

SUMMARY
The PutA flavoprotein from Escherichia coli is a transcriptional repressor and a bifunctional enzyme that regulates and catalyzes proline oxidation. PutA represses transcription of genes putA and putP by binding to the control DNA region of the put regulon. The objective of this study is to define and characterize the DNA binding domain of PutA. Previously, the DNA binding activity of PutA, a 1320 amino acid polypeptide, was localized to N-terminal residues 1-261. After exploring a potential DNA-binding region and an N-terminal deletion mutant of PutA, residues 1-90 (PutA90) were determined to contain DNA binding activity and stabilize the dimeric structure of PutA. Cell-based transcriptional assays demonstrate that PutA90 functions as a transcriptional repressor in vivo. The dissociation constant of PutA90 with the put control DNA was estimated to be 110 nM, which is slightly higher than that of the PutA-DNA complex (Kd ∼ 45 nM). Primary and secondary structure analysis of PutA90 suggested the presence of a ribbon-helix-helix DNA binding motif in residues 1-47. To test this prediction, we purified and characterized PutA47. PutA47 is shown to purify as an apparent dimer, exhibit in vivo transcriptional activity and bind specifically to the put control DNA. In gel-mobility shift assays, PutA47 was observed to bind cooperatively to the put control DNA with an overall dissociation constant of 15 nM for the PutA47-DNA complex. Thus, N-terminal residues 1-47 are critical for DNA-binding and the dimeric structure of PutA. These results are consistent with the ribbon-helix-helix family of transcription factors which are comprised of a two-stranded antiparallel β-sheet that recognizes DNA and a dimeric core of four α-helices.
Proline utilization A (PutA) from Escherichia coli is a multifunctional enzyme that catalyzes the flavin-dependent oxidation of proline to Δ1-pyrroline-5-carboxylate (P5C) and the NAD-dependent oxidation of P5C to glutamate (1, 2). In the dehydrogenation of proline to P5C two electrons are transferred from proline to a non-covalently bound FAD. Electrons from reduced FAD are then transferred to an acceptor in the electron transport chain to complete the catalytic cycle (3). Next, P5C, the product of the first reaction, is hydrolyzed to glutamate-γ-semialdehyde followed by subsequent transfer of two electrons to an NAD+ cofactor by the P5C dehydrogenase (P5CDH) domain to yield glutamate. In addition to enzymatic roles in proline utilization, PutA is responsible for the transcriptional regulation of the proline utilization genes, putA and putP (1, 4-8). PutP encodes a high affinity sodium-proline transporter (9). PutA represses the expression of the put genes, which are transcribed in opposite directions, by binding to the put intergenic DNA region (1, 7, 8). The transcriptional repression of the put genes is relieved in the presence of proline by the translocation of PutA to a peripheral position on the membrane where proline is efficiently converted to glutamate. Reduction of the flavin causes a conformation change in PutA that is thought to enhance PutA membrane associations thereby disrupting PutA-DNA binding (8, 10, 11).
Insights into the organization of the functional domains in PutA have been gained from molecular dissection and characterization of truncated PutA proteins. PutA is a polypeptide of 1320 amino acids and purifies predominately as a dimer though minor amounts of monomer species are observed. The dimeric form of PutA has an estimated molecular mass of ∼293 kDa (1). A truncated form of PutA containing residues 1-669 (PutA669) was shown to exhibit proline dehydrogenase (PRODH) and DNA binding activities but lack P5CDH activity (12). The X-ray crystal structure of PutA669 complexed to the competitive inhibitor L-lactate was solved to 2.0 Å resolution (13). The crystal structure included residues 87-612 of PutA669 and revealed that the PRODH domain is a β8α8 barrel comprised of residues 261-612, with the FAD bound at the C-terminal ends of the β-strands of the barrel. A putative helix-turn-helix (HTH) DNA-binding motif from residues 139-258 was also identified from the structure of the PutA669-lactate complex. The recognition helix (residues 230-241) of the HTH motif from the PutA669 structure contains basic residues that appear properly positioned for ionic and hydrogen bond interactions with DNA such as Arg230, Arg234 and Lys238. A truncated PutA protein containing residues 1-261, PutA261, was shown to bind the put intergenic DNA region demonstrating that DNA-binding activity can be separated from the PRODH domain and further implied that residues 139-258 were involved in DNA binding (13).
The goal of this study was to identify and characterize the DNA-binding domain of PutA. By engineering and examining several truncation and site-directed PutA mutants, we have identified the DNA-binding domain of PutA. In this report we establish that N-terminal residues 1-47 comprise a domain that is responsible for the DNA binding activity of PutA. Surprisingly, N-terminal residues 1-47 are also essential for dimerization of PutA suggesting that this region is a key element for the versatility of PutA. The properties of the isolated DNA-binding domain (PutA47) are described and suggest that PutA is a member of the ribbon-helix-helix (RHH) family of DNA binding proteins. The implications of these findings are discussed in terms of the multifunctional properties of PutA.
EXPERIMENTAL PROCEDURES
Materials - All chemicals and buffers were purchased from Fisher Scientific and Sigma-Aldrich, Inc. Restriction endonucleases and T4 DNA ligase were purchased from Fermentas and Promega, respectively. Molecular size standards used for calibrating size exclusion columns were purchased from Sigma. BCA reagents used for protein quantitation were obtained from Pierce. All experiments used Nanopure water. E. coli strains XL-Blue and BL21 DE3 pLysS were purchased from Novagen. E. coli strain JT31 putA- lacZ- was a generous gift from J. Wood (University of Guelph, Guelph, ON). The vector pET-23b (Novagen) containing a hexahistidine encoded sequence was used for expression of PutA669 R230/R234, PutAΔ85, and PutA261 as C-terminal hexahistidine tagged proteins. The vector pET-14b was used for the expression of PutA139-258 as a N-terminal hexahistidine tagged protein. The vector pET-3a (Novagen) was used for the expression of PutA90 and PutA47 which results in the addition of a GSGC amino acid sequence at the C-terminal end. Sequence specific synthetic oligonucleotides were purchased from Integrated DNA Technologies. The put intergenic DNA (419 bp) used for DNA binding assays was prepared as described previously using genomic DNA from E. coli strain JT31 (14).
