were then centrifuged for 2 minutes at 100×g and unbound material removed. The beads were washed extensively with HB (supplemented additionally with 0.1% NP-40 and 0.01% Tween). The bound proteins were then eluted by boiling with Laemmli SDS-sample buffer (62.5 mM Tris-HCl at pH 6.8, 10% glycerol, 2% SDS, 0.1% bromophenol blue and 600 mM 2-mercaptoethanol) for 5 minutes. Samples were separated on denaturing 8, 10 or 4–15% gradient gels by SDS-PAGE and further analyzed by Western blots and overlay assays.
Immunofluorescence studies of glioma cells
Glioma cells were plated at 20% confluency onto 12-mm diameter coverslips in appropriate tissue-culture media. After 48 hour incubation at 37°C, media was aspirated and the cells gently rinsed with PBS to remove excess serum and media components. The cells were then incubated for 10 minutes at 4°C with 5 μg ml−1 FITC-CTxB. Cells were then rinsed twice in cold PBS and fixed for 10 minutes at room temperature in PBS containing 4% paraformaldehyde. Cells were rinsed, after fixation, with PBS (3 × 5 minutes). After overnight incubation at 4°C with blocking buffer (PBS containing 3% goat serum, 0.1% azide and 0.3% Triton X-100), recommended dilutions of primary antibodies (αClC-3, 1:500; α-BK, 1:500) in blocking buffer were added to the cells and incubated for 1 hour at room temperature. Cells were then rinsed (3 × 10 minutes) with PBS containing blocker and then incubated with Alexa Fluor 546 (1:750) secondary antibodies in blocker for 1 hour at room temperature. Following washes (2 × 10 minutes) in PBS, DAPI (10–4 mg ml−1) in PBS was added to the cells and incubated for 5–10 minutes. Coverslips were mounted using gelmount following 2 × 10 minute washes in PBS. Immunofluorescence was carried out using a Zeiss epifluorescence microscope (63× oil) and images were captured using a digital camera.
Lipid-raft isolation
Lipid-raft isolation was performed using a protocol adapted from Rujoi et al. (2003). Briefly, glioma cells were grown to >85% confluence on 10-cm Petri dishes. Cells were placed on ice and rinsed with cold TNE buffer (15 mM Tris-HCL, 150 mM NaCl, 5 mM EDTA, pH 7.6) before scraping off the plates in 0.5 mL cold TNE buffer. The cells were lysed using a glass homogenizer with nylon pestle. Lysates were then centrifuged at 5000 rpm for 1 hour at 4°C. Supernatant was then carefully removed and saved as the water-soluble fraction. The pellet was resuspended in 500 μl TNE buffer + 1% Trition X-100 and incubated at 4°C for 15 minutes. The lysate was then centrifuged at 5000 rpm for 30 minutes at 4°C. The supernatant was saved as the detergent-soluble fraction, whereas the detergent-insoluble pellet was resuspended in 500 μl of 40% Optiprep solution made in TNE buffer + 1% Triton. This 40% Optiprep solution, which contains insoluble material, was placed at the bottom of an ultracentrifuge tube. Carefully layered on top of the 40% solution was 3.5 ml of a 30% Optiprep solution in the same TNE + Triton buffer. Finally, a 500 μl layer of 5% Optiprep was added to the top of the tube before spinning in an SW-40 rotor at 36 000×g for 16 hours at 4°C in a Beckman L8-M Ultracentrifuge. Fractions, 500 μl each, were taken carefully from the top to the bottom of the tube and samples were run on 4–20% SDS-PAGE gels to assay for proteins of interest using Western blots. Successful isolation was indicated by the concentrated presence of caveolin-1 in the buoyant fractions, predominantly fraction 2, at the 5%:30% density interface.
Internalization assay
Glioma cells were plated at a density of 5×105 cells in 10-cm diameter tissue-culture dishes in serum-containing media. After overnight incubation at 37°C, cells were washed and then incubated with serum-free media containing 300 nM His-Cltx, irrelevant His-protein or 50 μM 1–10 phenanthroline for 30 minutes at 37°C to allow sufficient internalization of His-Cltx. Cells were then washed and cell-surface biotinylation carried out as described above at 10 minutes, 30 minutes and 24 hours post-incubation with Cltx and other reagents. Western blots were used to quantitfy Cltx in biotinylated cell-surface fractions and intracellular fractions. Alternatively, cells were treated with 5 μg ml−1 Filipin for 15 minutes at 4°C to flatten the caveolae before treatment with Cltx. Cells were then processed for cell-surface biotinylation as described above to analyze the level and route of internalization of His-Cltx and associated channel proteins.
RESULTS
Do glioma cells shrink as they invade?
The fundamental hypothesis that glioma cells shrink as they invade is supported primarily by a series of light microscopic and electron microscopic images that capture glioma cells in the process of cell invasion. The hypothesized efflux of salt (i.e. K+ and Cl−) in concert with water is depicted schematically in Fig. 1, and illustrates the requirement for salt efflux as cells shrink driving water out of the cell. Indeed, the characteristic elongation in cell shape is observed readily when human glioma cells are confronted with tissue slices of rat brain or spheroids of fetal rat brain tissue. Examples are illustrated in Fig. 2 at the light microscopic and EM level. In Fig. 2A glioma cells that express EGFP were placed on the surface of a brain slice and, 6 hours later, a series of images were captured to allow 3-D reproduction of entire cells. The representative cell assumes an elongated wedge or spindle-like shape, which is conducive for cell migration through narrow extracellular spaces in the brain (Fig. 2A). A similar wedge shape is apparent at the EM level for representative examples of two glioma cells invading a fetal rat-brain spheroid (Fig. 2B). Because these are static images, it is possible that the changes in cell shape might not requie a concomitant change in cell volume. Indeed, cells might simply translocate cytoplasm from the leading end of the cell to the trailing end. Until the process of invasion can be imaged quantitatively with a 3-D assessment of cell volume, it is impossible to answer unequivocally whether the volume of a cell changes as it invades. Nevertheless, below we provide a compelling set of data that supports the presumption that a flux of ions and cytoplasm across the membrane is required for glioma-cell invasion.
Fig. 2. Glioma cells shrink as they invade.
