

Abstract
Objective: To construct a cDNA library from human liver tissue with chronic hepatitis B and check its quality for investigating the expression level of liver tissue infected by hepatitis B virus. This will then be used to find the relevant genes and interesting proteins associated with the development of hepatitis B. Methods: The total RNA from liver tissue with chronic hepatitis B was extracted and the mRNA was purified using TRIZOL method. Switching mechanism at 5′ end of the RNA transcript (SMART) technique and CDS III/3′ primer were used for first-strand cDNA synthesis. Long distance polymerase chain reaction (LD PCR) was then used to synthesize the double-strand cDNA that was then digested by Sfi I and fractionated by CHROMA SPIN-400 column. The longer than 0.4 kb cDNAs were collected and ligated to λTriplEx2 vector. Then λ phage packaging reaction and library amplification were performed. The qualities of both unamplified and amplified cDNA libraries were strictly checked by conventional titer determination. Fourteen plaques were randomly picked and tested using PCR with universal primers derived from the sequence flanking the vector. Results: The titers of unamplifed and amplified libraries were 1.94×106 pfu/ml and 1.49×109 pfu/ml respectively. The percentages of recombinants from both libraries were 98.15% in unamplified library and 98.76% in amplified library. The lengths of the inserts were 1.23 kb in average, 1–2 kb in 64.29%, and 0.5–1.0 kb in 35.71%. Conclusion: A high quality cDNA library from human liver tissue with chronic hepatitis B was successfully constructed.
Keywords: cDNA library, Human liver tissue, Chronic hepatitis B, Construction and characterization
INTRODUCTION
Chronic hepatitis B virus (HBV) infection is a serious clinical problem because of its wide distribution and possible adverse consequences, such as hepatic decompensation, cirrhosis and/or primary liver cancer (PLC). The natural course of chronic HBV infection is characterized by a series of hepatitic flares or exacerbations and remissions (Ganem and Prince, 2004). The severity, extent, duration and frequency of hepatic histopathological changes in hepatitic flares are determinants for the development of liver cirrhosis, which is the most frequent background for PLC development (Liaw et al., 1988; Chin and Locarnini, 2003). However, the molecular pathogenesis of chronic hepatitis B is not very clear and the eradication therapy against chronic hepatitis B is not yet available. The molecular pathogenesis from the initial stage of chronic hepatitis B to end-stage liver disease with liver failure remains largely unknown.
Constructing a cDNA library will lay solid foundation for finding relevant genes and investigating their functions. Unlike genome DNA due to the introns in it, cDNA is not difficult to be expressed. So a cDNA library can be used not only to screen the target genes required, but also to express them (Sambrook and Russell, 2002). Screening of a breast cancer cDNA library from SKBR3 human breast cancer cells by use of a breast cancer patient’s preselected serum, INT-MI-1 and INT-MI-2 which encode novel gene products was reported (Forti et al., 2002). In the yeast 2-hybrid system, DBCCR1 (deleted in bladder cancer chromosome region 1), HIV-2 gag, CRP2 (LIM-domain protein cysteine- and glycine-rich protein 2) expressed in stellate cells and PA subunit of the influenza virus polymerase complex were respectively used to screen an adult human bladder cDNA library, a T cell library and a kidney library, and new gene proteins of ASML3a (acid sphingomyelinase-like phosphodiesterase 3a), Tsg101, PIAS1 (protein inhibitor of activated STAT1) and CLE which interacted with DBCCR1, HIV-2 gag, CRP2 and PA respectively were reported (Wright et al., 2002; Myers and Allen, 2002; Weiskirchen et al., 2001; Huarte et al., 2001).
