

Abstract
Our understanding of prokaryote biology from study of pure cultures and genome sequencing has been limited by a pronounced sampling bias towards four bacterial phyla - Proteobacteria, Firmicutes, Actinobacteria and Bacteroidetes - out of 35 bacterial and 18 archaeal phylum-level lineages. This bias is beginning to be rectified by the use of phylogenetically directed isolation strategies and by directly accessing microbial genomes from environmental samples.
It is a common misconception that microorganisms isolated in pure culture from an environment represent the numerically dominant and/or functionally significant species in that environment. In fact, microorganisms isolated using standard cultivation methods are rarely numerically dominant in the communities from which they were obtained: instead, they are isolated by virtue of their ability to grow rapidly into colonies on high-nutrient artificial growth media, typically under aerobic conditions, at moderate temperatures. Easily isolated organisms are the 'weeds' of the microbial world and are estimated to constitute less than 1% of all microbial species (this figure was estimated by comparing plate counts with direct microscopic counts of microorganisms in environmental samples; it has been called the "great plate-count anomaly" [1]).
Given that the study of a microorganism is simpler if you have it in pure culture on an agar plate, it is not surprising that most of what we know about microbiology comes from the study of microbial weeds. For example, approximately 65% of published microbiological research from 1991 to 1997 was dedicated to only eight bacterial genera, Escherichia (18%), Helicobacter (8%), Pseudomonas (7%), Bacillus (7%), Streptococcus (6%), Mycobacterium (6%), Staphylococcus (6%) and Salmonella (5%) [2], all of which are relatively simple to grow on agar plates. Intuitively, it seems unlikely that this handful of organisms can be representative of the approximately 5,000 validly described prokaryotic species [3], but exactly how unrepresentative are they? And if more than 99% of microorganisms in the environment are unculturable using standard techniques, how representative are cultivated microorganisms of prokaryotic diversity as a whole? To answer these questions, we need a framework for placing prokaryotic species and genera in a broader evolutionary context.
A molecular-phylogenetic framework for mapping biodiversity
The pioneering work of Carl Woese and colleagues [4,5] on comparative analysis of small-subunit ribosomal RNAs (16S and 18S rRNAs) provided an objective framework for determining evolutionary relationships between organisms and thereby 'quantifying' diversity as sequence divergence on a phylogenetic tree. Woese found that cellular life can be divided into three primary lineages (domains), one eukaryotic (Eucarya, also called Eukaryota) and two prokaryotic (Bacteria and Archaea), and he also defined 11 major lineages (phyla or divisions) within the bacterial domain on the basis of 16S rRNA sequences obtained from cultivated organisms [5]. This analysis revealed distant relationships not suspected from phenotypic characterization, such as the association between the genera Bacteroides and Flavobacterium.
The leading reference source in prokaryotic taxonomy, Bergey's Manual of Systematic Bacteriology, has adopted a 16S rRNA framework to classify prokaryotes [6], replacing the previous ad hoc scheme that was based on traditional phenotypic characterization [7]. The Manual proposes a standardized prokaryote nomenclature that has mostly been fitted to a classical taxonomic hierarchy (species, genus, family, order, class, phylum); I will adhere to this system as far as possible in this article (see the taxonomic outline available at [8]). The phylum is the highest-level grouping in the bacterial and archaeal domains [9] and, therefore, is a useful rank for overviewing prokaryotic diversity.
The eight most intensively studied prokaryotic genera listed in the introduction are members of only three bacterial phyla: Proteobacteria (Escherichia, Helicobacter, Pseudomonas, Salmonella), Firmicutes (Bacillus, Streptococcus, Staphylococcus) and Actinobacteria (Mycobacterium). Moreover, the top 25 most-studied genera are all members of these three phyla, with the exceptions of Chlamydia and Borrelia (clinically important genera of the bacterial phyla Chlamydiae and Spirochaetes, respectively) [2]. In a recent study, 177 environmental, veterinary and clinical isolates that were not identifiable by traditional phenotypic characterization were evaluated by comparative 16S rRNA analysis [10]. The isolates included a large number of different genera and species, but at the phylum level all except one of the 177 were members of only four bacterial phyla: Proteobacteria (82 isolates), Firmicutes (61), Actinobacteria (29) and Bacteroidetes (4). This cultivation bias towards four bacterial phyla (the 'big four') is also reflected in microbial culture collections; for example, 97% of prokaryotes deposited in the Australian Collection of Microorganisms [11] are members of the big four (Figure 1a). In fact, it is a challenge to obtain isolates that do not belong to the big four, and these four phyla therefore dominate our present understanding of microbiology. A logical question to ask is how many prokaryotic phyla there are altogether, in order to estimate how biased a sampling of four may be.