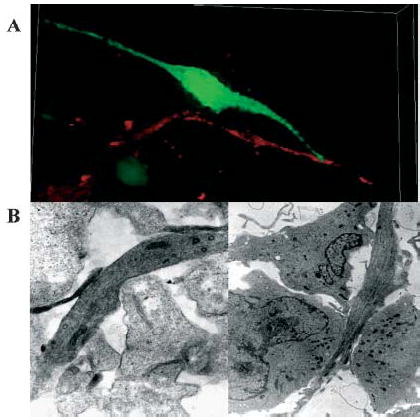
Images obtained by either light (A) or electron (B) microscopy. (A) To demonstrate the polarized, wedge-shape of invading glioma cells, which indicates cell shrinkage, we stably transfected D54-MG glioma cells with EGFP. These cells were placed on the surface of a 400-μm thick slice from rat brain and allowed to invade for 6 hours in a fully oxygenated chamber at 37°C. A series of confocal images was obtained (400 nm optical sections), which allows complete, 3-D reproduction of invading cells. Blood vessels are stained with CD31-Abs conjugated to phycoerythrin. (B) For electronmicroscopy of invading cells, D54-MG cells were grown in a spheroid and confronted with a spheroid of fetal rat brain cells. 20-nm sections are shown at 12 000× magnification. Images in (B) are reproduced, with permission, from Soroceanu et al. 1999.
Glioma cells have a high resting Cl−conductance that contributes to invasion via Cl− efflux
For glioma cells to shrink when they encounter tight extracellular spaces, it is necessary to secrete some of their cytoplasm. In all living systems, this occurs via secretion of salt, with water following passively. In glioma cells, secretion of cytoplasm appears to be driven by efflux of Cl− from the cell. The experimental findings that support this notion are as follows: (1) Glioma cells accumulate Cl− above their electrochemical gradient and, therefore, opening of Cl− channels leads to the efflux of Cl−; (2) either inhibition of Cl− efflux or replacement of Cl−with impermeant anions impairs cell invasion.
To backup the first argument experimentally, we obtained whole-cell recordings from several human glioma cell lines. To maintain intact, physiological ionic gradients, we used the perforated patch-clamp method in which the antibiotic amphoterecin was included in the patch pipette which then perforates the underlying membrane patch allowing electrical access to the cell without dialyzing the recorded cell. Glioma cells recorded this way had an average resting membrane potential of −16 mV (SD, 15 mV) (Ransom et al., 2001) and input resistance values of ~100 MΩ. When maintained in voltage-clamp, a negative holding current of ~200 pA was required to maintain the cells at −40 mV. Application of the Cl−-channel inhibitor NPPB (Fig. 3), but not the K+-channel inhibitor TEA, caused the negative holding current to disappear and the input resistance to increase dramatically. Similarly, the input resistance of glioma cells was elevated drastically when Cl− is replaced with either glutamate or gluconate, which are much less permeant anions (data not shown). These experiments indicate that the resting conductance for Cl− contributes significantly to the resting membrane potential of glioma cells.
Fig. 3. Glioma cells take advantage of a resting Cl− conductance as they invade.
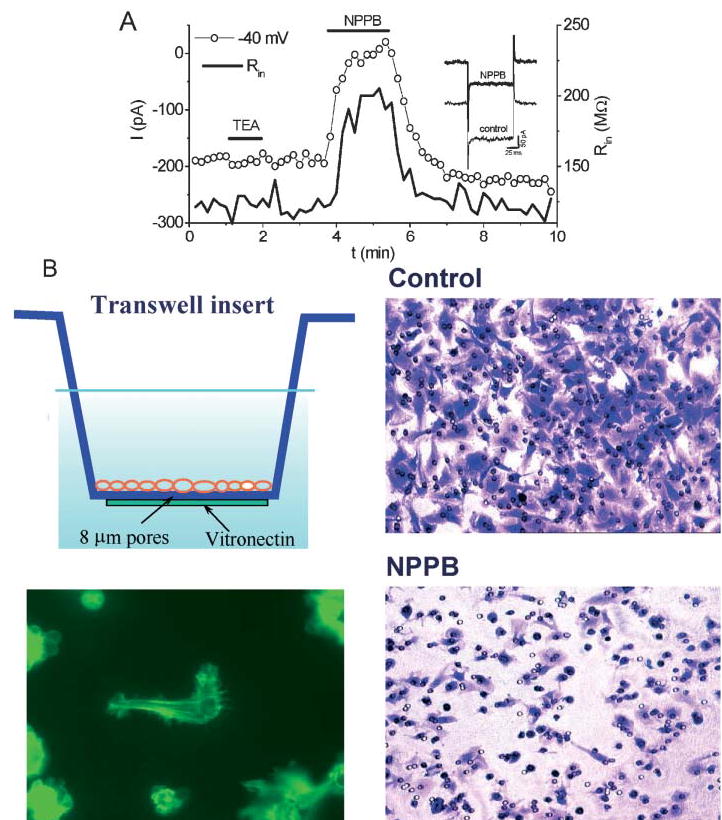
(A) To demonstrate the presence of a resting Cl− conductance, recordings were made from glioma cells using amphotericin-perforated, whole-cell patch-clamp recordings. A holding current of −200 pA maintained the cell at −40 mV (left axis). Cells had input resistances of ~100 MΩ (right axis). Application of the Cl−-channel inhibitor NPPB inhibited the holding current and increased cell resistance to ~200 MΩ, which is consistent with inhibition of a resting Cl− conductance. By contrast, TEA did not alter the input resistance or the holding current. (B) To show that a NPPB-sensitive Cl− conductance is required for successful migration across a spatial barrier, D54-MG glioma cells were plated on the upper surface of a Transwell insert with 8 μm pores and allowed to migrate for 4 hours towards vitronectin coated on the bottom of the filter insert (top left). Under control conditions, most cells migrated successfully, indicated by crystal violet staining of cells at the bottom (top right). In the presence of 30 μM NPPB only few cell migrated to the bottom of the filter (bottom right). Instead, most cells extend a process through the filter, but failed to move the entire cell through (bottom left). Modified, with permission, from Ransom et al. (2001).
The next important aspect pertains to the role that Cl−channels have in supporting cell invasion. To address this question, we confronted glioma cells with Transwell membrane barriers (Fig. 3B), and monitored their chemotactic migration across the barrier towards vitronectin the presence or absence of the Cl−-channel inhibitor NPPB, other channel blockers or after replacement of Cl− with impermeant anions. After 6 hours, fewer cells successfully migrated in the presence of NPPB (Fig. 3, right). An individual cell process is visualized as a cell penetrates the barrier (Fig. 3, bottom left). We obtained similar migration data when Cl− was replaced with either I− or Br−, which are more permeant than Cl− through glioma Cl− channels, and with gluconate and glutamate, which are poorly permeable (Ullrich and Sontheimer, 1996). These studies indicate that only ions with excellent permeability support efficient transwell migration of glioma cells (Soroceanu et al., 1999).
