

Abstract
Objective: To determine the protective effect of monosialoganglionside (GM1) and evaluate the influence of GM1 on expression of N-methyl-D-aspartate receptor subunit 1 (NMDAR1) in Sprague-Dawley (SD) rats with focal cerebral ischemia-reperfusion (I/R). Methods: Left middle cerebral artery (MCA) was occluded by an intraluminal suture for 1 h and the brain was reperfused for 72 h in SD rats when infarct volume was measured, GM1 (10 mg/kg) was given ip (intraperitoneally) at 5 min (group A), 1 h (group B) and 2 h (group C) after MCA occlusion (MCAo). Expression of NMDAR1 was detected by Western blot at various time after reperfusion (4 h, 6 h, 24 h, 48 h and 72 h) in ischemic hemispheres of the rats with or without GM1 administered. Results: (1) Adjusted relative infarct volumes of groups A and B were significantly smaller than that of group C and the control group (P<0.01 and P<0.05, respectively). (2) Expression level of NMDAR1 was temporally high at 6 h after reperfusion, and dipped below the normal level at 72 h after reperfusion. GM1 at 5 min after MCAo significantly suppressed the expression of NMDAR1 at 6 h after reperfusion (P<0.05 vs the control). At 72 h after reperfusion, the NMDAR1 expression level of rats treated with GM1 administered (at 5 min or 2 h after MCAo) was significantly higher than that of the control (P<0.05). Conclusion: GM1 can time-dependently reduce infarct volume in rats with focal cerebral I/R partly through stabilizing the expression of NMDAR1.
Keywords: G(M1) ganglioside, Middle cerebral artery occlusion, Reperfusion, N-methyl-D-aspartate receptors, Rats
INTRODUCTION
GM1 ganglioside (GM1) is the main kind of gangliosides in mammalia, and most abundant in brain tissue (Duchemin et al., 2002). It was reported that GM1 could protect cerebral ischemia in vivo and in vitro, one protective mechanism of which is that GM1 could reduce neural injury induced by toxicity of excitatory amino acid via N-methyl-D-aspartate receptor (NMDAR) (Kharlamov et al., 1993; Simon et al., 1993; Garofalo and Cuello, 1994; de Erausquin et al., 1990). NMDAR is a kind of ionotrophic glutamate receptor, a receptor gated Ca2+ channel, and is extensively distributed in the central nervous system (CNS). NMDAR subunit is coded by two gene families: NMDAR1 and NMDAR2; NMDAR1 subunit is rigueur to form the NMDAR (Luo et al., 1997). In the mammalian CNS, NMDAR serves prominent roles in the pathophysiologic process of cerebral ischemia; NMDAR becomes over excited during cerebral ischemia to induce Ca2+ influx and exacerbate neural injury (Monaghan et al., 1989; Collingridge and Singer, 1990).
GM1 could reduce the neurotoxicity of NMDAR, but could not suppress the function of the NMDAR gated Ca2+ channel directly (de Erausquin et al., 1990; Manev et al., 1990). Is it possible that the suppressive effect of GM1 on excitatory amino acid toxicity is via the influence of GM1 on the expression of NMDAR? Brandoli et al.(1998) found brain-derived neurotrophic factor (BDNF) elicited a time-dependent decrease in the expression of NMDAR2A and NMDAR2C subunits (two subunits mainly distributing in the cerebellum) and reduced the NMDA-mediated [Ca2+] i increase with a time dependency that correlated with their abilities to decrease NMDAR2A and NMDAR2C subunit expression, suggesting that BDNF may protect cerebellar granule cells against excitotoxicity by altering the NMDAR-Ca2+ signaling via a downregulation of NMDAR subunit expression. GM1 can enhance the protective effect of BDNF and nerve growth factor (NGF) on neurons (Duchemin et al., 2002; Brandoli et al., 1998). However it is not clear whether GM1 can influence the expression of NMDAR subunits in ischemic neurons. Therefore, we investigated in this study whether GM1 could modulate the expression of NMDAR1 in the ischemic hemisphere of the focal cerebral ischemia/reperfusion rat model.