Preparation of PutA Protein and Reporter Constructs – A PutAΔ85 truncation construct was prepared by introducing an NdeI site at amino acid codon 84 of the putA gene in pET-23b, respectively (15). NdeI digestion of the modified putA gene resulted in PutAΔ85. PutA47 (residues 1-47) and PutA90 (residues 1-90) constructs were engineered by positioning a BamHI site at amino acid codons 48 and 91 of the putA gene in pET3a, respectively (14). BamHI digestion then removed the corresponding PutA amino acid codons 48-1320 in PutA47 and codons 91-1320 in PutA90. The PutA139-258 construct (residues 139 through 258) was prepared by introducing NdeI and Bpu1102 sites at amino acid codons 137 and 256 of the putA gene in pET3a, respectively. The NdeI-Bpu1102 fragment containing residues 139-258 of PutA was subcloned into pET-14b. The insertion of the NdeI, BamHI, and Bpu1102 sites during the subcloning procedures were introduced using synthetic oligonucleotides and the QuikChange (Stratagene) site-directed mutagenesis kit. A G256A substitution in PutA139-258 was generated from the introduction of the Bpu1102 site. The PutA669 mutant R230A/R324A was prepared in pUTA669 (construct in pET-23b encoding residues 1-669 of PutA with a C-terminal hexahistidine tag) using QuikChange site-directed mutagenesis and oligonucleotides 5'-GAAGCCAGCCTCTCCGCCTCGCTGAACC-3' and 5'-CCCGCTCGCTGAACGCCATTATCGGTAAAAGCG-3' for R230A and R234A, respectively (12). Nucleic acid sequencing confirmed each of the PutA constructs and PutA669 mutant R230A/R234A.
A reporter construct containing the lacZ gene (3.5 kb) under control of the put regulatory region (putC, 419 bp) was prepared as a derivative of pACYC184 (New England Biolabs). The lacZ gene, obtained from a PROTet.333 vector (ClonTech), was positioned downstream of the put control DNA region in pUTC01 (a pUC18 vector containing the put intergenic DNA sequence, putC) using PstI and SalI to generate pUTC02 (14). A partial digest of pUTC02 with PstI and EcoRI generated a 3.9 kb fragment that contained putC and the lacZ gene (putC:lacZ). The 3.9 kb fragment was inserted into pACYC184 using PstI and EcoRI resulting in a low copy construct containing the lacZ reporter gene under control of the put control DNA (pUTC03). Constructs to test in vivo transcriptional repressor activity of PutA, PutAΔ85, PutA669, PutA90, and PutA47 for were prepared by subcloning the corresponding putA genes from pET3a into pUC18 using XbaI and HindIII. Thus, none of the PutA proteins have a hexahistidine tag in the reporter gene experiments. Unique XbaI and HindIII sites were introduced at the 5' and 3' ends, respectively, of each putA gene using PCR and the appropriate primers. After digestion with XbaI and HindIII, the PCR products were inserted into pUC18 positioning the putA genes downstream of the lac promoter (pUC18-PutA). Nucleic acid sequencing of the pUC18-PutA constructs confirmed the correct orientation of each putA gene.
Purification and Characterization of PutA Proteins - PutA669 mutant R230A/R234, PutAΔ85, PutA261, and PutA139-258 were overexpressed in E. coli strain BL21 DE3 pLysS and purified using a Ni2+ NTA affinity column (Novagen) as previously described for wild-type PutA669 and PutA261 (12, 13). The hexahistidine tags were retained after purification. The purified proteins were stored in 50 mM Tris (pH 7.5) containing 10% glycerol at −70 °C.
PutA90 and PutA47 were also overexpressed in E. coli strain BL21 DE3 pLysS at 37 °C as previously described but without a hexahistidine tag (13). PutA90 was purified by anion exchange chromatography as described for PutA (14). Eluted PutA90 was identified by SDS-PAGE (15%) analysis and gel-mobility shift assays. PutA90 was then applied to a heparin column (Sigma) equilibrated with 10 mM Tris-HCl (pH 7.5-8.0) and eluted in the same buffer containing 1.5 M NaCl. Fractions containing PutA90 were identified by SDS-PAGE analysis and dialyzed into 50 mM Tris (pH 7.5) containing 10% glycerol. Because the pI for the PutA47 denatured polypeptide is around pH = 7.9, PutA47 was purified using cation exchange chromatography. PutA47 cell supernatant was applied to a CM-sepharose column (Sigma) equilibrated with 20 mM Mes (pH 6.5) containing 5% glycerol. A 1 L gradient from 0 to 500 mM NaCl in 20 mM Mes (pH 6.5) and 5% glycerol eluted PutA47. Fractions containing PutA47 were identified by SDS-PAGE (18%) and gel-mobility shift assays. PutA47 was then concentrated using a 50 ml Amicon ultrafiltration cell with a 10 kDa molecular weight cutoff and purified further by gel-filtration using a Superdex-75 column (Amersham Biosciences) equilibrated with 50 mM Tris (pH 7.5). Purified PutA90 and PutA47 were stored in 50 mM Tris (pH 7.5) containing 10% glycerol at −70 °C.