Figure 1.
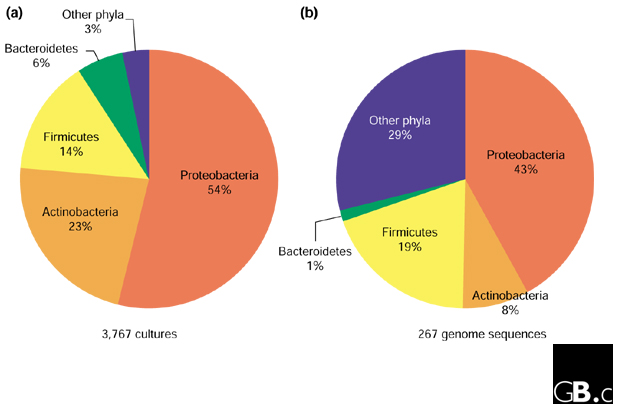
Pie charts showing the phylum-level distribution of prokaryotic isolates (a) in the Australian Collection of Microorganisms [11] and (b) in the prokaryote genome sequences completed or in progress as of 20 August 2001 [29].
Prokaryotic diversity beyond the weeds
In the mid 1980s, Norman Pace and colleagues outlined a molecular approach that bypassed the need to cultivate a microorganism in order to determine the sequence of its 16S rRNA gene (16S rDNA) [12]. Essentially, bulk nucleic acids are extracted directly from environmental samples, 16S rDNA sequences are isolated from the bulk DNA, typically via PCR (using primers broadly targeting 16S rDNAs) and cloning, and these sequences are compared with known sequences (Figure 2). Gene sequences obtained in this manner ('environmental clone sequences') can then be assigned a location in a phylogenetic tree and can thus act as a marker for the organism from which they were obtained. The approach can be brought full circle by applying 16S rRNA-targeted nucleic-acid probes specific for the organisms of interest to visualize and quantify the target group in the environmental sample using techniques such as whole-cell fluorescence in situ hybridization (FISH) and membrane hybridization [13] (Figure 2).
Figure 2.
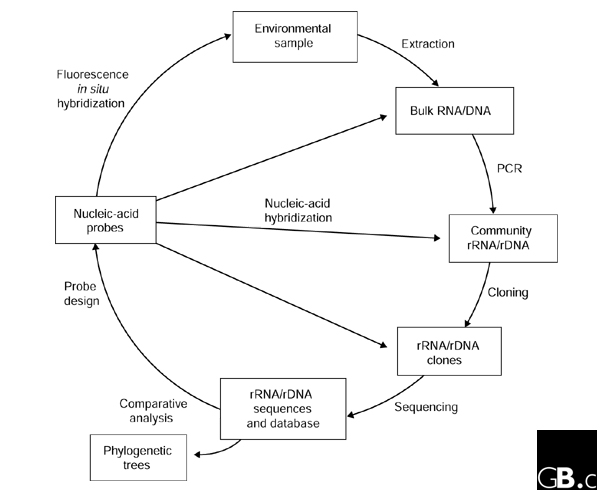
'Full-cycle' rRNA approach to characterizing microorganisms in their natural settings without the need for cultivation. Access to whole genomes of uncultivated organisms is also possible using the same basic approach but with large-insert cloning vectors, such as BACs, which remove the need for PCR.
Many researchers have applied the rRNA approach to a wide variety of environmental samples over the past decade and, perhaps not surprisingly given the great plate-count anomaly, the number of recognized bacterial phyla has exploded from the original estimate of 11 in 1987 [5] to 36 in 1998 [14]. This increase is due not only to environmental sequences that have filled out the tree, but also to a steady trickle of sequences from 'exotic' cultured organisms, particularly thermophiles, that highlight new lineages. Figure 3a presents a recent conservative estimate of bacterial diversity at the phylum level; it is conservative because it includes only phyla for which at least four near-full-length 16S rDNA sequences (over 1,300 nucleotides) are known. The total number of phylum-level lineages in this tree is 35, 22 (63%) of which have one or more cultivated representatives and 13 (37%) of which are known only from environmental sequences. There are at least another ten phylum-level lineages, however, that are present in the bacterial domain but are not shown in Figure 3a because they are represented by too few and/or only partial sequences. These lineages include cultivated bacteria such as Chrysiogenes and Dictyoglomus, which are recognized as representing independent phyla in the taxonomic outline of Bergey's Manual of Systematic Bacteriology [8]. The latest tally of bacterial phyla is therefore probably nearer 45.