METHODS
Male Sprague-Dawley (SD) rats, weighing 250–300 g, were bought from the Medical Institute of Zhejiang Province, China (Certificate No. 2001001). Rats for evaluating infarct volume were divided into the following groups: Control (n=7); groups with GM1 administrated at 5 min (Group A, n=7); 1 h (Group B, n=6); and 2 h (Group C, n=6) after 1 h’s MCAo. Rats of all groups underwent reperfusion for 72 h after MCAo. For NMDAR1 Western blot, rats underwent 4 h, 6 h, 24 h, 48 h, 72 h reperfusion after MCAo (n of group at each time point and the control were both 6). The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH Publication No. 85–23, revised 1996).
GM1 (TRB PHARMA S.A. of Argentina) was given ip at 5 min, 1 h and 2 h after MCAo, and repeated once at 24 h and 48 h after reperfusion respectively; every dose was 10 mg/kg.
Rats were anesthetized with intraperitoneal injection of 400 mg/kg chloral hydrate. During the surgical procedure, temperature was continuously monitored with a rectal probe and maintained at 37 °C with a thermostatically controlled heating pad. Focal cerebral ischemia was induced by the intraluminal suture MCAo method as described in Longa et al.(1989). Briefly, the left common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA) were exposed through a midline incision of the neck. A 3-0 silica gel-coated nylon suture was used as an occluder and was inserted via the ECA. The occluder was inserted through a stump of the ECA, and the CCA was kept intact. The occluder was advanced into the ICA about 20 mm beyond the carotid bifurcation. Mild resistance indicated that the occluder was properly lodged in the anterior cerebral artery and thus blocked blood flow to the middle cerebral artery. At 1 h after MCAo, reperfusion began with the suture withdrawn. In sham-operated animals, the suture was inserted 5 mm from the incision.
Immediately after the rats totally recovering from anesthesia, the neurological scores were determined by the method described by Longa et al.(1989). After 72 h reperfusion, the rats were reanesthetized with an intraperitoneal injection of 400 mg/kg chloral hydrate and decapitated. The brains were coronally sectioned into 6 slices 2 mm thick from the frontal polar. The fourth slice was divided at the midline into two parts for NMDAR1 detection. Other brain slices were incubated for 30 min in a 2% solution of triphenyltetrazolium chloride at 37 °C, then fixed by immersion in a 10% buffered formalin solution and photographed with the use of a charge-coupled device camera. The white unstained areas were defined as infarcted. Infarct area of the posterior surface of each brain slice times 2 mm was the infarct volume; the total infarct volume (V) was the sum of the infarct volume of each slice. The posterior surface of the fourth brain slice was substituted by the anterior surface of the fifth brain slice because they were continuous sections. Total volume of ischemic hemisphere (V TL) and non-ischemic hemisphere (V TR) were obtained by the same way. The adjusted relative infarct volume (ARV) was calculated by the following formula:
| ARV=[VTR−(VTL−V)]/VTR |
Brain tissue sample of the fourth brain slices obtained as mentioned above was washed twice in PBS and pelleted. Preparation of cell membrane protein, detection of protein concentrations, procedure of SDS-PAGE and Western blot, and source of NMDAR1 antibody were described before (Luo et al., 1996; Zang and Wei, 2003). Protein of left hemisphere of a normal rat was set as the control. Ratio of optical density (OD) of the band of each sample to that of the control was calculated as the relative OD.
All data were expressed as mean±SD. Neurological deficit scores and adjusted relative infarct volume were analyzed using one-way analysis of variance (ANOVA). Band density ratio between 2 groups was analyzed using t-test. P<0.05 was considered significant.
RESULTS
There were no significant differences in neurological outcome of rats among the four groups immediately after MCAo (P>0.05) (Fig.1).
Fig. 1.