The concentrations of the PutA proteins were determined using the BCA method (Pierce) with bovine serum albumin as the standard and spectrophotometrically using a molar extinction coefficient at 451 nm of 12,800 M−1 cm−1 for PutA669 mutant R230/R234 and PutAΔ85 and newly estimated molar extinction coefficients at 280 nm for PutA90 and PutA47 (12,14). The molar extinction coefficients for PutA90 and PutA47 at 280 nm were determined by recording the spectrum of the proteins denatured in 6 M guanidinium chloride from 350 to 250 nm. The total amount of PutA90 and PutA47 polypeptides was determined using the predicted molar extinction coefficient of 6970 M−1 cm−1 for denatured PutA90 and PutA47. The molar extinction coefficients at 280 nm for native PutA90 and PutA47 were estimated to be 4530 ± 200 M−1 cm−1 and 4690 ± 200 M−1 cm−1, respectively. PRODH activity was measured using the proline:DCPIP oxidoreductase assay performed at 25 °C as previously described (14). Circular dichroism spectra of PutA and PutA47 were recorded from 260 to 195 nm in a 1 mm path length cuvette using an Olis DSM 1000 CD spectrophotometer. The α-helix content was estimated from the molar ellipticity at 222 nm as described previously (16).
The molecular sizes of PutAΔ85, PutA90, and PutA47 were estimated by size exclusion chromatography. PutAΔ85 was applied to a Superose 6 column equilibrated in 50 mM Tris (pH 7.5) containing 50 mM NaCl and calibrated as previously described (14). PutA139-258, PutA90 and PutA47 were applied to a Superdex-75 column equilibrated in 50 mM Tris (pH 7.5) containing 50 mM NaCl and calibrated using the molecular mass standards cytochrome c (14 kDa), carbonic anhydrase (29 kDa), bovin serum albumin (66 kDa), alcohol dehydrogenase (150 kDa), and β-amylase (200 kDa).
DNA Binding Assays - Nondenaturing gel electrophoretic mobility shift assays were used to test the binding of the PutA proteins to the put control intergenic DNA as previously described (14). The overall dissociation constant (Kd) of PutA90 with the put intergenic DNA was estimated from nitrocellulose filter binding assays. The put intergenic DNA was labeled at the 5' end with [γ-32P]ATP as described (14). In a total binding reaction volume of a 200 μl, varying concentrations of PutA90 (0-2 μM) were incubated with 32P-labeled put intergenic DNA (20,000 cpm) for 15 min in 50 mM Tris (pH 7.5). The binding reactions were then vacuum filtered through a Slot-Blot apparatus (Bio-Rad, Hercules, CA) and washed with 300 μl of the binding reaction buffer. After the filters were dried, the radioactivity on the filter from the PutA90-DNA complexes was quantitated using a Storm 860 Phosphoimager (Molecular Dynamics). The radioactivity from binding reactions containing only 32P-labeled DNA was used to correct for the background contribution of free DNA to the total radioactivity at each PutA90 concentration. Data were averaged from different experiments and analyzed by plotting the fraction of bound DNA vs PutA90 dimer concentration. A macroscopic dissociation constant (Kd) was estimated by fitting the data to equation (1) where θ is the fraction of DNA bound (i.e. radioactivity retained on the filter), [P] is the total protein concentration, αH is the Hill coefficient, and n
| (1) |
is the maximum fraction of DNA bound.
In gel-shift assays with PutA47, fluorescently labeled put intergenic DNA was used in the binding reactions. The synthetic oligonucleotide 5' end-labeled with IRdye™-700 (LI-COR, Inc.) was used as one of the primers in a PCR reaction to amplify the put intergenic DNA. The resulting IRdye-700 labeled put intergenic DNA was purified and quantitated by measuring the nucleic acid concentration at 260 nm and the absorbance of the IRdye-700 at 685 nm using an extinction coefficient of 170 mM−1 cm−1 according to the recommendations of the manufacturer. PutA47 (0-200 nM) was incubated with 2 nM put intergenic DNA in a total volume of 25 μl in 50 mM Tris (pH 7.5) containing 10% glycerol for 20 min prior to electrophoresis. Calf thymus competitor DNA (100 μg/ml) was added to the binding mixtures to prevent nonspecific PutA47-DNA interactions. The PutA47-DNA complexes were separated using a polyacrylamide (8%) native gel at 4 °C. The gels were visualized and the fraction of DNA bound was determined using a LI-COR Odyssey Imager. Data from three independent binding experiments were averaged and analyzed using equation 1.
LacZ Reporter Assays – E. coli strain JT31 putA- lacZ- containing putC:lacZ (pUT03) and various putA gene constructs (pUC18-PutA) were grown at 37 °C in minimal medium with ampicillin (50 μg ml−1) and chloramphenical (30 μg ml−1) to O.D. at 600 nm ∼ 1.0. Pelleted cells were then resuspended and broken using the Bacterial Protein Extraction Reagent from Pierce (20 mM Tris-HCl, pH 7.5). β-galactosidase activity assays were performed in a 1 ml volume of 100 mM sodium phosphate (pH 7.3) containing 1 mM MgCl2, 50 mM β-mercaptoethanol and 2 mM o-nitrophenyl-β-D-galactopyranoside (ONPG). The initial velocity was determined by measuring the increase in absorbance at 420 nm. Expression of each construct was confirmed by gel-shift or PRODH activity assays with the cell extracts. The results in Table I are averaged values from four independent experiments.
Table I.