A cDNA library can be constructed from human liver tissue with chronic hepatitis B and be used to find the relevant genes and interesting proteins associated with the development of hepatitis B. For example, single strand cDNAs reverse transcribed from mRNA extracted from liver tissue with chronic hepatitis B, can be used to hybridize with excessive cDNAs from healthy liver tissue. Then the remainder of the cDNAs (differentially expressed in chronic hepatitis B) can be used as substruct cDNA probes for screening a cDNA library from human liver tissue with chronic hepatitis B to search for the homologous clone. By this means, we could obtain gene specially expressed in chronic hepatitis B. Also in the yeast 2-hybrid system, cDNA library from human liver tissue with chronic hepatitis B can be used to screen the protein which interacted with the given protein, such as HBsAg. Thus, construction of a cDNA library from human liver tissue with chronic hepatitis B will help in gaining better understanding of the molecular pathogenesis of chronic hepatitis B. We constructed a cDNA library from liver tissue with chronic hepatitis B in order to find relevant genes and proteins of interest that might play important roles in the development of chronic hepatitis B. And this information will then be used to find the relevant genes and interesting proteins associated with the development from chronic hepatitis B to the end-stage liver disease by comparison with other 3 cDNA libraries from healthy human, cirrhosis and PLC liver tissues.
METHODS
The liver tissue was sampled from a patient with chronic hepatitis B who was admitted to the Second Affiliated Hospital of Zhejiang University and gave consent to the operation of excising part of the liver. The excised liver tissue was placed into liquid nitrogen immediately after being excised and the active chronic hepatitis B was pathologically confirmed.
Extraction of total RNA
Eighty-nine milligram of fresh liver tissue was ground in liquid nitrogen in an autoclaved mortar into a pile of powder. Trizol, chloroform and isopropyl alcohol were used respectively to homogenize the sample, and separate the aqueous phase of RNA and sendiment RNA. The RNA pellet was washed once with 75% ethanol and briefly vacuum-dried. In the end, the RNA pellet was dissolved in 30 μl diethyl pyrocarbonate (DEPC) H2O. The integrity of the total RNA was analyzed by 1.1% agarose gel electrophoresis alongside RNA marker (SM0421, Fermentas, Lithuania), and the purity of the total RNA was checked by the ratio of OD260/OD280.
Purification of mRNA
The total RNA sample was processed according to the protocol of the mRNA purification kit (Sangon, China). At the end of the procedure, the mRNA was dissolved in 8 μl R-Elution Buffer. The integrity of the mRNA was analyzed by 1.1% agarose gel electrophoresis alongside RNA marker as described above.
cDNA synthesis
1. First-strand cDNA synthesis: First-Strand cDNA was synthesized according to the protocol of SMART™ cDNA Library Construction Kit (Clontech, USA). Three μl mRNA sample, SMART IV Oligonucleotide and CDS III/3′ PCR Primer were incubated at 72 °C for 2 min. Five×First-Strand buffer, DTT, dNTP Mix and PowerScript Reverse Transcriptase were incubated at 42 °C for 1 h.
2. Amplification of cDNA by LD PCR: Two μl First-Strand cDNA, deionized H2O, Advantage 2 PCR Buffer, dNTP Mix, 5′ PCR Primer, CDS III/3′ PCR Primer and Advantage 2 Polymerase Mix were added into a new pre-chilled 0.5 ml EP tube and was amplified by the following program: 95 °C 20 s; 24 cycles of 95 °C 5 s, 68 °C 6 min. Five μl of the PCR product was taken for analysis by running 1.1% agarose/EtBr gel alongside DNA marker (SM0331, Fermentas, Lithuania) and the concentration of the double strand (ds) cDNA was roughly estimated.
3. Proteinase K digestion: Fifty μl of amplified ds cDNA and 2 μl of proteinase K (20 μg/μl) were added into a 0.5 ml sterile EP tube and incubated at 45 °C for 20 min.
4. PCR product purification: PCR product purification was carried out according to the protocol of Qiaquick PCR Purification Kit (Qiagen, Germany). Finally, 30 μl of deionized H2O (pH 8.0) was used to elute purified DNA. Deionized H2O was added up to 79 μl.
5. Sfi I digestion: Sfi Buffer, Sfi I Enzyme and BSA were added into a 0.5 ml EP tube containing 79 μl cDNA and incubated at 50 °C for 2 h; and then xylene cyanol FF was added into and mixed well in the tube.