Figure 3.
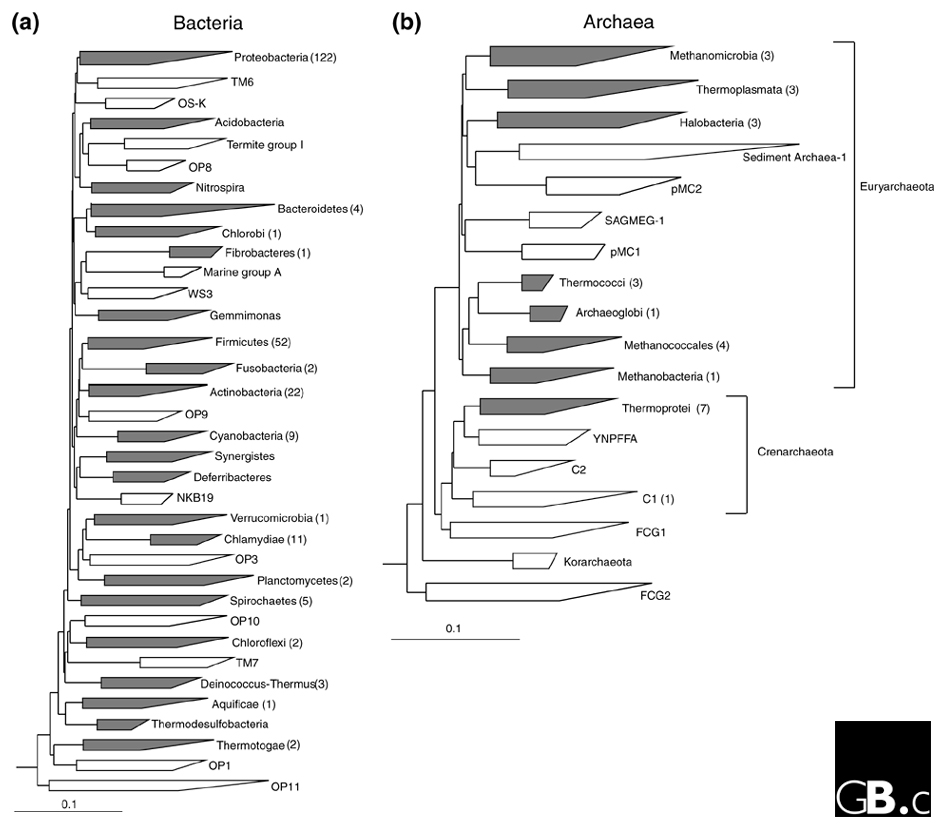
Evolutionary distance dendrograms of (a) bacterial and (b) archaeal diversity derived from comparative analysis of 16S rRNA gene sequences. The trees were constructed using the ARBsoftware package and a sequence database modified from the March 1997 ARB database release [39] using 50% consensus sequence filters for each domain and the Olsen correction and neighbor-joining options. This modified database will be available from the Ribosomal Database Project [40] user-submitted alignments download site [41]. Major lineages (phyla) are shown as wedges with horizontal dimensions reflecting the known degree of divergence within that lineage. Phyla with cultivated representatives are in gray and, where possible, named according to the taxonomic outline of Bergey's Manual [8]. Phyla known only from environmental sequences are in white; because they are not formally recognized as taxonomic groups, they are usually named after the first clones found from within the group [14,20]. Note that environmental groups E2 and E3 defined in [20] are part of the Thermoplasmata phylum in the archaeal tree in (b). The number of genome sequences completed or in progress for each phylum is given in brackets after the phylum name, with the exception of Methanopyrus kandleri, which is not included in the tree because it is represented by a single sequence. The scale bar represents 0.1 changes per nucleotide.
As more 16S rDNA sequences accumulate from both cultured and uncultured prokaryotes, the boundaries of existing phyla are being challenged and need to be re-evaluated. For example, the bacterial phylum Firmicutes, as currently defined [8], may not be monophyletic and may comprise at least four distinct phylum-level lineages that include the Haloanaerobiales, Thermoanerobacteriales, and Sulfobacillus groups [9]. Higher-level associations between bacterial phyla have not been resolved in 16S rDNA trees, with the exceptions of the sister-group affiliations of the Bacteroidetes and Chlorobi, and of the Chlamydiae and Verrucomicrobia [14]. This is presumably because such relationships are beyond the resolution that can be obtained from the 16S rRNA molecule and/or the current inference methods [9,14]. Recently, trees based on concatenated ribosomal proteins obtained from complete genome sequences have suggested higher-order associations between Chlamydiae and Spirochaetes, between Thermotogae and Aquificae, and between Actinobacteria, Deinococcus-Thermus and Cyanobacteria [15]. The phylum Verrucomicrobia is also likely to be a member of the same group as Chlamydiae and Spirochaetes, given that it is a sister group to Chlamydiae; this prediction can be tested when a completed genome sequence becomes available for the Verrucomicrobia.