Glioma cells express Cl− channels sensitive to NPPB and Cltx
The lack of selective inhibitors for Cl− channels does not permit an unequivocal identification of the underlying Cl− channels by pharmacological means. For example, NPPB blocks several Cl− channels including volume-dependent anion channels, CFTR, ClC-2 and ClC-3 (d’Anglemont et al., 2003). Therefore, initially we used PCR, Western blots and immunohistochemistry to narrow the search for candidate genes. These studies indicate that the channel most likely to mediate the NPPB/Cltx sensitive Cl− currents in glioma cells is ClC-3 (Olsen et al., 2003). Representative immunogold labeling of ClC-3 channels is illustrated in Fig. 4A, showing clusters of channels intracellularly and on the cell surface. To show more directly that ClC-3 channels give rise to outwardly rectifying currents, we treated D54-MG glioma cells for 48 hours with antisense oligonucleotides that specifically suppress the expression of ClC-3 channels. This procedure selectively reduces channel expression, judged by Western blots (Fig. 4B) and biophysical recordings (Fig. 4C,D). The residual Cl− current in cells in which CLC-3 is eliminated is small and no longer sensitive to NPPB (data not shown). Based on these data, we are confident that outwardly rectifying, NPPB-sensitive Cl− channels that facilitates Cl− efflux from cells as they either invade or migrate through narrow spaces are most likely to be ClC-3 channels.
Fig. 4. Glioma cells express functional ClC-3 Cl− channels.
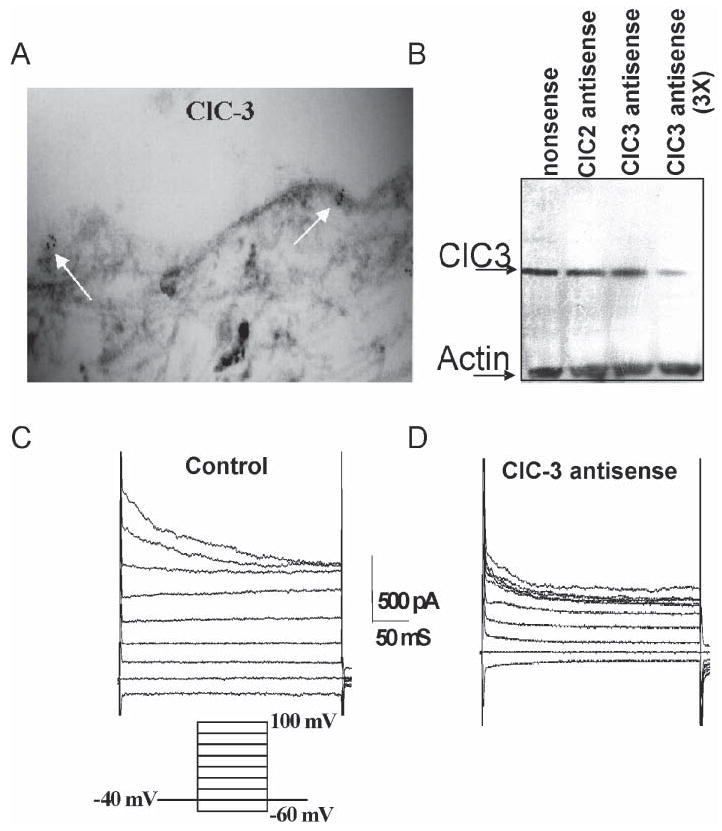
(A) D54-MG glioma cells in culture labeled using antibodies to ClC-3. Channels were identified with a secondary antibody conjugated to 6-nm immuno-gold particles. (B) Lysates of cultured D54-MG cells contain ClC-3. To suppress expression of this protein, sister cultures were treated for 48 hours with antisense oligonucleotides against ClC-3. This suppressed the concentration of ClC-3 significantly but did not affect expression of ClC-, another Cl− channel. (C) Whole-cell patch clamp recordings reveal the presence of outwardly rectifying Cl− currents with time-dependent inactivation in control D54-MG cells. These currents are sensitive to NPPB, DIDS and Cltx. (D) Sister cultures treated with ClC-3 antisense show a significant (>50%) suppression of these currents. Modified, with permission, from Olsen et al. (2003).
Ca2+-activated BK channels might provide K+ efflux during cell invasion
The Cl− efflux is hypothesized to be accompanied by the movement of K+ ions. The principal pathway for K+ efflux from glioma cells appears to be via Ca2+-activated BK channels, which have the unique ability to couple changes in intracellular Ca2+ to changes in membrane K+ conductance and are expressed highly in glioma cells (Ransom and Sontheimer, 2001). Glioma cells express a splice variant of the hslo gene that forms BK channels with enhanced sensitivity to intracellular Ca2+ (Liu et al., 2002). BK channels are inhibited by 1 mM TEA, which also blocks Transwell glioma migration (Soroceanu et al., 1999). More specific BK-channel inhibitors include scorpion toxins charybdotoxin and iberiotoxin. Both drugs inhibit glioma cell migration in a dose-dependent fashion (Weaver et al., submitted), whereas blockers of other K+ channels have no effect.