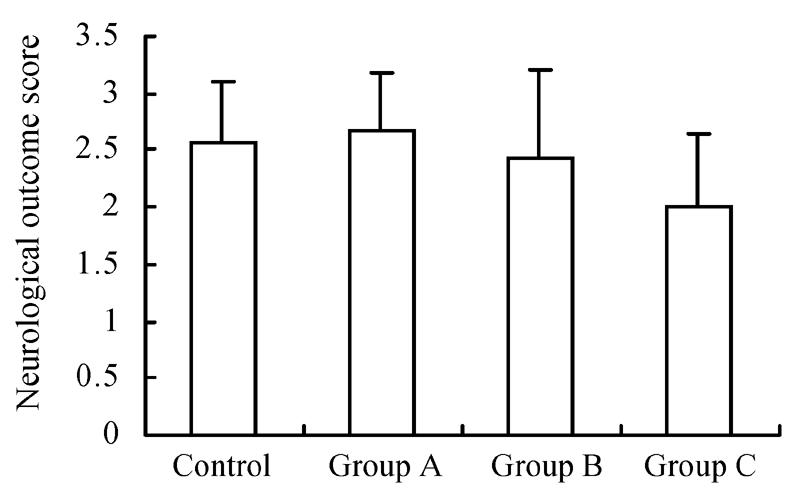
Neurologic outcome score of rats immediately recovering from anesthesia among different groups
Rats were separated as control (n=7), groups with GM1 administrated at 5 min (Group A, n=7), 1 h (Group B, n=6), and 2 h (Group C, n=6) after MCAo. There were no significant differences in neurological outcome of rats among the four groups immediately after MCAo (P>0.05)
Infarct size and ARV of groups with GM1 administered at 5 min and 1 h after MCAo were significantly lower than those of the group with GM1 administered at 2 h after MCAo and the control (Fig.2, Fig.3).
Fig. 2.




Influence of GM1 on the infarct size of the posterior surface of the third rat brain slice at 72 h after reperfusion
Rats were separated as control (n=7) (a), groups with GM1 administrated at 5 min (Group A, n=7) (b), 1 h (Group B, n=6) (c), and 2 h (Group C, n=6) (d) after MCAo. Infarct size of the posterior surface of the third rat brain slices of Groups A and B was significantly lower than that of Group C and the control
Fig. 3.
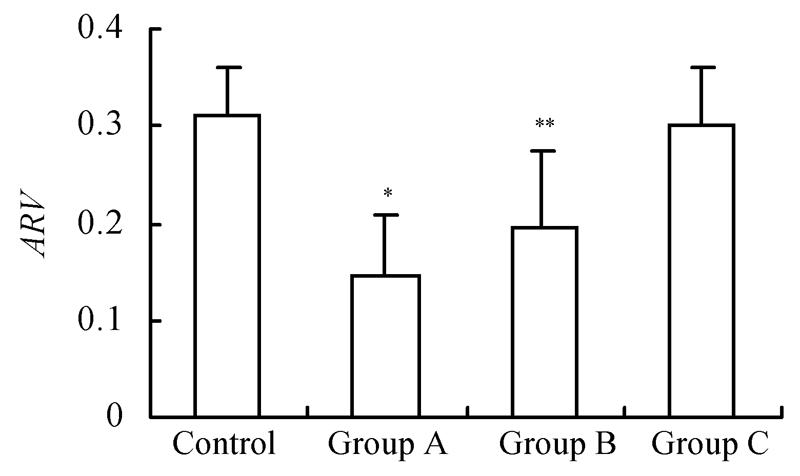
Influence of GM1 on adjusted relative infarct volume of rats at 72 h after reperfusion
Rats were separated as control (n=7), groups with GM1 administrated at 5 min (Group A, n=7), 1 h (Group B, n = 6), and 2 h (Group C, n=6) after MCAo. The adjusted relative infarct volume (ARV) of Groups A and B was significantly lower than that of Group C and the control (* P<0.05 and ** P<0.05 vs control and the Group C)
The expression of NMDAR1 peaked at 6 h after reperfusion, then decreased gradually and came back to around the level of the sham at 48 h after reperfusion. GM1 (administered at 5 min after MCAo) made the expression of NMDAR1 more smooth along with the reperfusion time; the expression level of NMDAR1 at 6 h after reperfusion in the GM1 treated group was significantly lower than that of the control (P<0.05) (Fig.4).
Fig. 4.