β-galactosidase activities from E. coli strain JT31 containing the lacZ reporter construct pUTC03 and various putA gene constructs
| PutA Protein | β-galactosidase activity (U OD−1)a | Percent Activity |
|---|---|---|
| none | 17.0 ± 0.8 | 100 |
| PutA | 3.3 ± 1.4 | 19 |
| PutA669 | 0.18 ± 0.01 | 1 |
| PutA90 | 0.27 ± 0.04 | < 2 |
| PutA47 | 0.17 ± 0.02 | 1 |
| PutAΔ85 | 12.9 ± 1.5 | 76 |
Optical density was measured at 600 nm prior to cell harvesting. E. coli strain JT31 has no β-galactosidase activity.
RESULTS
Localization of DNA-Binding to the N-terminal Region – A potential HTH motif was identified from the PutA669 X-ray crystal structure involving residues 139-258 (13). A truncated PutA protein that contains the HTH motif (PutA261) was shown to bind specifically to the put control DNA (Figure 1). To test whether the HTH motif was involved in DNA binding, the HTH domain was engineered and purified as PutA139-258. PutA139-258, however, failed to show DNA binding activity in gel mobility shift assays (Figure 1). It was not clear whether the lack of DNA binding was due to misfolding or to the inability of PutA139-258 to form a dimer. Size-exclusion chromatography indicated PutA139-258 purified as a polypeptide of molecular mass around 20 kDa. The predicted molecular weight of PutA139-258 is 15.6 kD indicating PutA139-258 is a monomer. To further address the role of the HTH domain in DNA-binding, residues R230 and R234 were replaced with Ala in PutA669. The spacing of the two guanidinium residues and their apparent solvent accessibility implied that R230 and R234 would be involved in important ionic interactions with the DNA phosphodiester backbone and perhaps hydrogen bond pair interactions with specific nucleotide bases (13). Simultaneous removal of R230 and R234, however, did not disrupt the DNA binding activity of PutA669 (Figure 1). PutA669 mutant R230A/R234A has similar PRODH activity and an FAD absorbance spectrum compared to wild-type PutA669 indicating proper folding of the PutA699 mutant. Thus, from the results with PutA139-258 and PutA669 mutant R230A/R234A, it appeared unlikely that the putative HTH domain identified in the X-ray crystal structure of PutA669 was involved in DNA binding.
Fig. 1.
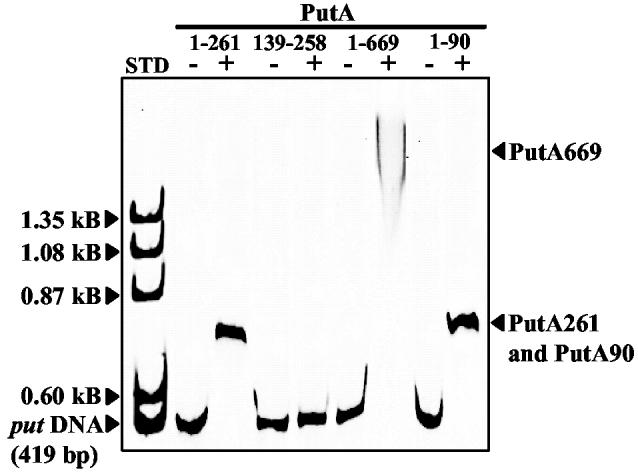
Gel mobility shift assays of truncated PutA proteins and the put control DNA. PutA261, PutA139-258, PutA669 R230A/R234A, and PutA90 (1 μM monomer each) were incubated in separate binding mixtures (50 mM Tris, pH 7.5) with IRdye-700 labeled put control DNA (5 nM) at 20 °C. The complexes were separated using an agarose/polyacrylamide (0.5%/3%) native gel at 4 °C. The position of uncomplexed put control DNA and the protein-DNA complexes are indicated. The left-hand lane (STD) shows the migration of uncomplexed put control DNA (0.419 kB) and ΦX174 ladder DNA (500 ng) with molecular size standards of 1.353 kB, 1.078 kB, 0.872 kB and 0.603 kB. The gel was stained with ethidium bromide to visualize the molecular size standards.
Attempts to locate the DNA-binding domain of PutA then focused on the N-terminal region of PutA. The role of the N-terminal region in PutA-DNA binding was first elucidated by engineering the N-terminal deletion mutant PutAΔ85. PutAΔ85 displays PRODH activity typical of PutA and has two main FAD absorbance bands at 452 and 377 nm in the UV-visible spectrum with a 452/377 ratio of 1.13 that is slightly higher than PutA (1.05) (14). PutAΔ85 contains 0.8 mole of FAD per mole of polypeptide, similar to PutA (0.83 mole of FAD per subunit). Analysis of PutAΔ85 by size exclusion chromatography showed a single species with an estimated molecular mass of 128 kDa (data not shown). PutAΔ85 is comprised of 1252 amino acids (including the C-terminal hexahistidine tag) and has a predicted molecular weight of 136,365 Da. Thus, PutAΔ85 purifies as a monomer in contrast to PutA which purifies mainly as a dimer. PutAΔ85 was also shown to lack DNA-binding activity. PutAΔ85 was not observed to bind the put control DNA even at 100-fold molar excess of PutAΔ85 to DNA (data not shown). Therefore, the deletion of the N-terminal residues 1-85 has profound effects on dimerization and DNA-binding activity of PutA. Conclusions concerning the DNA binding domain, however, are not clear since PutAΔ85 does not purify as a dimer. The inability of PutAΔ85 to form a stable dimer may be the principal reason that PutAΔ85 lacks DNA-binding activity. Furthermore, the molecular dissection of PutA at amino acid codon 85 may divide a functional domain.