ClC-3 Cl− channels colocalize with BK K+ channels to lipid-raft domains on invadipodia
One assumes that the processes of invading cells should be the first part of the cell to invade the narrow extracellular spaces in brain, as illustrated in Fig. 3 (bottom left). Hence, the volume regulatory machinery (i.e. channels for Cl−, K+ and water) are likely to be located on the invading cellular processes, the so called invadipodia. This appears to be the case for ClC-3 Cl−channels and for BK K+ channels. ClC-3 immunostaining with specific antibodies decorate predominantly the outermost processes of the cells (Olsen et al., 2003). These processes also stain with the cholera toxin B subunit, a marker for specialized raft domains called lipid rafts (Hering et al., 2003) and, indeed, ClC-3 channels co-localize with lipid rafts (Fig. 5A,C). Staining for BK channels show these channels to be enriched on the edges of the lamellipodia and to co-localize with the lipid-raft marker cholera toxin (Fig. 5B). Hence, ClC-3 and BK channels localize to the same cellular domains. Many membrane proteins are organized on cell membranes by lipid-raft domains and lipid rafts define front–rear cell polarity in MCF-7 adenocarcinoma cells (Manes et al., 1999). To see whether ClC-3 and BK channels are generally enhanced in lipid-raft domains, we also used a biochemical approach to separate these lipid domains. Specifically, we used a gradient-centrifugation approach that has been described previously (Rujoi et al., 2003) to separate the lipid-raft-rich fractions from intracellular fractions. These fractions were separated by SDS page and probed with antibodies to ClC-3, BK and the caveolae marker caveolin-1. These studies show that ClC-3 and BK channels localize to the caveolar raft fraction (fraction 2 on the gel in Fig. 5C). Often, caveolar rafts are associated with protein trafficking, for example AMPAR (Hering et al., 2003) and Kv1.5 K+ channels (Martens et al., 2001) traffic via caveolar rafts.
Fig. 5. ClC-3 and BK channels localize to lipid raft domains on invadipodia of glioma cells.
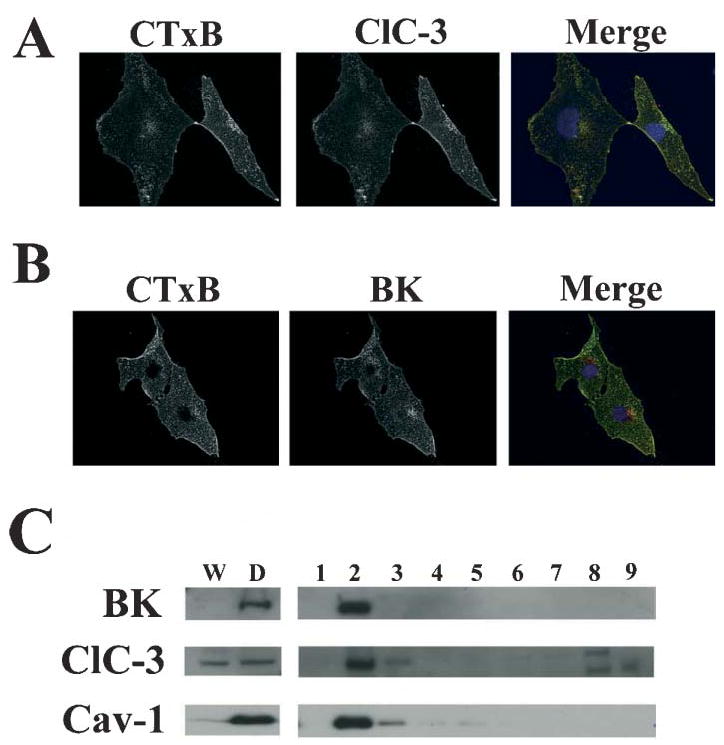
(A) Fluorescein-conjugated cholera toxin B subunit was used to label lipid-raft domains of D54-MG cells grown on glass coverslips. Cells were fixed and labeled with polyclonal, rabbit anti-BK K+ primary antibodies followed by secondary labeling with Alexa 546 goat anti-rabbit antibodies to illustrate the localization of BK K+ to lipid-raft domains. (B) Sister cultures were labeled with fluorescein-conjugated cholera toxin B subunit followed by labeling with polyclonal anti-ClC-3 antibodies, which exhibited similar localization patterns. (C) Lipid rafts were isolated from D54-MG cells by subcellular fractionation followed by density-gradient centrifugation. Proteins in each fraction were separated by SDS-PAGE and Western blotted for BK channels, ClC-3 channels and the caveolar lipid raft marker caveolin-1. Fractions W and D represent water-soluble and detergent-soluble fractions, respectively. Fractions 1–9 represent the fractions from the top to the bottom following density-gradient centrifugation (5–40% Optiprep) of the detergent insoluble fraction. Fraction 2 contains the buoyant lipid-raft fraction, as evidenced by caveolin-1 controls. BK and ClC-3 channels partition into this lipid-raft fraction.
A putative Cl− channel inhibitor (Cltx) is currently in clinical trials for treatment of glioma
Before the molecular characterization of glioma Cl− channels, which we now presume to be primarily ClC-3 channels, we found (Ullrich et al., 1996; Ullrich and Sontheimer, 1996) that Cltx, a scorpion-derived 36-residue peptide that was isolated as a putative Cl−-channel blocker (DeBin et al., 1993) inhibits whole-cell Cl− currents in glioma cells in vitro (Lippiat et al., 1998) and in situ (Ullrich et al., 1998). Moreover, Cltx inhibits glioma-cell invasion in several in situ invasion models, as does the Cl−-channel inhibitor NPPB (Soroceanu et al., 1999). Moreover, using an animal model in which human glioma cells are xenografted into the cerebrum of scid mice, we show the selective homing of the peptide onto the tumor (Soroceanu et al., 1998). A thorough analysis of biopsy tissues from patients demonstrates the selective binding of Cltx to all grades of malignant gliomas and to tumors that are embryologically related (Lyons et al., 2002). These studies have paved the way for a Phase I/II clinical study in which a single dose of [131]I-Cltx was administered to 18 patients. Detailed results from this study will be published elsewhere, but initial imaging data that shows selective homing of the peptide to tumors in patients are published (Hockaday et al., 2005). An open-label, multi-center, dose-escalating study in which multiple doses of Cltx will be administered opened for patient enrollment in 2005.
While these clinical studies were progressing, laboratory studies continued to further identify the target of Cltx on glioma cells, which is presumed to be a Cl− channel (possibly ClC-3) and to further examine the interactions of Cltx with Cl− channels and the resulting anti-invasive response. These results were surprising because they indicate that Cltx, unlike the related scorpion peptides, does not bind directly to a Cl−channel but that it binds to a cell-surface protein complex that contains MMP-2 and MT1-MMP (Deshane et al., 2003). Binding of Cltx causes internalization of this complex, along with ClC-3 channels, into caveolar rafts, which depletes the available membrane-associated Cl− channels.
Application of Cltx results in the near complete inhibition of glioma Cl− currents (Fig. 6A), as described previously (Ullrich et al., 1998). Of note, unlike the block by NPPB, which develops within seconds, complete block by Cltx requires 10–15 minutes, and the example illustrated is recorded 15 minutes after application of 1 μM Cltx. Interestingly, a similar, slow time-course of drug action has been reported for Cltx blockade of single Cl− channels recorded from reactive astrocytes (Dalton et al., 2003). This is unlike the effects of scorpion peptides on other ion channels, which typically occur within milliseconds (Possani et al., 2000).