Several 'candidate' phyla [16], comprising only environmental clone sequences, have developed into large groups with sequence divergences similar to or greater than those within the big four phyla (examples include OP11 [14] and WS6 [16]), and yet we know nothing about these lineages beyond a crude outline of their environmental distribution. Most have not even been (knowingly) observed under the microscope. In a preliminary investigation of one candidate phylum, TM7, we determined that representatives of the group had typical Gram-positive cell envelopes and that they may have Archaea-like streptomycin resistance [17]. Detailed study of lineages like this one may yield insights into the evolutionary history of Gram-positive bacteria (including, perhaps, a radical proposal that Gram-positive bacteria are related to Archaea [18]), which so far appear to have a restricted phylum-level distribution within the bacterial domain (Actinobacteria and Firmicutes). TM7 bacteria have also been implicated in human subgingival (gum) disease, which might promote their study [19].
The Archaea are formally divided into two phyla, Crenarchaeota and Euryarchaeota, from 16S rRNA phylogeny [8], but these groupings may be artifacts because analysis of concatenated ribosomal protein sequences suggests that Euryarchaeota, at least, is not a monophyletic group [15]. Figure 3b presents a current estimate of the major lineages in the archaeal 16S rDNA tree below the level of the Crenarchaeota and Euryarchaeota (indicated to the right of the tree), using the same criteria and annotation used for the bacterial tree (Figure 3a). The total number of phylum-level lineages in the archaeal tree is 18, of which 8 (44%) have cultivated representatives and 10 (56%) have none. A higher tally of 23 phyla is arrived at if lineages not meeting the selection criteria are included in the estimate. These include Methanopyri [8], currently represented by a single sequence, and environmental group C3 [20], which has no full-length representatives. Most archaeal research has concentrated on the cultivated methanogenic (such as Methanococci) and thermophilic (such as Thermoprotei and Thermococci) lineages (Figure 3b). As is the case with the Bacteria, most candidate archeal phyla are completely uncharacterized at this point. A notable exception is candidate phylum C1 (Figure 3b), which contains Cenarchaeum symbiosum, an uncultured archaeon that has been amenable to detailed study, including partial genome sequencing, because it exists as a near monoculture in a marine sponge [21]. Members of the C1 group are particularly prevalent in marine habitats [22].
The bumpy transition from gene phylogeny to genome phylogeny
The advent of large-scale DNA sequencing has provided unprecedented access to molecular data for inferring the tree of life. Currently, complete genome sequences of prokaryotes have been obtained only from pure cultures and hence, at the phylum level, microbial genomics reflects the bias towards the big four phyla (Figures 1b,3). This bias (71% from the big four) is not as extreme as in culture collections (97%; Figure 1) because phyla containing human pathogens, such as Chlamydiae and Spirochaetes, are better represented by genome sequences (Figure 3a) [23], as are Archaea (Figure 3b). Increasing efforts are being made to select phylogenetically diverse prokaryotes (Archaea for example) for genome sequencing, using the 16S rRNA phylogeny as a guide [24].
But is selection solely on the basis of an exotic location in a 16S rRNA tree justified? The implicit assumption is that the evolutionary history of 16S rRNA represents the evolutionary history of the whole organism (the whole genome), but the concept of a unified organismal phylogeny has been significantly compromised by the finding of widespread lateral gene transfer (LGT) between organisms [25]. LGT appears to affect the informational genes (those involved in transcription and translation) to a lesser extent than metabolic and other operational genes, leading to the hypothesis that a core set of vertically transmitted informational genes define organismal phylogeny [26]. Recent evidence suggests that this may not be the case for the Euryarchaeota, however; here, informational genes are apparently no less subject to LGT than operational genes [27]. Reliable detection of LGT by comparison of gene trees is complicated by gene duplication and loss [23], and different methods for detecting LGT are not particularly consistent [28]. The extent to which LGT blurs organismal phylogenies is therefore unclear at this point. At one extreme, if genomes are largely chimeric assemblages of genes with different histories, then any random sampling of organisms should provide a representative 'window' into genome space. On the other hand, if a core of vertically transmitted genes (which includes 16S rDNA) defines the organism, then striving to obtain genome sequences from all major lineages in the 16S rRNA tree [24] seems justified. Either way, a more complete sampling of phyla defined using 16S rRNA should help to resolve the issue.