Influence of GM1 (administered at 5 min after MCAo) on the expression of NMDAR1 in ischemic hemisphere of rat after various reperfusion time
(a) Western blot showed NMDAR1 expression level peaked at 6 h after reperfusion, then decreased gradually and came back around the level of the sham at 48 h after reperfusion, while GM1 (delivered at 5 min after MCAo) made the expression of NMDAR1 more smooth along with the reperfusion time (RP: Reperfusion); (b) The expression level of NMDAR1 at 6 h after reperfusion in the group with GM1 administered was significantly lower than that of the control (n=6 rats, mean±SD, * P<0.05 vs control, t-test)
At 72 h after reperfusion, the expression level of NMDAR1 of rats with GM1 (administered at 5 min or 2 h after MCAo) was significantly higher than that of the control (P<0.05); there was no significant difference in the expression level of NMDAR1 of non-ischemic hemispheres among groups (P>0.05) (Fig.5).
Fig. 5.
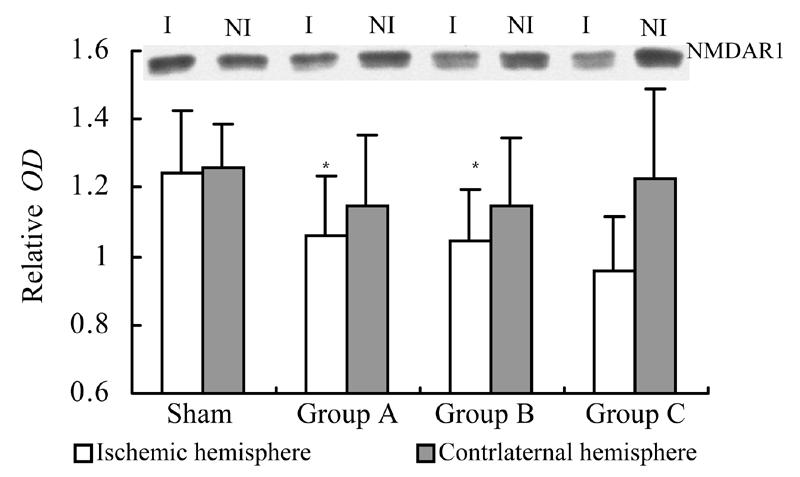
Influence of GM1 on the expression of NMDAR1 in ischemic and nonischemic hemispheres of rats at 72 h after reperfusion
Rats were separated as control (n=7), groups with GM1 administrated at 5 min (Group A, n=7), 1 h (Group B, n=6), and 2 h (Group C, n=6) after MCAo. At 72 h after reperfusion, the expression level of NMDAR1 of rats with GM1 (administered at 5 min or 2 h after MCAo) was significantly higher than that of the control (* P<0.05 vs control); there was no significant difference in the expression level of NMDAR1 of non-ischemic hemispheres among groups (P>0.05). (I: Ischemic hemisphere, NI: Nonischemic hemisphere)
DISCUSSION
Simon et al.(1993) reported that the protective effect of GM1 on focal cerebral ischemia was time-dependent in the permanent MCAo rat model. They considered GM1 administered at 5 min after MCAo could reduce the infarct volume at 24 h after MCAo while GM1 administered at 15 min after MCAo could not. In the present study, we found that GM1 could reduce the infarct volume with a time-dependency in rats which underwent MCAo (for 1 h) and reperfusion (for 72 h), that is, GM1 administered at 5 min or 1 h after MCAo could significantly reduce the infarct volume while GM1 administered at 2 h after MCAo could not. Anyway, early usage of GM1 may provide better protection for cerebral ischemia.
The mechanisms underlying the neuroprotection of GM1 on cerebral ischemia are not yet completely understood. One of which was considered to be the blockade of excitatory amino acid-mediated neurotoxicity. It is well established that NMDAR is involved in the brain injury associated with acute ischemic stroke. The large amount of glutamate accumulated in the extracellular space in the ischemic brain is probably due to glutamate release and impairment of its uptake (Rossi et al., 2000), with this high level of glutamate being thought to induce the hyperactivation of NMDARs followed by excess Ca2+ influx into neurons, and cell death (Choi and Rothman, 1990). Consistent with this view, NMDAR antagonists attenuate the Ca2+ influx and resulting neuronal death elicited by glutamate or NMDA in vitro (Michaels and Rothman, 1990; Wahlestedt et al., 1993). GM1 could reduce neural injury induced by toxicity of excitatory amino acid via NMDAR, but GM1 did not suppress function of NMDAR gated Ca2+ channel directly (de Erausquin et al., 1990; Manev et al., 1990). So, in this study we try to determine whether GM1 could modulate the expression of NMDAR1 subunit (NMDAR1) in the ischemic hemisphere of MCAo reperfusion rat.