Fig. 6. Cltx inhibits glioma Cl− currents by inducing endocytosis of channels into caveoli.
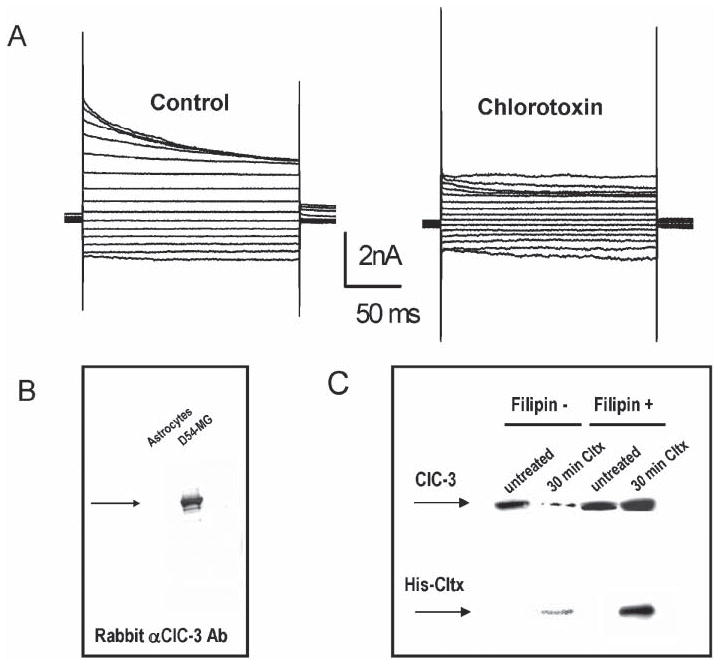
(A) Outwardly rectifying, inactivating Cl− currents recorded by whole-cell patch clamp recording in D54-MG glioma cells using voltage steps ranging from −120−160 mV (20 mV increments). Application of Cltx causes a slow and irreversible reduction of measurable currents. The example illustrated was recorded after 15 minutes. (B) Recombinant 6×-His-Cltx peptide was used to affinity purify interacting proteins from glioma lysates. The affinity-purified fraction was run on SDS-PAGE and probed with antibodies to ClC-3 to detect the channel in lysates from glioma cells but not from astrocytes. (C) A surface biotinylation approach, previously described by Ye et al. (1999) was used to assess the surface expression of ClC-3 protein. Separation over avidin beads isolated the cell-surface proteins that were accessible to the biotinylation reagent at the time of exposure. This fraction was probed by Western blot with antibodies to ClC-3 and anti-His to detect bound His-Cltx. ClC-3 protein was present in the membrane of untreated glioma cells. Treatment with Cltx for 30 minutes before exposure to the biotinylation reagent reduced the amount of ClC-3 on the membrane surface. Disrupting caveolar endocytosis with 5 μg ml−1 Filipin (a sterol-binding drug) prevents Cltx-mediated reduction in surface expression of ClC-3 in these cells, which indicates that Cltx causes endocytosis of ClC-3 into caveoli.
In light of this unusual finding we approached the question of how Cltx interacts with Cl− channels by search for its binding partner using the ligand as bait. Specifically, we generated a recombinant Cltx with a 6×His tag that could be conjugated to avidin beads for affinity purification of potentially interacting proteins. The purified proteins were separated on SDSPage and identified by mass spectrometry. Unexpectedly, these studies isolated a protein complex consisting of MT1-MMP, MMP-2 and TIMP-2 (Deshane et al., 2003), which are all involved in the enzymatic degradation of the extracellular matrix (Chen and Wang, 1999; Forsyth et al., 1999). This protein complex is instrumental in facilitating the invasion of glioma cells (Belien et al., 1999; Nakada et al., 2003). Most surprisingly, however, it also interacts with ClC-3 channels. This is illustrated in Fig. 6B, which shows affinity-purified fractions of glioma membrane isolated by binding to His-Cltx on Western blots. Probing with antibodies to ClC-3 demonstrates the presence of the channel in the affinity-purified membrane fractions from D-54MG glioma cells but not astrocytes. The interaction of Cltx with MMP-2/MT1-MMP and ClC-3 channels therefore raises the possibility that Cltx binds to a macromolecular protein complex on the cell surface and that its binding affects Cl−-channel function indirectly.
Based on the finding that inhibition of currents requires ~15 minutes, we hypothesized that Cltx binding might alter the presence of ClC channels on the cell surface. To examine this possibility directly we treated cells for 30 minutes with 1 μM Cltx after exposure to a biotinylation reagent that allows us to separate surface membrane proteins from intracellular protein fractions. The experiment illustrated in Fig. 6C shows a Western blot of the biotinylated surface fraction before and after 30-minute treatment of cells with Cltx. ClC-3 channels were reduced by exposure to Cltx, which indicates internalization of ClC-3 channels. Similar experiments with antibodies to MMP-2 show that MMP-2 also decreases on the cell surface (Deshane et al., 2003). The internalization of ClC-3 channels induced by Cltx is prevented by simultaneously treating the cells with the sterol-binding drug filipin (5 μg ml−1) (Fig. 6C). Filipin disrupts the formation of caveoli (Pol et al., 2000), which are a subset of lipid-raft vesicles that are involved in membrane trafficking in glioma cells (Cameron et al., 2002).