Characterization of PutA90 - PutA90 was then designed to test the role of residues 1-85 in DNA binding. Figure 1 shows that PutA90 binds to the put control DNA demonstrating that the DNA binding activity is at the N-terminal region of PutA. PutA90 was estimated to have a molecular mass of around 50 kDa indicating PutA90 self associates to a high molecular weight species during purification since the predicted molecular weight of PutA90 is only 9962 Da. The large estimated molecular mass of PutA90 suggests it contains residues involved in dimerization of PutA as initially observed with PutAΔ85. The overall dissociation constant of the PutA90-DNA complex (Kd) was estimated by nitrocellulose filter binding assays. Cooperative interactions became apparent from the binding analysis of PutA90 to the put control DNA. A Scatchard plot showed a positive curve and a best fit of the binding data was obtained by incorporating a Hill coefficient parameter. A macroscopic dissociation constant of ∼ 110 ± 8 nM was determined by fitting the data to eq 1 (Figure 2). The Kd for the PutA90-DNA complex is about two-fold higher than the Kd determined previously for the PutA-DNA complex (Kd ∼ 45 nM).
Fig. 2.
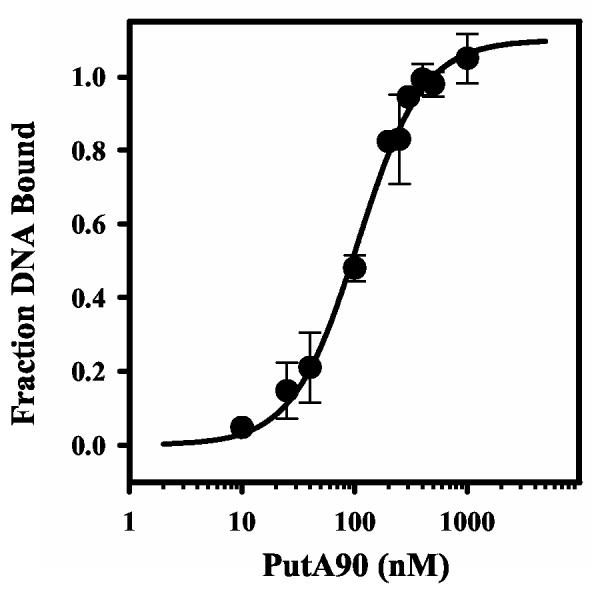
Nitrocellulose filter binding analysis of PutA90 and put control DNA. A plot of fraction of bound DNA vs PutA90 dimer concentration from nitrocellulose filter binding assays. 32P-labeled put control DNA and varying concentrations of PutA90 (0- 1000 nM dimer) were incubated in binding mixtures (50 mM Tris, pH 7.5) at 20 °C for 20 min. The uncomplexed DNA and PutA90-complexed DNA were then separated by nitrocellulose filter binding and the DNA bound to the filter was quantitated. The data were fit to eq 1 (n = 1.1 and αH = 1.5) to yield an overall dissociation constant of 110 ± 8 nM.
The ability of PutA90 to bind put control DNA and repress transcription in vivo was tested using β-galactosidase reporter assays in the E. coli strain JT31 putA- lacZ-. The lacZ gene was positioned downstream of the put control DNA region in a low copy construct vector (pUTC03). Different truncated PutA proteins were encoded on a second plasmid (pUC18-PutA) to test transcriptional repressor activity. Wild type PutA reduced β-galactosidase activity to about 20% relative to control cells that lacked PutA protein. PutA669 and PutA90 repressed lacZ expression more strongly with levels of β-galactosidase activity diminished to <2% relative to controls cells. In contrast, PutAΔ85 reduced activity to only about 75% compared to control cells. Thus, the reporter assays demonstrate that PutA90 acts as a transcriptional repressor of the put control DNA in vivo. The greater repression of lacZ expression by PutA669 and PutA90 relative to PutA is most likely due to differences in membrane binding. PutA669 and PutA90 are not anticipated to interact with the membrane while PutA associates with both the DNA and the membrane (12). Partitioning of PutA onto the membrane would presumably relieve lacZ repression.
Assignment of a RHH motif - The primary structure of PutA90 was then analyzed for any conserved DNA binding domains. A conserved domain architectural retrieval search identified a RHH domain of the CopG family in PutA residues 1-47. Figure 3 shows the predicted secondary structure of PutA47 characterized by a β-strand from residues 4-10 and a helix-helix domain from residues 12-42. The alignment of PutA47 with the RHH consensus sequence of the CopG family in Figure 3 demonstrates a 33% sequence identity between PutA47 and the CopG family. CopG is a small transcriptional repressor of only 45 amino acids (17). The DNA recognition element in the CopG family is the β-strand which forms an antiparallel β-sheet in the dimer complex to generate β-sheet-DNA interactions (17, 18). The role of the helix-helix fold in the RHH motif is to form key hydrophobic dimerization interactions (17). Extensive mutagenesis of the RHH repressor Arc of bacteriophage P22, has shown that substitution of aromatic residues in helix A and helix B often results in disruption of the dimer structure (19-21). In PutA, Trp31 and Tyr40 appear to be positioned as important aromatic residues in a putative hydrophobic dimerization core similar to the RHH family. Conserved hydrophobic residues in PutA that are potentially important for the stability of the RHH dimeric structure are highlighted in Figure 3. Thus, the RHH motif contains structural elements involved in DNA-binding and dimerization which match our observations with PutAΔ85 that lacks the DNA-binding and dimeric properties of PutA (17).
Fig. 3.

Predicted strand-helix-helix fold of PutA47 and sequence alignments. The secondary structure of PutA47 was predicted using the PSIPRED protein structure prediction server and is illustrated directly above the PutA47 amino acid sequence. The alignment of PutA47 with the RHH consensus sequence (RHH) of the CopG family is shown. A conserved domain search revealed the RHH fold at the N-terminal region of PutA from S. typhimurium (P10503), K. aerogenes (O52485), Y. pestis KIM (NP_669761), P. putida (AAF25000), R. solanacearum (NP_521420), and B. fungorum (ZP_00029196) as shown by the sequence alignments. Conserved hydrophobic residues are shaded in grey. The sequence alignments were obtained using the conserved domain architecture retrieval tool from NCBI.