It was reported that cerebral ischemia could cause change of the expression of NMDAR subunits in the gerbil and rat brain in vivo (Kang et al., 2001; Won et al., 2001; Friedman et al., 2001), but changes of the expression of NMDAR1 in rats after focal cerebral ischemia/reperfusion in vivo have not been reported. In the present study, we observed expression of NMDAR1 at 4 h, 6 h, 24 h, 48 h and 72 h after reperfusion in ischemic hemispheres of MCAo rats, and investigated the influence of GM1 on the expression of NMDAR1, and found that the expression level of NMDAR1 was elevated temporally at 6 h after reperfusion, and dipped below the normal level at 72 h after reperfusion, but GM1 can suppress the temporally high expression of NMDAR1 at 6 h after reperfusion and prevent exceptionally low expression of NMDAR1 at 72 h after reperfusion, i.e., GM1 can stabilize the expression of NMDAR1 after focal cerebral I/R in rats.
In the early stage of focal cerebral I/R in rats, NMDAR1 was highly expressed, which caused greater NMDA receptor-related neurotoxicity, while GM1 can inhibit the high expression of NMDAR1, and reduce the neurotoxicity to protect the ischemic neuron. NMDAR also plays important role in the physiologic process of the brain including learning and memory, overly low expression of which in the late stage after focal cerebral ischemia/reperfusion indicates abnormal neural function (Monaghan et al., 1989; Collingridge and Singer, 1990), while GM1 can inhibit the overly low expression of NMDAR1 to sustain the normal physiologic function of the neurons. So, GM1 can reduce infarct volume with a time dependency in focal ischemia reperfusion rat through stabilizing the expression of NMDAR1.
Footnotes
Project (No. 2004QN012) supported by the Youth Talent Special Fund of Health Bureau of Zhejiang Province, the National Basic Research Program (973) of China (No. G1999054000) and the National Natural Science Foundation of China (No. 30371637)
References
- 1.Brandoli C, Sanna A, De Bernardi MA, Follesa P, Brooker G, Mocchetti I. Brain-derived neurotrophic factor and basic fibroblast growth factor downregulate NMDA receptor function in cerebellar granule cells. J Neurosci. 1998;18(19):7953–7961. doi: 10.1523/JNEUROSCI.18-19-07953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- 3.Collingridge GL, Singer W. Excitatory amino acid receptors and synaptic plasticity. Trends Pharmacol Sci. 1990;11(7):290–296. doi: 10.1016/0165-6147(90)90011-v. [DOI] [PubMed] [Google Scholar]
- 4.de Erausquin GA, Manev H, Guidotti A, Costa E, Brooker G. Gangliosides normalize distorted single-cell intracellular free Ca2+ dynamics after toxic doses of glutamate in cerebellar granule cells. Proc Natl Acad Sci USA. 1990;87(20):8017–8021. doi: 10.1073/pnas.87.20.8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duchemin AM, Ren Q, Mo L, Neff NH, Hadjiconstantinou M. GM1 ganglioside induces phosphorylation and activation of Trk and Erk in brain. J Neurochem. 2002;81(4):696–707. doi: 10.1046/j.1471-4159.2002.00831.x. [DOI] [PubMed] [Google Scholar]
- 6.Friedman LK, Ginsberg MD, Belayev L, Busto R, Alonso OF, Lin B, Globus MY. Intraischemic but not postischemic hypothermia prevents non-selective hippocampal downregulation of AMPA and NMDA receptor gene expression after global ischemia. Brain Res Mol Brain Res. 2001;86(1-2):34–47. doi: 10.1016/s0169-328x(00)00252-7. [DOI] [PubMed] [Google Scholar]