6. cDNA size fractionation by CHROMA SPIN-400: The cDNA size fractionation was carried out using CHROMA SPIN-400 columns according to the protocol of CHROMA SPIN-400. Sixteen fractions were collected in the separated tubes. Three μl of each fraction was taken to run 1.1% agarose/EtBr gel alongside 0.1 μg of 1 kb DNA size marker (N3232V, NEW ENGLAND Biolabs, USA) at 150 V for 3 min. The peak fractions were determined by visualizing the intensity of the bands under UV; the first three tubes of fractions containing cDNA were put together into a clean 1.5 ml EP tube. Sodium acetate, glycogen and 95% ethanol (−20 °C) were added and placed in −20 °C freezer overnight and then centrifuged. The supernatant was carefully removed, and the pellet was washed by 70% ethanol and vacuum-dried. Five μl deionized H2O was added to resolve the pellet, which was then kept at −20 °C.
Construction of a cdna library
1. Ligation of cDNA to λTriplEx2 vector: cDNA and λTriplEx2 vector (Clontech, USA) were ligated according to the ratios of 1:2, 1:1 and 2:1; the reaction mixture of cDNA, λTriplEx2 vector, 10×ligation buffer, T4 DNA ligase, ATP and deionized H2O was incubated at 16 °C overnight.
2. λ phage packaging: λ phage packagings in vitro for each ligation to produce unamplified cDNA libraries were set up according to the protocol of MaxPlax™ Packaging Extract (Epicentre, USA), and the products were stored at 4 °C.
3. Titering the unamplified library: XL1-Blue working stock plate was prepared using LB/tet, from which a single isolated colony was picked up and used to inoculate LB/MgSO4/maltose broth to prepare the XL1-Blue overnight culture. The culture was centrifuged, the pellet was resuspended in MgSO4. Made a 1:10 dilution of each of the packaging products, from which 1 μl of the diluted phage was taken and added to 200 μl of the XL1-Blue overnight culture, and the phage was allowed to be preabsorbed at 37 °C for 15 min. Two ml of melted LB/MgSO4 top agar was added to each mixture of XL1-Blue and phage, and were poured onto 90 mm LB-agar/MgSO4 plates pre-warmed to 37 °C. The plates were inverted after being cooled and incubated at 37 °C for 8 h. The plaques were counted and the titer of the phage (pfu/ml) was calculated as: pfu/ml=(number of plaques)×(dilution factor)×(103 μl/ml)/(μl of diluted phage for plating).
4. Determining the percentage of recombinant clones in unamplified library: Two ml of melted LB/MgSO4 top agar was added to each mixture of 200 μl of the XL1-Blue overnight culture, 1 μl of the diluted phage, 50 μl IPTG stock solutions (0.1 mol/L) and 50 μl X-gal stock solutions (0.1 mol/L) into a sterilized tube and then poured onto 90 mm LB/MgSO4 plates pre-warmed to 37 °C. The plates were inverted after being cooled and were incubated at 37 °C for 8 h. The ratio of white plaques (recombinants) to blue plaques (non-recombinants) rapidly estimated as the recombination efficiency. The percentage of the recombinants=(number of white plaques)×(100%)/(number of white plaques+number of blue plaques).
5. Library amplification: The λ lysate packaged product with the most efficient ligation ratio of cDNA and λTriplEx2 vector were put into 5 ml sterilized tube with 500 μl of XL1-Blue overnight culture and incubated in a 37 °C water bath for 15 min. After 4.5 ml of melted LB/MgSO4 top soft agar was added into each tube, the mixture was poured onto LB/MgSO4 agar plates. The plate was inverted after being cooled and incubated at 37 °C for 8 h. Twelve ml of 1×lambda dilution buffer was added to each plate. The plates were stored at 4 °C overnight, and then incubated on a platform shaker at 50 rpm at room temperature for 1 h. The phage lysates were mixed well and then poured into a sterile 50 ml polypropylene screw-cap microcentrifuge tube containing 10 ml of chloroform, vortexed for 2 min, centrifuged (Beckman J2-21 centrifuge) at 7 000 rpm for 10 min. The supernatant was transfered into another sterilized 50 ml centrifuge tube, and DMSO was added (final concentration 7%). The amplified library was put into 1.5 ml sterilized microcentrifuge tubes and stored at −70 °C.