An alignment of selected PutA sequences with PutA47 is also shown in Figure 3. PutA is known to have transcriptional repressor activity from Samonella typhimurium, Klebsiella aerogenes and Pseudomonas putida (7, 22, 23). In the remaining bacteria such as Yersinia pestis, Ralstonia solanacearum and Burkholderia fungorum, the regulation of the put regulon has not been described but on the basis of the alignment in Figure 3 it appears PutA most likely acts as a transcriptional repressor as well. The inability to identify an RHH domain in PutA from other bacteria such as Bradyrhizobium japonicum and Sinorhizobium mellitus corroborates previous genetic studies with these microbes that have shown PutA is not involved in transcriptional regulation of the put genes (24, 25). Thus, primary structure comparisons using PutA47 from E. coli should be valuable for identifying whether PutA from other organisms is endowed with DNA-binding activity. An interesting variation between PutA and the CopG family is the lack of the conserved Gly mediated turn between helix A and helix B. A Gly mediated turn is conserved in RHH DNA-binding proteins except for a few members such as MetJ. From the sequence alignment, Pro rather than Gly is predicted to direct the turn between helix A and helix B in PutA. Figure 3 shows Pro29 is conserved in PutA from various bacteria. Recently, a classification of RHH proteins has been proposed on the basis of whether positively charged residues occur at the beginning (Type I) or the end (Type II) of the β-strand (26). Accordingly, Lys10 is anticipated to reside toward the end of the β-strand classifying PutA as a Type II RHH protein.
PutA47 was purified to test the prediction of the RHH motif in PutA. Circular dichroism spectra of PutA and PutA47 are shown in Figure 4. From the degree of ellipticity at 222 nm the α-helix content was estimated to be around 45% in PutA and about 56% in PutA47. The estimate of α-helix content in PutA47 resembles members of the RHH family which typically exhibit > 50% α-helix content. The molecular mass of PutA47 was estimated to be around 12 kDa by size exclusion chromatography. Because the predicted molecular weight of the PutA47 polypeptide is 5698 Da, PutA47 purifies as a dimer. Figure 5 demonstrates that PutA47 binds specifically to the put control DNA. A lower mobility protein-DNA complex forms upon increasing the concentration of PutA47 in the DNA binding reactions (Figure 5, top). An intermediate PutA47-DNA band is also observed due possibly to dissociation of the PutA47-DNA complex during electrophoresis or partial occupancy of binding sites in the put control DNA. Similar to PutA90-DNA interactions, PutA47 binding to the put control DNA displays positive cooperativity. A Scatchard plot exhibited a positive curve and the binding data were best described using a Hill coefficient parameter (data not shown). An overall dissociation constant of about 15 nM was estimated for the PutA47-DNA complex using equation 1 (Figure 5, bottom). PutA47 was also shown to function as a transcriptional repressor in whole cells with only about 1% β-galactosidase activity relative to control cells (Table I). Thus, characterization of PutA47 establishes that residues 1-47 contain the DNA binding and dimerization domain of the PutA protein.
Fig. 4.
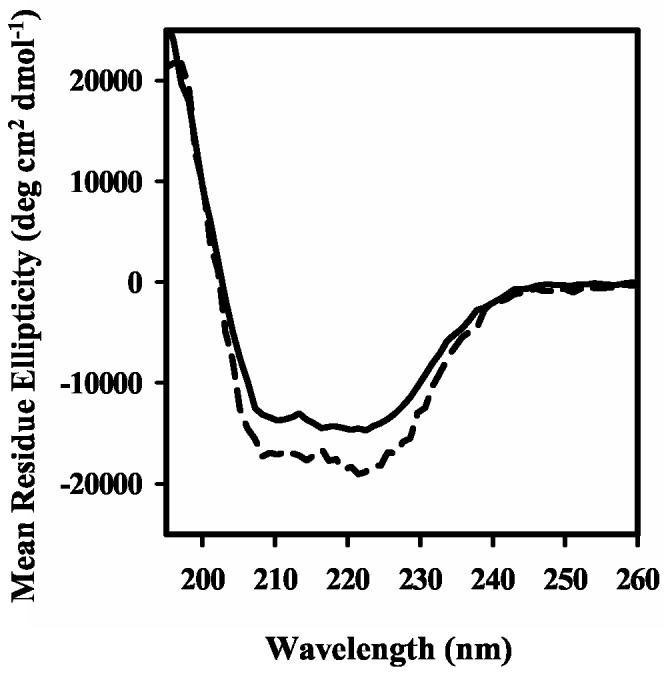
Circular Dichroism Spectra of PutA. Far-UV CD spectra of PutA (0.2 mg/ml) (solid line) and PutA47 (0.095 mg/ml) (broken line) in 10 mM potassium phosphate (pH 7.5) obtained with a 1 mm path length cuvette.
Fig. 5.