The number of prokaryote genome-sequencing projects completed or in progress as of 20 August 2001 [29] is shown for each phylum-level lineage in the bacterial (Figure 3a) and archaeal (Figure 3b) domains. Several bacterial phyla that have cultivated representatives have no sequenced genomes (Table 1). These should provide compelling targets for future genome-sequencing projects. Phylum-level lineages comprising only environmental clone sequences (Figure 3) also need to be sampled for genome sequences; this could best be achieved by obtaining one or more representatives of each phylum in pure culture.
Table 1.
Bacterial phyla with cultured representatives but without representative sequenced genomes
| Phylum* | Genera* |
| Acidobacteria | Acidobacterium, Geothrix, Holophaga |
| Chrysiogenetes | Chrysiogenes |
| Coprothermobacter† | Coprothermobacter |
| Deferribacteres | Deferribacter, Denitrovibrio, Flexistipes, Geovibrio |
| Dictyoglomus | Dictyoglomus |
| Gemmimonas | Gemmimonas |
| Nitrospira | Nitrospira, Leptospirillum, Magnetobacterium, Thermodesulfovibrio |
| Synergistes† | Synergistes, Aminobacterium, Aminomonas, Anaerobaculum, Dethiosulfovibrio, Thermanaerovibrio |
| Thermodesulfobacteria | Thermodesulfobacterium, Desulfatotherma, Geothermobacterium |
* Where possible, the nomenclature of the taxonomic outline for Bergey's Manual of Systematic Bacteriology [8] has been used. †These phyla are currently misclassified as part of the Firmicutes phylum [8].
Cultivating the uncultivated
The classical approach to cultivating microorganisms is to prepare a solid or liquid growth medium containing an appropriate carbon source, energy source and electron acceptor depending on the physiological type of organism being isolated. The medium is then inoculated with a suitable source of microorganisms and left to incubate at a desired growth temperature until organisms multiply to the point at which we become aware of their presence by colony formation or increased turbidity. This approach is not phylogenetically directed, however, and, as discussed above, typically ends up collecting fast-multiplying microbial weeds. To isolate representatives of novel environmental lineages, a directed form of cultivation is required. In one such approach, the first step is to select a target group and design group-specific oligonucleotide probes [30] to detect or visualize the target organisms in environmental samples (Figure 2). The probes can be used to screen a range of samples and, hopefully, to identify a habitat that is a rich source of the target group. The target organisms then need to be either selectively enriched on the basis of their phenotype or physically isolated from other non-target organisms present in the sample. As it is likely that we know nothing about the physiology of the target environmental group, physical isolation is the preferred route.
Several methods have been used successfully to physically isolate microorganisms, including sample dilution, filtration, micromanipulators and optical tweezers, density-gradient centrifugation, and cell sorting using flow cytometry (for an excellent review, see [31]). Sample dilution may work when the target organism is numerically dominant in a microbial community. The sample is simply diluted until only the target organism remains, albeit at a much lower cell density than in the starting material. Sample filtration separates cells according to size, so if the target group is particularly large or small, this might be useful for initial sorting away from the primary inoculum. Micromanipulators and optical tweezers are instruments for physically moving single cells or tight clusters of cells from a mixture of cells to fresh growth medium, where the cell(s) can grow in isolation. These methods are most suitable for isolation of large, morphologically conspicuous microorganisms, such as filaments. Density-gradient centrifugation separates cells according to buoyant density and may be useful for initial sorting of communities to enrich for the target organisms. Cell sorting by flow cytometry is a high-throughput method for quickly isolating target cells from a mixed culture; it is most suitable for singly-occurring cells because cell aggregates can interfere with the hydrodynamic focusing in the apparatus. When individual cells are being isolated (by micromanipulators or optical tweezers), the isolation procedure cannot be directly monitored by FISH because cells are killed (by fixation with paraformaldehyde) and isolated cells must be viable for the next step in culturing; procedures in which subsamples can be sacrificed (such as filtration or density-gradient centrifugation) can be monitored by FISH.