Identification of the amplified library
1. Titering the amplified library: Ten μl of 1:100 000 diluted phage and 200 μl of the XL1-Blue overnight culture were added into a 5 ml sterilized tube, and the phage was allowed to be adsorbed at 37 °C for 15 min. Three ml of melted LB/MgSO4 top soft agar was added into the tube, and the mixture was poured onto a 90 mm LB/MgSO4 plate preheated to 37 °C. The plate was cooled, inverted and incubated at 37 °C for 7 h. The plaques were counted and the titer of the phage (pfu/ml) was calculated as: pfu/ml=(number of plaques)×(dilution factor)×(103 μl/ml)/(μl of diluted phage plated).
2. Determining the percentage of recombinant clones of amplified library: The percentage of recombinant clones in amplified library was determined in the same way as that for unamplified library as described above.
3. Identification of the cDNA inserts of the recombinants: Fourteen plaques were randomly picked with sterilized tooth sticks from plate into PCR tube with 20 μl deionized H2O and incubated at 100 °C for 10 min. And then Pyrobest DNA polymerase (TaKaRa, Japan), Pyrobest buffer, dNTP mixture, upstream primer (synthesised by Sangon Company, sequence: 5′-CTCCGAGATCTGGACGAGC-3′) and downstream prime (5′-TAATACGACTCACTATAGGG-3′) were added into the tube. PCR was then carried out according to the following program: 95 °C 3 min; 94 °C 30 s, 55 °C 30 s, 72 °C 2 min for 30 cycles; 72 °C 7 min. PCR products were checked by running 1.1% agarose gel alongside DNA marker (DL2000, TaKaRa, Japan).
RESULTS
Analysis of total RNA and mRNA
High quality isolation of total RNA and purification of mRNA are critical steps for constructing a cDNA library. In our study the total RNA (Fig.1) appeared as a long smear with clear bands of 28 S and 18 S. The ratio of OD260/OD280 to the total RNA was 1.89, and the concentration was 1.178 μg/μl. Therefore the conclusion could be drawn that the total RNA obtained from liver tissue did not degrade obviously and that the purity was high. Fig.2 showing that the smear of mRNA was so long indicated integrity of mRNA had been achieved.
Fig. 1.
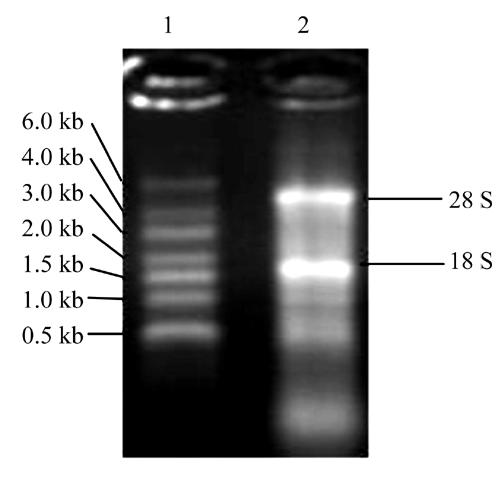
1.1% agarose gel electrophoresis of total RNA
1: RNA Marker SM0421; 2: Total RNA
Fig. 2.

1.1% agarose gel electrophoresis of mRNA
1: RNA Marker SM0421; 2: mRNA
Analysis of LD PCR product
The concentration of ds cDNA was 0.049 μg/μl. The long smear of ds cDNA (Fig.3) distributed from 0.2 kb to 10.0 kb or longer, which indicated LD PCR was successful.
Fig. 3.