Previously, we suggested that the ability of glioma cells to cross Transwell barriers depends on their ability to regulate their cell volume, which requires the activity of Cl− channels, most likely ClC-3. In addition we showed that treatment with Cltx inhibits Transwell migration. Based on the above findings, which indicate that Cltx causes internalization of ClC-3 channels into caveolar rafts, we did not expect Cltx to block Transwell migration in the presence of filipin because Cl−channels can no longer be internalized. This is the case (Deshane et al., 2003). Thus, the mechanisms by which NPPB and Cltx inhibit Transwell migration differ because NPPB inhibits the function of ClC-3 channes, whereas Cltx depletes functional ClC-3 channels from the membrane. These data indicate a novel action of this scorpion toxin, which was presumed to be a Cl−-channel-specific peptide (DeBin et al., 1993). Rather than acting directly on the ion channel, it appears to interfere with protein trafficking. Through interaction with MMP-2/MT1-MMP it induces the internalization into caveolar rafts and, within 10–30 minutes depletes Cl− channels from the cell surface. Internalization occurs in conjunction with the Cltx peptide (Deshane et al., 2003) and this feature has been exploited recently to manufacture a Cltx-based multi-functional nanoprobe that either targets glioma cells or detecting them by magnetic resonance imaging and fluorescence microscopy (Veiseh et al., 2005). Moreover, the finding that the molecule is captured intracellularly by tumor cells and, hence, cannot be cleared explains why [131]I-labeled Cltx is still visualized in tumors 8 days after administration in clinical trials (Hockaday et al., 2005). By contrast, how binding of Cltx to MMP-2/MT1-MMP leads to enhanced caveolar internalization of a macromolecular complex remains unknown. However, even in the absence of Cltx, MMP-2, MT-1MMP and ClC-3 channels co-localize to lipid rafts (data not shown). Hence, the association of an enzyme complex that degrades the extracellular matrix with Cl− channels appears to be an important biological feature of these tumors.
CONCLUSIONS
In glioma cells, invasion appears to involve a coordinated reduction in cell volume, which is mediated by the efflux of Cl− and K+ through ion channels. Similar mechanisms involving ion channels might also facilitate the movement of other cell types, such as progenitor cells and stem cells. Further study and direct comparison with glioma cells appears warranted.
DISCUSSION
Cell movement accompanies normal brain development and occurs in response to injury and in diseases such as glioma. The biophysics of cell movement is understood poorly, and the spatial constraints and the requirement for cells to adapt their size and volume to fit into narrow spaces have been examined little. We suggest that the cell shrinkage described here is a prerequisite of many migratory cells. To the best of our knowledge, most immature cells that can migrate also accumulate intracellular Cl− and are, thus, well equipped to release KCl and to shrink, whereas there is no net force that drives Cl− out of the cell in most differentiated neurons. Because of the active accumulation of Cl−, immature neurons (Owens et al., 1996) and neuronal stem cells in the ventricular zone (LoTurco et al., 1995) depolarize in response to GABA, whereas differentiated neurons, which no longer move, either hyperpolarize or stabilize their membrane potential (Bormann et al., 1987). Although it is proposed that this depolarizing GABA response might be important physiologically in the context of synapse development (Ben Ari et al., 2004) we suggest that these cells might utilize GABA-gated Cl− channels to adjust their cell volume because they are still migrating through the brain. In this context, GABA would trigger the efflux of Cl− and, hence, cause cells to shrink. Other migration-competent cells, for example microglia (Eder et al., 1998; Farber and Kettenmann 2005; Zierler and Kerschbaum, 2005), O2A progenitors (Berger et al., 1992), astrocytes (Bormann and Kettenmann, 1988), neural stem cells (LoTurco et al., 1995) and glioma cells (Synowitz et al., 2001) also accumulate Cl− and show an efflux in response to opening of either ligand or voltage-gated Cl− channels. It is possible that this is an archetypical mechanism by which immature and migration-competent cells establish the appropriate parameters that allow them to shrink on demand. Of course, this begs the question of how Cl− is pumped into these cells. It is believed that Cl− uptake generally occurs via the NKCC transporter (Russell, 2000), which is the primary transporter in glioma cells (Ernest and Sontheimer, 2005), and is balanced by Cl− efflux via KCC transporters. In neurons, the shift from accumulation of Cl− in immature, migratory cells to a passive Cl− distribution in differentiated, post-migratory cells occurs by the developmental insertion of KCC2 transporters (Lee et al., 2005). In glioma cells, the Cl− equilibrium is maintained by NKCC and KCC1 and KCC3a transporters (Ernest et al., 2005; Ernest and Sontheimer, 2005). Accordingly, cell migration should be disrupted if the Cl− gradient of these cells is disrupted, and that is indeed observed in glioma cells when the permeant Cl− ion is replaced with impermeant anions (Soroceanu et al., 1999).
Although much of our attention has focused on Cl− channels as the determinant of cell volume, the efflux of K+ and water is equally important. Our data indicate that Ca2+-activated K+ channels might participate in this process (Weaver et al., 2004) and we have recently identified two water channels (AQP 1 and 4) in glioma cells (McCoy and Sontheimer, 2005). In most cells, water permeability does not limit the regulation of cell size and cell volume (King et al., 2000) and we assume that this is also the case here.
One of the major unanswered questions is how the processes of cell movement and invasion are coupled to the controlled activation of Cl− and K+ channels. In cerebellar granule cells migration is accompanied by oscillatory changes in intracellular Ca2+ (Rakic and Komuro, 1995; Komuro and Rakic, 1996) and these changes are mediated by activation of NMDA receptors. In migratory glioma cells, similar Ca2+ oscillations are reported in response to activation of ACh-R (Bordey et al., 2000), which activate charybdotoxin-sensitive BK K+ channels. Moreover, glioma cells express Ca2+-permeable AMPA receptors, and Ca2+ influx through these receptors is required for cell migration (Ishiuchi et al., 2002). Hence, receptor-mediated activation of Ca2+ influx into either migratory granule cells or glioma cells appears to be a prerequisite for cell migration: we suspect that this Ca2+ signal is instructive with regards to cell-volume changes that occur down-stream. Whether Ca2+-activated K+ channels are a target of this Ca2+ influx is unknown. Future studies should focus on identifying the signaling events that transduce cell motility into a coordinated change in cell volume and how these changes facilitate the translocation of cells through narrow spaces. Moreover, understanding the pathways that control the biophysics of cell movement might allow drugs to be targeted at cells that show aberrant cell migration, most notably, cancer cells.
Acknowledgments
The authors wish to acknowledge the valuable contributions made by Jessy Deshane who made recombinant His-tagged Cltx and helped with studies of ClC-3 trafficking and affinity purification of glioma proteins. Susan A. Lyon contributed Fig. 2A. This work was supported by grants RO1-NS-36692, NS-31234, P50-CA-97247 and P30-HD-038985-06 from the National Institutes of Health.