Once individual target cells have been physically isolated, a range of growth conditions can be tested to try to promote growth without the complication of overgrowth by non-target cells. Strategies include using habitat-simulating growth media, diffusion-gradient enrichments and longer incubation times (reviewed in [31,32]). Common growth media, such as tryptic soy agar, poorly simulate most natural habitats because they are overly substrate-rich; media that more closely resemble the inoculum habitat will therefore have a greater chance of supporting target-organism growth. The use of cell-free filtrates of the inoculum habitat as the basis for the growth medium is one way of achieving this. Diffusion-gradient enrichments facilitate rapid determination of the optimal growth conditions for two parameters at a time, such as pH and nutrient concentrations, usually applied as gradients over a solid or semisolid medium at right angles to each other. Finally, simply allowing inoculated growth media to incubate for longer periods than the standard overnight to two-week period may increase the chance of successful isolation of target organisms (see below). Throughout the process, progress can be monitored in subsamples using FISH or PCR.
A phylogenetically directed isolation approach has been successfully demonstrated for an archaeal clone sequence, pSL91, obtained from a hot spring [33]. Sequence-specific FISH probes were designed and applied to an enrichment from the hot spring, and grape-like cell clusters were highlighted by FISH. Clusters demonstrating this morphotype were then physically isolated using optical tweezers, grown in pure culture in a liquid medium and confirmed as the target archaeon by FISH. The pSL91 sequence represents a member of the Thermoprotei, however [8] (Figure 3b), and this phylum contains other cultivated representatives, including one genus, Desulfurococcus, relatively closely related to pSL91 (96% 16S rDNA sequence identity). This may have provided physiological clues as to how to grow the target organism, given that close phylogenetic relatives often (but not always) have similar phenotypes [32].
In some instances, cultivation of novel groups with unknown physiology may not be as difficult as imagined. For example, we discovered that micromanipulated filaments belonging to candidate phylum TM7 [17] (Figure 3a) could form colonies visible to the naked eye on low-nutrient solid media (R2A [34]) under aerobic conditions; the only catch was that they took 50 days to do so (P.H., G.W. Tyson, and L.L. Blackall, unpublished observations). This may be the case for a wide range of uncultivated organisms, with simple removal of the target organism from the weeds in the inoculum, and a little patience, being all that is required for success. There are likely to be many prokaryotes that will never be brought into pure culture, however, such as organisms that live in obligately interdependent relationships, because the conditions for their growth are too exacting (and thus cannot be reproduced in the laboratory). For such organisms, direct access to their genomes may be the only feasible approach.
Directly accessing microbial genomes from the environment
Genomes of uncultured prokaryotes can be accessed by a relatively straightforward adaptation of the rRNA approach (Figure 2). High-molecular-weight DNA extracted from environmental samples can be cloned directly into large-insert cloning vectors, such as cosmids or bacterial artificial chromosomes (BACs) [35]. With careful handling of the environmental DNA, this results in access to large contiguous portions of microbial genomes - 35-40 kilobases (kb) for cosmids and up to 200 kb for BACs - without the need for cultivation. BACs have the additional advantage that heterologous expression of some of the insert genes may be possible in the Escherichia coli host harboring the vector [35]. Clones can be sequenced using shotgun or chromosome-walking methods and comparatively analyzed (Figure 2). If a 16S rRNA gene or another conserved gene is identified in a clone then the phylogenetic identity of the genome segment can be determined.
Perhaps the most impressive application of this approach to date is the discovery of proteorhodopsin in an uncultured lineage of marine bacterioplankton belonging to the Gammaproteobacteria [36]. An open reading frame encoding proteorhodopsin was found on a 130 kb genomic fragment together with a 16S rDNA sequence identifying its owner as a member of the 'SAR86' group in the Gammaproteobacteria. Members of the SAR86 group had been detected on numerous occasions in culture-independent surveys of marine habitats, but no function could be inferred for them because there are no close cultivated representatives for the group. The discovery of proteorhodopsin, which is phylogenetically related to the light-driven proton pump bacteriorhodopsins, suggests that the SAR86 lineage lives phototrophically in the marine environment [36].
Ideally, we would like to reconstruct entire genomes from uncultured prokaryotes using large-insert cloning-vector approaches. This is a daunting task given the species complexity of most microbial communities and the genomic microheterogeneity within prokaryotic populations [21]. It will probably be an impossible task for habitats such as soil, containing thousands of individual genomes [37]. It remains to be seen, however, whether it is possible to reconstruct complete genomes from a low-diversity microbial community.