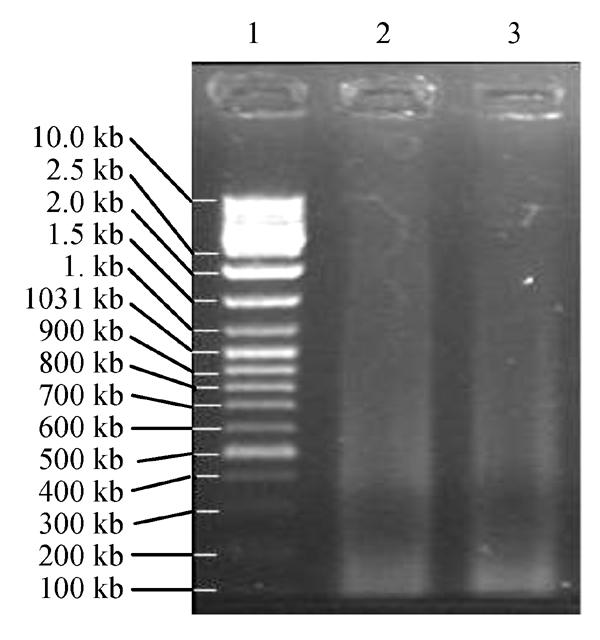
1.1% agarose gel electrophoresis of LD PCR products. 1: DNA Marker SM0331; 2, 3: LD PCR Products
Analysis of fractionated cDNA
The smear of the fractionated cDNA (Fig.4) mostly distributed longer than 0.5 kb.
Fig. 4.

1.1% agarose gel electrophoresis of fractionated cDNA. 1: DNA Marker N3232V; 2: Fractionated cDNA
CONSTRUCTION AND CHARACTERIZATION OF THE cDNA LIBRARY
Titering the unamplified library and determining the percentage of the recombinants
The titers of three unamplifed libraries corresponding to the cDNA and λ phage ligation ratio of 1:2, 1:1 and 2:1, were 1.9×105 pfu/ml, 1.94×106 pfu/ml and 2.7×105 pfu/ml respectively. And the percentages of the recombinants for the three different ligation ratios were 93.42%, 98.15% and 96.38% respectively.
Titering the amplified library and determining the percentage of the recombinants
The λ lysate package with the ligation ratio of 1:1 was taken and amplified (Fig.5), and tittered then (Fig.6). The titer of the amplified library was 1.49×109 pfu/ml, and the percentage of the recombinants was 98.76% according to the Blue and White screening (Fig.7).
Fig. 5.
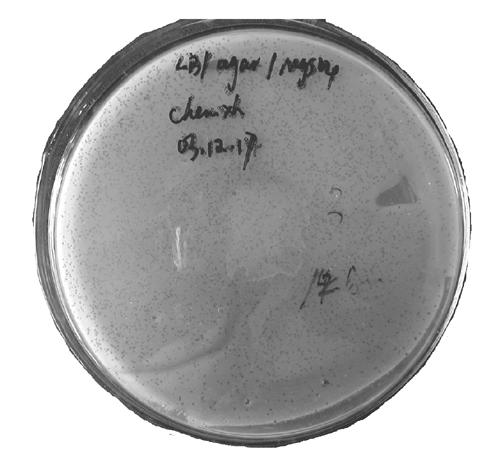
Library amplification. The dark dots bestrewed on the culture densely are plaques, some of them fused to small pieces
Fig. 6.

Titering the amplified library. The grey dots on the culture are plaques, the total of them is 149
Fig. 7.
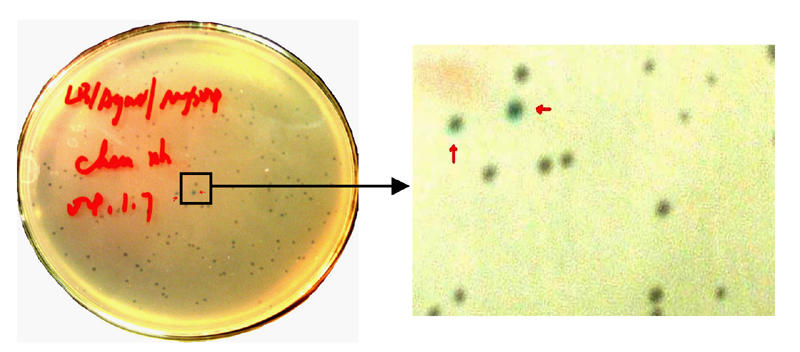
Blue and white screening of the amplified library. The blue plaques marked by red arrows were uncombinants
Identification of the cDNA inserts of the recombinants
Fourteen plaques were randomly picked and amplified by PCR using the universal primers. The sizes of PCR products were between 1–2 kb for 9 samples (64.29%), 0.5–1.0 kb for 5 samples (35.71%) (Fig.8).