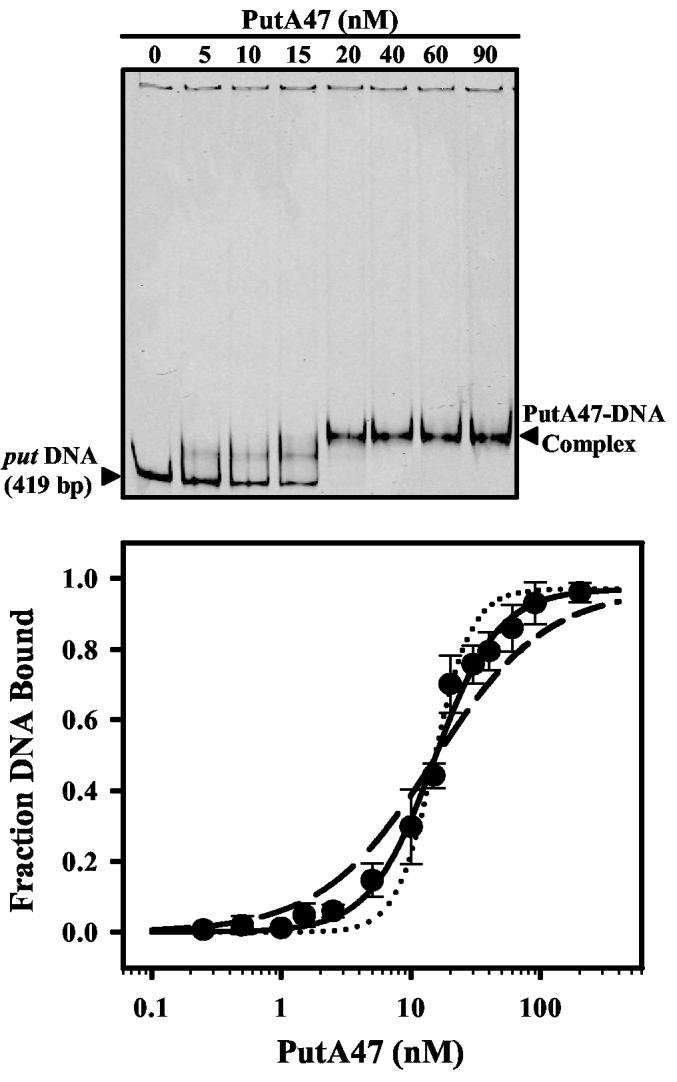
Gel mobility shift analysis of PutA47 DNA binding. (Top) Representative gel-shift assay with increasing concentrations of PutA47 (0-90 nM dimer) added to binding mixtures containing IRdye-700 labeled put control DNA and 100 μg/ml of nonspecific calf thymus DNA at 20 °C. The complexes were separated using a nondenaturing polyacrylamide gel (8%). (Bottom) Plot of fraction of bound DNA vs PutA47 dimer concentration from the gel shift assays. Solid curve is the fit of the data to eq 1 (n = 0.96 and αH = 1.75) to yield an overall dissociation constant of 15 ± 1 nM. Theoretical curves using n =0.96 and Kd = 15 nM but with αH = 1 (broken line) and αH = 3 (dotted line) are shown for comparison.
DISCUSSION
Knowledge of the domain architecture of the PutA flavoprotein is necessary for understanding its unique multifunctional properties. After examination of a putative DNA-binding domain in residues 139-258 our focus shifted to the N-terminal region of PutA. The N-terminal deletion mutant PutAΔ85 was shown to lack DNA-binding activity and purify only as a monomer suggesting the presence of a DNA-binding domain and a dimerization core in the N-terminal region of PutA. Subsequent characterization and conserved domain analysis of PutA90 suggested a RHH DNA-binding domain was located in residues 1-47. In a RHH domain, a helix-helix hydrophobic dimerization core is formed while the β-strands form an antiparallel β-sheet that recognizes DNA. Our characterization of PutA47 clearly shows that residues 1-47 contain the entire DNA binding domain of PutA and are involved in stabilizing the dimeric structure of PutA. PutA47 binds specifically to the put control DNA region and acts as a transcriptional repressor in vivo. PutA47 also purifies as a dimer and has α-helix content consistent with the RHH superfamily. Thus, even though PutA is a relatively large protein, the dimeric structure of PutA appears to primarily rely on a hydrophobic dimerization core analogous to members of the RHH family which are considerably smaller proteins such as the 53-residue Arc repressor (19).
Members of the RHH family such as the dimeric repressors MetJ, Arc, and CopG exhibit cooperative DNA binding through dimer-dimer contacts on DNA (17, 27, 28). Mnt, a tetrameric repressor, also binds DNA cooperatively via dimer-dimer interactions (29). Formation of the PutA47-DNA complex appears to involve cooperativity consistent with properties of some RHH DNA-binding proteins (17, 29). In previous studies with PutA, cooperative DNA binding has not been described although multiple PutA binding sites exist in the put control DNA (7, 12, 14). Detailed analysis of PutA-DNA interactions will be required to determine whether the observed cooperative DNA binding of PutA47 is relevant to the function of PutA as a transcriptional repressor or is unique to PutA47. Mapping of the PutA-DNA binding sites in the put control DNA region is in progress which will help address cooperativity of PutA-DNA interactions. Coinciding with cooperative DNA binding, RHH members have been observed to cause DNA bending. In the X-ray crystal structure of the CopG-operator complex, the DNA is bent 60° when bound by the CopG tetramer (17). Similarly, PutA binding has been shown to induce significant curvature in the put control DNA (7). Thus, the cooperative DNA binding noted with PutA47 and the DNA bending observed with PutA are consistent with the DNA binding properties of the RHH family.
As a result of our observations, we propose PutA is a member of the RHH family of transcriptional repressors although definitive classification of PutA as a RHH member awaits structure determination. PutA would become the largest member of the RHH family of repressors with a unique appendage of a RHH motif onto two large catalytic domains. Members of the RHH family are much smaller proteins ranging from the 45-residue CopG repressor to the 133-residue NikR from E. coli with variations due to additional N-terminal or C-terminal domains (17, 30). Examples of C-terminal functional regions in RHH members include protein tetramerization in the Mnt repressor, binding of the corepressor S-adenosylmethionine in MetJ, binding of the ParE toxin protein in the ParD repressor, and nickel binding by the repressor NikR (31-36). In NikR, a C-terminal nickel binding domain is attached to an N-terminus RHH fold (36). NikR regulates nickel uptake in bacteria by binding to promoter regions of the nik operon in the presence of excess nickel to repress transcription of genes nikABCDE (35). In the absence of nickel, NikR-DNA interactions are significantly weakened allowing derepression of the nik operon (35). Activation of NikR DNA-binding has been proposed to involve nickel-dependent conformational changes that are propagated from the nickel regulatory domain to the RHH domain (36). PutA would join NikR in which a RHH domain is affected by a regulatory site but in the case of PutA, the regulation of DNA-binding involves FAD redox signaling. It seems that the RHH motif is quite adaptable to regulation by various sensory domains.