In conclusion, several major lineages of Bacteria (but not Archaea) containing isolated representatives lack even a single sequenced genome. Over a third of phylum-level prokaryotic lineages are represented exclusively by sequences of uncultured prokaryotes that have been repeatedly detected in culture-independent habitat surveys over the past decade. The mere existence of such large phylogenetically conspicuous groups, about which we know virtually nothing, should be reason enough to study them. Yet there remains a reluctance amongst many microbiologists to accept these 'virtual bacteria' [38] as bona fide members of the microbial world. By analogy, imagine that we were unaware of the Metazoa until a few years ago, when we began detecting them in environmental surveys using phylogenetic markers. Imagine that Metazoa-specific probes were designed to allow us to see this new group under the 'macroscope'. Our first viewing reveals a beetle, an octopus and an elephant. What do these creatures do for a living? What other organisms remain to be discovered in this group? This is approximately the stage we are at in the description of candidate prokaryotic phyla. At the very least, uncharacterized prokaryotic phyla will probably contain members with impressive physiological repertoires and interesting evolutionary histories, worthy of study and of genome sequencing.
Acknowledgments
Acknowledgements
I thank Mark Ragan, Norman Pace and George Garrity for providing comments on the manuscript.
References
- Staley JT, Konopka A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol. 1985;39:321–346. doi: 10.1146/annurev.mi.39.100185.001541. [DOI] [PubMed] [Google Scholar]
- Galvez A, Maqueda M, Martinez-Bueno M, Valdivia E. Publication rates reveal trends in microbiological research. ASM News. 1998;64:269–275. [Google Scholar]
- DSMZ Bacterial Nomenclature Up-to-date http://www.dsmz.de/bactnom/bactname.htm
- Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci USA. 1977;74:5088–5090. doi: 10.1073/pnas.74.11.5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR. Bacterial evolution. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone DR, Castenholz RW, Garrity GM, (eds) Bergey's Manual of Systematic Bacteriology 2nd edn New York: Springer, 2001.
- Holt JG, (ed) Bergey's Manual of Systematic Bacteriology 1st edn Baltimore: Williams and Wilkins, 1 1984. [Google Scholar]
- Bergey's Manual Trust http://www.cme.msu.edu/bergeys/
- Ludwig W, Klenk H-P. Overview: a phylogenetic backbone and taxonomic framework for procaryotic systematics. In: Boone DR, Castenholz RW, Garrity GM, editor. In Bergey's Manual of Systematic Bacteriology. 2. Vol. 1. New York: Springer;; 2001. pp. 49–65. [Google Scholar]
- Drancourt M, Bollet C, Carlioz A, Martelin R, Gayral JP, Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 2000;38:3623–3630. doi: 10.1128/jcm.38.10.3623-3630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Australian Collection of Microorganisms http://www.biosci.uq.edu.au/micro/culture/culture.htm
- Pace NR, Stahl DA, Lane DJ, Olsen GJ. Analyzing natural microbial populations by rRNA sequences. ASM News. 1985;51:4–12. [Google Scholar]
- Amann RI, Ludwig W, Schleifer KH. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev. 1995;59:143–169. doi: 10.1128/mr.59.1.143-169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 1998;180:4765–4774. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Grishin NV, Tatusov RL, Koonin EV. Genome trees constructed using five different approaches suggest new major bacterial clades. BMC Evol Biol. 2001;1:8. doi: 10.1186/1471-2148-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dojka MA, Harris JK, Pace NR. Expanding the known diversity and environmental distribution of an uncultured phylogenetic division of bacteria. Appl Environ Microbiol. 2000;66:1617–1621. doi: 10.1128/aem.66.4.1617-1621.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugenholtz P, Tyson GW, Webb RI, Wagner AM, Blackall LL. Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives. Appl Environ Microbiol. 2001;67:411–419. doi: 10.1128/AEM.67.1.411-419.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RS. What are archaebacteria: life's third domain or monoderm prokaryotes related to Gram-positive bacteria? A new proposal for the classification of prokaryotic organisms. Mol Microbiol. 1998;29:695–707. doi: 10.1046/j.1365-2958.1998.00978.x. [DOI] [PubMed] [Google Scholar]
- Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, Sahasrabudhe A, Dewhirst FE. Bacterial diversity in human subgingival plaque. J Bacteriol. 2001;183:3770–3783. doi: 10.1128/JB.183.12.3770-3783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong EF, Pace NR. Environmental diversity of bacteria and archaea. Syst Biol. 2001;50:470–478. [PubMed] [Google Scholar]
- Schleper C, DeLong EF, Preston CM, Feldman RA, Wu KY, Swanson RV. Genomic analysis reveals chromosomal variation in natural populations of the uncultured psychrophilic archaeon Cenarchaeum symbiosum. J Bacteriol. 1998;180:5003–5009. doi: 10.1128/jb.180.19.5003-5009.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrman JA, Campbell L. Marine ecology: microbial microdiversity. Nature. 1998;393:410–411. [Google Scholar]
- Brown JR. Genomic and phylogenetic perspectives on the evolution of prokaryotes. Syst Biol. 2001;50:497–512. doi: 10.1080/10635150117729. [DOI] [PubMed] [Google Scholar]
- Woese CR. A manifesto for microbial genomics. Curr Biol. 1998;8:R781–R783. doi: 10.1016/s0960-9822(07)00498-8. [DOI] [PubMed] [Google Scholar]
- Doolittle WF. Phylogenetic classification and the universal tree. Science. 1999;284:2124–2128. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- Woese CR. Interpreting the universal phylogenetic tree. Proc Natl Acad Sci USA. 2000;97:8392–8396. doi: 10.1073/pnas.97.15.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbo CL, Boucher Y, Doolittle WF. Defining the core of nontransferable prokaryotic genes: the euryarchaeal core. J Mol Evol. 2001;53:340–350. doi: 10.1007/s002390010224. [DOI] [PubMed] [Google Scholar]
- Ragan MA. On surrogate methods for detecting lateral gene transfer. FEMS Microbiol Lett. 2001;201:187–191. doi: 10.1111/j.1574-6968.2001.tb10755.x. [DOI] [PubMed] [Google Scholar]
- GOLD: Genomes OnLine Database Homepage http://wit.integratedgenomics.com/GOLD/
- Hugenholtz P, Tyson GW, Blackall LL. In Methods in Molecular Biology, Gene Probes: Principles and Protocols Edited by Aquino de Muro M, Rapley R Totowa: Humana, Vol. 179. 2001. Design and evaluation of 16S rRNA-targeted oligonucleotide probes for fluorescence in situ hybridization. pp. 29–42. [DOI] [PubMed] [Google Scholar]
- Liesack W, Janssen PH, Rainey FA, Ward-Rainey NL, Stackebrandt E. In Modern Soil Microbiology Edited by van Elsas JD, Trevors JT, Wellington EMH New York: Marcel Dekker, 1997. Microbial diversity in soil: the need for a combined approach using molecular and cultivation techniques. pp. 375–439. [Google Scholar]
- Zinder SH, Salyers AA. Microbial ecology - new directions, new importance. In Bergey's Manual of Systematic Bacteriology Edited by Boone DR, Castenholz RW, Garrity GM 2nd edn New York; Springer, 2001;1:101–109. [Google Scholar]
- Huber R, Burggraf S, Mayer T, Barns SM, Rossnagel P, Stetter KO. Isolation of a hyperthermophilic archaeum predicted by in situ RNA analysis. Nature. 1995;376:57–58. doi: 10.1038/376057a0. [DOI] [PubMed] [Google Scholar]
- Reasoner DS, Geldreich EE. A new medium for the enumeration and subculture of bacteria from potable water. Appl Environ Microbiol. 1985;49:1–7. doi: 10.1128/aem.49.1.1-7.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondon MR, August PR, Bettermann AD, Brady SF, Grossman TH, Liles MR, Loiacono KA, Lynch BA, MacNeil IA, Minor C, et al. Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorgan-isms. Appl Environ Microbiol. 2000;66:2541–2547. doi: 10.1128/aem.66.6.2541-2547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beja O, Aravind L, Koonin EV, Suzuki MT, Hadd A, Nguyen LP, Jovanovich S, Gates CM, Feldman RA, Spudich JL, et al. Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science. 2000;289:1902–1906. doi: 10.1126/science.289.5486.1902. [DOI] [PubMed] [Google Scholar]
- Torsvik V, Goksoyr J, Daae FL. High diversity in DNA of soil bacteria. Appl Environ Microbiol. 1990;56:782–787. doi: 10.1128/aem.56.3.782-787.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gest H. Letters: Gest's postulates. ASM News. 1999;65:123. [Google Scholar]
- TheARB project http://www.arb-home.de/
- Maidak BL, Cole JR, Lilburn TG, Parker CT, Saxman PR, Farris RJ, Garrity GM, Olsen GJ, Schmidt TM, Tiedje JM. The RDP-II (Ribosomal Database Project). Nucleic Acids Res. 2001;29:173–174. doi: 10.1093/nar/29.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribosomal Database Project II http://www.cme.msu.edu/RDP/html/index.html