Fig. 8.
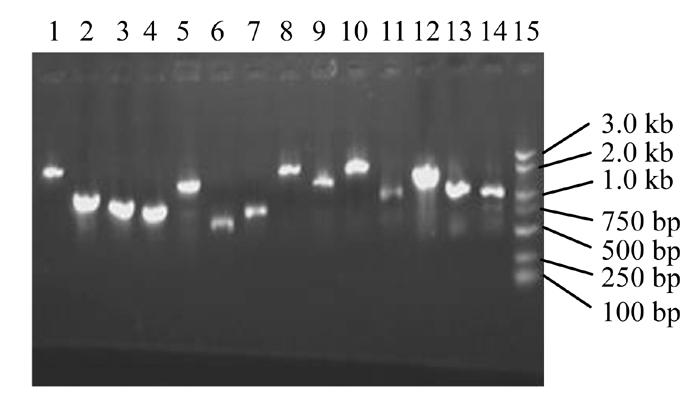
1.1% agrose gel electrophoresis of PCR products of inserts. 1: DNA Marker DL2000; 2~12: Products of inserts
DISCUSSION
The exploitation and utilization of genetic resources play an important role in research on structure and function of genes. A cDNA library constructed from human liver tissue with chronic hepatitis B will be of help in acquiring the expression spectrum of the liver tissue infected by HBV and finding the interesting genes associated with the development of hepatitis B.
SMART technique is a novel and useful method for constructing cDNA libraries. Its important characteristic is that it provides a method for producing high-quality and full-length cDNA libraries that preserve the complete 5′ terminal sequence of mRNA. Conventional methods for constructing cDNA library have two defects: Firstly, they contain fewer cDNA clones preserving the complete 5′ terminal sequence of mRNA. Secondly, it is difficult to determine which cDNA clone preserves the complete 5′ terminal sequence of mRNA by conventional cDNA library methods.
cDNA libraries constructed by SMART method contain full-length cDNA preserving the complete 5′ terminal sequence of mRNA. It was achieved because of 3 reasons: firstly, although PowerScript™ Reverse Transcriptase lacks RNase H activity, it retains wild-type polymerase activity so that it can synthesize longer first-strand cDNA than wild-type. Secondly, LD PCR (Barnes, 1994) overcame the difficulty of intermingling the un-matched bases into the 3′ terminal of the extending strand, which made it possible to amplify the long distance template DNA. Moreover, the SMART IV Oligo serves as a short, extension template at the 5′ end of the mRNA and a modified oligo (dT) primer (CDS III/3′ PCR Primer) is used to lead the first-strand synthesis. The sequence complementary to the SMART IV Oligo then serves as a universal priming site (SMART anchor) in the amplification by LD PCR. Only those ss cDNAs having a SMART anchor sequence at the 5′ end can be exponentially amplified. Incomplete cDNAs or cDNA synthesized through only polyA could not be amplified in the next experiment as they lacked of the SMART anchor. Thirdly, Sfi I restriction enzyme cutting the sites at both 5′ and 3′ ends of cDNA are extremely rare in mammalian DNA, almost all SMART cDNAs therefore, remain intact after Sfi I digestion.
It is convenient expressing insert gene in all three reading frames or transferring inserts from phage into plasmid via Cre-lox-mediated subcloning after cDNA library successfully used SMART technique and λTriplEx2 that containing two translation start sites.
Low abundance mRNA (<14 copy/cell) exists in about 30% of all mRNA. According to Clareke-Carbon’s formula, a cDNA library should contain at least 1.7×105 independent clones (Sambrook and Russell, 2002), so that a clone derived from low abundance mRNA would be screened out with 99 percent probability from the library. The capacity of the cDNA library we constructed was 1.49×109 pfu/ml, which could meet almost all requirements of finding a cDNA clone derived from low abundance mRNA. So it was a successful cDNA library.