References
- Belien AT, Paganetti PA, Schwab ME. Membrane-type 1 matrix metalloprotease (MT1-MMP) enables invasive migration of glioma cells in central nervous system white matter. Journal of Cell Biology. 1999;144:373–384. doi: 10.1083/jcb.144.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Ari Y, Khalilov I, Represa A, Gozlan H. Interneurons set the tune of developing networks. Trends in Neurosciences. 2004;27:422–427. doi: 10.1016/j.tins.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Berger T, Walz W, Schnitzer J, Kettenmann H. GABA- and glutamateactivated currents in glial cells of the mouse corpus callosum slice. Journal of Neuroscience Research. 1992;31:21–27. doi: 10.1002/jnr.490310104. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H, Trouslard J. Muscarinic activation of bk channels induces membrane oscillations in glioma cells and leads to inhibition of cell migration. Journal of Membrane Biology. 2000;176:31–40. doi: 10.1007/s00232001073. [DOI] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurons. Journal of Physiology London. 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bormann J, Kettenmann H. Patch-clamp study of gamma-aminobutyric acid receptor Cl− channels in cultured astrocytes. Proceedings of the National Academy of Sciences of the U.S.A. 1988;85:9336–9340. doi: 10.1073/pnas.85.23.9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron PL, Liu C, Smart DK, Hantus ST, Fick JR, Cameron RS. Caveolin-1 expression is maintained in rat and human astroglioma cell lines. Glia. 2002;37:275–290. doi: 10.1002/glia.10036. [DOI] [PubMed] [Google Scholar]
- Chen WT, Wang JY. Specialized surface protrusions of invasive cells, invadopodia and lamellipodia, have differential MT1-MMP, MMP-2 and TIMP-2 localization. Annals New York Academy of Science. 1999;878:361–371. doi: 10.1111/j.1749-6632.1999.tb07695.x. [DOI] [PubMed] [Google Scholar]
- d’Anglemont dT, Souktani R, Ghaleh B, Henry P, Berdeaux A. Structure and pharmacology of swelling-sensitive chloride channels, I (Cl,swell) Fundamental and Clinical Pharmacology. 2003;17:539–553. doi: 10.1046/j.1472-8206.2003.00197.x. [DOI] [PubMed] [Google Scholar]
- Dalton S, Gerzanich V, Chen M, Dong Y, Shuba Y, Simard JM. Chlorotoxin-sensitive Ca2+-activated Cl− channel in type R2 reactive astrocytes from adult rat brain. Glia. 2003;42:325–339. doi: 10.1002/glia.10212. [DOI] [PubMed] [Google Scholar]
- DeBin JA, Maggio JE, Strichartz GR. Purification and characterization of chlorotoxin, a chloride channel ligand from the venom of the scorpion. American Journal of Physiology. 1993;264:C361–9.26. doi: 10.1152/ajpcell.1993.264.2.C361. [DOI] [PubMed] [Google Scholar]
- Deshane J, Garner CC, Sontheimer H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. Journal of Biological Chemistry. 2003;278:4135–4144. doi: 10.1074/jbc.M205662200. [DOI] [PubMed] [Google Scholar]
- Eder C, Klee R, Heinemann U. Involvement of stretch-activated Cl− channels in ramification of murine microglia. Journal of Neuroscience. 1998;18:7127–7137. doi: 10.1523/JNEUROSCI.18-18-07127.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernest N.J. and Sontheimer H. (2005) chloride chloride cotransporters maintain intracellular chloride and cell size in human glioma cells. Society for Neuroscience Abstracts
- Ernest NJ, Weaver AK, Van Duyn LB, Sontheimer HW. Relative contribution of chloride channels and transporters to regulatory volume decrease in human glioma cells. Americal Journal of Physiology. Cell Physiology. 2005;288:C1451–C1460. doi: 10.1152/ajpcell.00503.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber K, Kettenmann H. Physiology of microglial cells. Brain Research. Brain Research Reviews. 2005;48:133–143. doi: 10.1016/j.brainresrev.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Fenster SD, Chung WJ, Zhai R, Cases-Langhoff C, Voss B, Garner AM, et al. Piccolo, a presynaptic zinc finger protein structurally related to bassoon. Neuron. 2000;25:203–214. doi: 10.1016/s0896-6273(00)80883-1. [DOI] [PubMed] [Google Scholar]
- Forsyth PA, Wong H, Laing TD, Rewcastle NB, Morris DG, Muzik H, et al. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. British Journal of Cancer. 1999;79:1828–1835. doi: 10.1038/sj.bjc.6990291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Lin CC, Sheng M. Lipid rafts in the maintenance of synapses, dendritic spines and surface AMPA receptor stability. Journal of Neuroscience. 2003;23:3262–3271. doi: 10.1523/JNEUROSCI.23-08-03262.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockaday DC, Shen S, Fiveash J, Raubitschek A, Colcher D, Liu A, et al. Imaging glioma extent with 131I-TM-601. Journal Nuclear Medicine. 2005;46:580–586. [PubMed] [Google Scholar]
- Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H, et al. Blockage of Ca (2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nature Medicine. 2002;8:971–978. doi: 10.1038/nm746. [DOI] [PubMed] [Google Scholar]
- King LS, Yasui M, Agre P. Aquaporins in health and disease. Molecular Medicine Today. 2000;6:60–65. doi: 10.1016/s1357-4310(99)01636-6. [DOI] [PubMed] [Google Scholar]
- Komuro H, Rakic P. Intracellular Ca2+ fluctuations modulate the rate of neuronal migration. Neuron. 1996;17:275–285. doi: 10.1016/s0896-6273(00)80159-2. [DOI] [PubMed] [Google Scholar]
- Lee H, Chen CX, Liu YJ, Aizenman E, Kandler K. KCC2 expression in immature rat cortical neurons is sufficient to switch the polarity of GABA responses. European Journal of Neuroscience. 2005;21:2593–2599. doi: 10.1111/j.1460-9568.2005.04084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippiat JD, Standen NB, Davies NW. Block of cloned BKCa channels (rSlo) expressed in HEK 293 cells by N-methyl d-glucamine. Pflugers Archives. 1998;436:810–812. doi: 10.1007/s004240050708. [DOI] [PubMed] [Google Scholar]
- Liu X, Chang Y, Reinhart PH, Sontheimer H, Chang Y. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. Journal of Neuroscience. 2002;22:1840–1849. doi: 10.1523/JNEUROSCI.22-05-01840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoTurco JJ, Owens DF, Heath MJS, Davis MBE, Kriegstein AR. GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron. 1995;15:1287–1298. doi: 10.1016/0896-6273(95)90008-x. [DOI] [PubMed] [Google Scholar]
- Lyons SA, O’Neal J, Sontheimer H. Chlorotoxin, a scorpionderived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia. 2002;39:162–173. doi: 10.1002/glia.10083. [DOI] [PubMed] [Google Scholar]
- Manes S, Mira E, Gomez-Mouton C, Lacalle RA, Keller P, Labrador JP, et al. Membrane raft microdomains mediate front-rear polarity in migrating cells. EMBO Journal. 1999;18:6211–6220. doi: 10.1093/emboj/18.22.6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens JR, Sakamoto N, Sullivan SA, Grobaski TD, Tamkun MM. Isoform-specific localization of voltage-gated K+ channels to distinct lipid raft populations. Targeting of Kv1.5 to caveolae. Journal of Biological Chemistry. 2001;276:8409–8414. doi: 10.1074/jbc.M009948200. [DOI] [PubMed] [Google Scholar]
- McCoy E.S. and Sontheimer H. (2005) Expression and Function of AQP1 in astrocytes. Society for Neuroscience Abstracts.