- 7.Garofalo L, Cuello AC. Nerve growth factor and the monosialoganglioside GM1: analogous and different in vivo effects on biochemical, morphological, and behavioral parameters of adult cortically lesioned rats. Exp Neurol. 1994;125(2):195–217. doi: 10.1006/exnr.1994.1024. [DOI] [PubMed] [Google Scholar]
- 8.Kang TC, Hwang IK, Park SK, An SJ, Yoon DK, Moon SM, Lee YB, Sohn HS, Cho SS, Won MH. Chronological changes of N-methyl-D-aspartate receptors and excitatory amino acid carrier 1 immunoreactivities in CA1 area and subiculum after transient forebrain ischemia. J Neurocytol. 2001;30(12):945–955. doi: 10.1023/a:1021832004954. [DOI] [PubMed] [Google Scholar]
- 9.Kharlamov A, Guidotti A, Costa E, Hayes R, Armstrong D. Semisynthetic sphingolipids prevent protein kinase C translocation and neuronal damage in the perifocal area following a photochemically induced thrombotic brain cortical lesion. J Neurosci. 1993;13(6):2483–2494. doi: 10.1523/JNEUROSCI.13-06-02483.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 11.Luo J, Bosy TZ, Wang Y, Yasuda RP, Wolfe BB. Ontogeny of NMDAR1 subunit protein expression in five regions of rat brain. Brain Res Dev Brain Res. 1996;92(1):10–17. doi: 10.1016/0165-3806(95)00191-3. [DOI] [PubMed] [Google Scholar]
- 12.Luo J, Wang Y, Yasuda RP, Dunah AW, Wolfe BB. The majority of n-methyl-d-aspartate receptor Complexes in adult rat cerebral cortex contain at least three different subunits (NMDAR1/NMDAR2A/NMDAR2B) Mol Pharmacol. 1997;51(1):79–86. doi: 10.1124/mol.51.1.79. [DOI] [PubMed] [Google Scholar]
- 13.Manev H, Favaron M, Vicini S, Guidotti A, Costa E. Glutamate-induced neuronal death in primary cultures of cerebellar granule cells: protection by synthetic derivatives of endogenous sphingolipids. J Pharmacol Exp Ther. 1990;252(1):419–427. [PubMed] [Google Scholar]
- 14.Michaels RL, Rothman SM. Glutamate neurotoxicity in vitro: antagonist pharmacology and intracellular calcium concentrations. J Neurosci. 1990;10(1):283–292. doi: 10.1523/JNEUROSCI.10-01-00283.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monaghan DT, Bridges RJ, Cotman CW. The excitatory amino acid receptors: their classes, pharmacology, and distinct properties in the function of the central nervous system. Annu Rev Pharmacol Toxicol. 1989;29:365–402. doi: 10.1146/annurev.pa.29.040189.002053. [DOI] [PubMed] [Google Scholar]
- 16.Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischemia is mainly by reversed uptake. Nature. 2000;403(6767):316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- 17.Simon RP, Chen J, Graham SH. GM1 ganglioside treatment of focal ischemia: a dose-response and microdialysis study. J Pharmacol Exp Ther. 1993;265(1):24–29. [PubMed] [Google Scholar]
- 18.Wahlestedt C, Golanov E, Yamamoto S, Yee F, Ericson H, Yoo H, Inturrisi CE, Reis DJ. Antisense oligodeoxynucleotides to NMDA-R1 receptor channel protect cortical neurons from excitotoxicity and reduce focal ischaemic infarctions. Nature. 1993;363(6426):260–263. doi: 10.1038/363260a0. [DOI] [PubMed] [Google Scholar]
- 19.Won MH, Kang T, Park S, Jeon G, Kim Y, Seo JH, Choi E, Chung M, Cho SS. The alterations of N-Methyl-D-aspartate receptor expressions and oxidative DNA damage in the CA1 area at the early time after ischemia-reperfusion insult. Neurosci Lett. 2001;301(2):139–142. doi: 10.1016/s0304-3940(01)01625-1. [DOI] [PubMed] [Google Scholar]
- 20.Zang LH, Wei EQ. Neuroprotective effect of ONO-1078, a leukotriene receptor antagonist, on transient global cerebral ischemia in rats. Acta Pharmacol Sin. 2003;24(12):1241–1247. [PubMed] [Google Scholar]