The liver tissue used to construct the cDNA library was sampled from a Chinese patient with chronic hepatitis B, and to our knowledge, the library was China’s first cDNA library from liver tissue of a Chinese patient with chronic hepatitis B. It may contribute to the research on molecular pathogenesis and gene therapy of patients with chronic hepatitis B.
Chronic hepatitis B virus infection is a serious clinical problem because of its wide distribution and possible adverse consequences, such as hepatic decompensation, cirrhosis and/or primary liver cancer. HBV is not itself cytopathic. Instead, chronic HBV infection is a dynamic state of interaction among HBV, hepatocytes and the immune system of the host. The molecular mechanism of why and how some patients with chronic hepatitis B infection develop to hepatic decompensation, cirrhosis and/or PLC still remains largely unknown. We successfully constructed this cDNA library from liver tissue with chronic hepatitis B as well as 3 other cDNA libraries from healthy human, cirrhosis and PLC liver tissues aimed at gaining further understanding of the mechanism and finding related genes and interesting proteins that may play important roles in the development of cirrhosis and PLC from chronic hepatitis B.
CONCLUSION
A high quality cDNA library from human liver tissue with chronic hepatitis B was successfully constructed.
Footnotes
Project (No. 30371270) supported by the National Natural Science Foundation of China
References
- 1.Barnes WM. PCR amplification of up to 35-kb DNA with high fidelity and high yield from λ bacteriophage templates. Proc Natl Acad Sci. 1994;91(6):2216–2220. doi: 10.1073/pnas.91.6.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chin R, Locarnini S. Treatment of chronic hepatitis B: current challenges and future directions. Rev Med Viro. 2003;13(4):255–272. doi: 10.1002/rmv.393. [DOI] [PubMed] [Google Scholar]
- 3.Forti S, Scanlan MJ, Invernizzi A, Castiglioni F, Pupa S, Agresti R, Fontancelli R, Morelli D, Old LJ, Pupa SM, et al. Identification of breast cancer-restricted antigens by antibody screening of SKBR3 cDNA library using a preselected patient’s serum. Breast Cancer Res Treat. 2002;73(3):245–256. doi: 10.1023/a:1015854415746. [DOI] [PubMed] [Google Scholar]
- 4.Ganem D, Prince AM. Hepatitis B virus infection–natural history and clinical consequences. N Engl J Med. 2004;350(11):1118–1129. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 5.Huarte M, Sanz-Ezquerro JJ, Roncal F, Ortin J, Nieto A. PA subunit from influenza virus polymerase complex interacts with a cellular protein with homology to a family of transcriptional activators. J Virol. 2001;75(18):8597–8604. doi: 10.1128/JVI.75.18.8597-8604.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liaw YF, Tai DI, Chu CM, Chen TJ. The development of cirrhosis in patients with chronic type B hepatitis: a prospective study. Hepatology. 1988;8(3):493–496. doi: 10.1002/hep.1840080310. [DOI] [PubMed] [Google Scholar]
- 7.Myers EL, Allen JF. Tsg101, an inactive homologue of ubiquitin ligase e2, interacts specifically with human immunodeficiency virus type 2 gag polyprotein and results in increased levels of ubiquitinated gag. J Virol. 2002;76(22):11226–11235. doi: 10.1128/JVI.76.22.11226-11235.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiskirchen R, Moser M, Weiskirchen S, Erdel M, Dahmen S, Buettner R, Gressner AM. LIM-domain protein cysteine and glycine-rich protein 2 (CRP2) is a novel marker of hepatic stellate cells and binding partner of the protein inhibitor of activated STAT1. Biochem J. 2001;359(3):485–496. doi: 10.1042/0264-6021:3590485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wright KO, Messing EM, Reeder JE. Increased expression of the acid sphingomyelinase-like protein ASML3a in bladder tumors. J Urol. 2002;168(6):2645–2649. doi: 10.1016/S0022-5347(05)64236-X. [DOI] [PubMed] [Google Scholar]
- 10.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd Ed. Cold Spring Harbor Lab Press; 2002. pp. 11.8pp. 11.26–11.35. [Google Scholar]