- Nakada M, Okada Y, Yamashita J. The role of matrix metalloproteinases in glioma invasion. Frontiers in Bioscience. 2003;8:E261–E269. doi: 10.2741/1016. [DOI] [PubMed] [Google Scholar]
- Olsen ML, Schade S, Lyons SA, Amarillo MD, Sontheimer H. Expresssion of voltage-gated chloride channels in human glioma cells. Journal of Neuroscience. 2003;23:5572–5582. doi: 10.1523/JNEUROSCI.23-13-05572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MB, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. Journal of Neuroscience. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pol A, Lu A, Pons M, Peiro S, Enrich C. Epidermal growth factor-mediated caveolin recruitment to early endosomes and MAPK activation. Role of cholesterol and actin cytoskeleton. Journal of Biological Chemistry. 2000;275:30566–30572. doi: 10.1074/jbc.M001131200. [DOI] [PubMed] [Google Scholar]
- Possani LD, Merino E, Corona M, Bolivar F, Becerril B. Peptides and genes coding for scorpion toxins that affect ion-channels. Biochimie. 2000;82:861–868. doi: 10.1016/s0300-9084(00)01167-6. [DOI] [PubMed] [Google Scholar]
- Rakic P, Komuro H. The role of receptor/channel activity in neuronal cell migration. Journal of Neurobiology. 1995;26:299–315. doi: 10.1002/neu.480260303. [DOI] [PubMed] [Google Scholar]
- Ransom CB, O’Neal JT, Sontheimer H. Volume-activated chloride currents contribute to the resting conductance and invasive migration of human glioma cells. Journal of Neuroscience. 2001;21:7674–7683. doi: 10.1523/JNEUROSCI.21-19-07674.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. BK channels in human glioma cells. Journal of Neurophysiology. 2001;85:790–803. doi: 10.1152/jn.2001.85.2.790. [DOI] [PubMed] [Google Scholar]
- Rujoi M, Jin J, Borchman D, Tang D, Yappert MC. Isolation and lipid characterization of cholesterol-enriched fractions in cortical and nuclear human lens fibers. Investigative Ophthalmology and Visual Science. 2003;44:1634–1642. doi: 10.1167/iovs.02-0786. [DOI] [PubMed] [Google Scholar]
- Russell JM. Sodium-potassium-chloride cotransport. Physiological Reviews. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- Soroceanu L, Gillespie Y, Khazaeli MB, Sontheimer H. Use of chlorotoxin for targeting of primary brain tumors. Cancer Research. 1998;58:4871–4879. [PubMed] [Google Scholar]
- Soroceanu L, Manning TJ, Jr, Sontheimer H. Modulation of glioma cell migration and invasion using Cl− and K+ ion channel blockers. Journal of Neuroscience. 1999;19:5942–5954. doi: 10.1523/JNEUROSCI.19-14-05942.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synowitz M, Ahmann P, Matyash M, Kuhn SA, Hofmann B, Zimmer C, et al. GABA (A)-receptor expression in glioma cells is triggered by contact with neuronal cells. European Journal of Neuroscience. 2001;14:1294–1302. doi: 10.1046/j.0953-816x.2001.01764.x. [DOI] [PubMed] [Google Scholar]
- Ullrich N, Bordey A, Gillespie GY, Sontheimer H. Expression of voltageactivated chloride currents in acute slices of human gliomas. Neuroscience. 1998;83:1161–1173. doi: 10.1016/s0306-4522(97)00456-9. [DOI] [PubMed] [Google Scholar]
- Ullrich N, Gillespie GY, Sontheimer H. Human astrocytoma cells express a unique chloride current. Neuroreport. 1996;7:1020–1024. doi: 10.1097/00001756-199604100-00013. [DOI] [PubMed] [Google Scholar]
- Ullrich N, Sontheimer H. Biophysical and pharmacological characterization of chloride currents in human astrocytoma cells. American Journal of Physiology. Cell Physiology. 1996;270:C1511–C1521. doi: 10.1152/ajpcell.1996.270.5.C1511. [DOI] [PubMed] [Google Scholar]
- Veiseh O, Sun C, Gunn J, Kohler N, Gabikian P, Lee D, et al. Optical and MRI multifunctional nanoprobe for targeting gliomas. Nano Letters. 2005;5:1003–1008. doi: 10.1021/nl0502569. [DOI] [PubMed] [Google Scholar]
- Weaver AK, Liu X, Sontheimer H. Role for calcium-activated potassium channels (BK) in growth control of human malignant glioma cells. Journal of Neuroscience Research. 2004;78:224–234. doi: 10.1002/jnr.20240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Rothstein JD, Sontheimer H. Compromised glutamate transport in human glioma cells: reduction- mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. Journal of Neuroscience. 1999;19:10767–10777. doi: 10.1523/JNEUROSCI.19-24-10767.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H. Cytokine modulation of glial glutamate uptake: A possible involvement of nitric oxide. Neuroreport. 1996;7:2181–2185. doi: 10.1097/00001756-199609020-00025. [DOI] [PubMed] [Google Scholar]
- Zierler S, Kerschbaum HH. Blockade of chloride conductance antagonizes PMA-induced ramification in the murine microglial cell line, BV-2. Brain Research. 2005;1039:162–170. doi: 10.1016/j.brainres.2005.01.086. [DOI] [PubMed] [Google Scholar]