The discovery that the N-terminal region of PutA is responsible for DNA-binding activity and dimerization provides important new insights into the functional domain arrangement of PutA. The PRODH domain (residues 261-612) has been clearly defined by X-ray crystallography while the location of the P5CDH domain (residues 650-1130) is based on sequence analysis (13, 37). Interestingly, limited proteolysis of PutA by chymotrypsin generates two major polypeptide products that originate at Ser58 demonstrating an accessible and flexible linker between the DNA-binding domain and the PRODH domain (11). The function of the region between the DNA binding and the PRODH domains is not known except that conformational changes have been mapped to residues in this region, specifically Ser216-Arg234 (11). Therefore, this flexible region may be involved in transmitting signals from the PRODH active site to the DNA binding domain which transform PutA from a DNA-binding protein to a membrane bound enzyme. Models for how PutA switches from a DNA-binding protein to a peripheral position on the membrane generally involve proline reduction of FAD and a conformational change that increases the overall hydrophobicity of PutA and PutA-membrane associations (8, 10). Proline and FAD reduction only slightly affect the overall dissociation constant of the PutA-DNA complex (about a two-fold increase) (14, 38). However, Maloy et al. (1997) have shown that in the presence of membrane vesicles proline disrupts PutA-DNA binding implying that the membrane has a critical influence on PutA-DNA interactions (39). Presently, the PutA-membrane binding domain is not known. Future studies will focus on identifying the membrane binding domain of PutA to further understand the regulation of PutA macromolecular associations.
Acknowledgements
We thank Prof. Cynthia Dupureur (University of Missouri-St. Louis, St. Louis, MO) for help with the nitrocellulose binding assays.
Footnotes
This work was supported in part by NSF (MCB0091664), NIH grants GM61068 (Becker) and GM65546 (Tanner), University of Nebraska Biochemistry Department and Redox Biology Center, and the Nebraska Agricultural Research Division (Journal Series No. XXXXX). This publication was also made possible by NIH Grant Number P20 RR-017675-02 from the National Center for Research Resources. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Abbreviations used are: FAD, flavin adenine dinucleotide; put, proline utilization; NAD+, nicotinamide adenine dinucleotide; PRODH, proline dehydrogenase; P5CDH, Δ1-pyrroline-5-carboxylate dehydrogenase; P5C, Δ1-pyrroline-5-carboxylate; DCPIP, dichlorophenolindophenol; EDTA, ethylenediaminetetraacetic acid; SDS-PAGE, sodium dodecyl sulfate polyacrylamide electrophoresis; RHH, ribbon-helix-helix; HTH, helix-turn-helix.
References
- 1.Brown E, Wood JM. J. Biol. Chem. 1992;267:13086–13092. [PubMed] [Google Scholar]
- 2.Menzel R, Roth J. J. Biol. Chem. 1981;256:9762–9766. [PubMed] [Google Scholar]
- 3.Abrahamson JLA, Baker LG, Stephenson JT, Wood JM. Eur. J. Biochem. 1983;134:77–82. doi: 10.1111/j.1432-1033.1983.tb07533.x. [DOI] [PubMed] [Google Scholar]
- 4.Menzel R, Roth J. J. Mol. Biol. 1981;148:21–44. doi: 10.1016/0022-2836(81)90233-3. [DOI] [PubMed] [Google Scholar]
- 5.Wood JM. J. Bacteriol. 1981;146:895–901. doi: 10.1128/jb.146.3.895-901.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maloy S, Roth JR. J. Bacteriol. 1983;154:561–568. doi: 10.1128/jb.154.2.561-568.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ostrovsky De Spicer P, O'Brian K, Maloy S. J. Bacteriol. 1991;173:211–219. doi: 10.1128/jb.173.1.211-219.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ostrovsky De Spicer P, Maloy S. Proc. Natl. Acad. Sci. USA. 1993;90:4295–4298. doi: 10.1073/pnas.90.9.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen CC, Tsuchiya T, Yamane Y, Wood JM, Wilson TH. J. Membr. Biol. 1985;84:157–164. doi: 10.1007/BF01872213. [DOI] [PubMed] [Google Scholar]
- 10.Brown ED, Wood JM. J. Biol. Chem. 1993;268:8972–8979. [PubMed] [Google Scholar]
- 11.Zhu W, Becker DF. Biochemistry. 2003;42:5469–5477. doi: 10.1021/bi0272196. [DOI] [PubMed] [Google Scholar]
- 12.Vinod MP, Bellur P, Becker DF. Biochemistry. 2002;41:6525–6532. doi: 10.1021/bi025706f. [DOI] [PubMed] [Google Scholar]
- 13.Lee YH, Nadaraia S, Gu D, Becker DF, Tanner JJ. Nat. Struct. Biol. 2003;10:109–114. doi: 10.1038/nsb885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becker DF, Thomas EA. Biochemistry. 2001;40:4714–4722. doi: 10.1021/bi0019491. [DOI] [PubMed] [Google Scholar]
- 15.Zhu W, Gincherman Y, Docherty P, Spilling CD, Becker DF. Arch. Biochem. Biophys. 2002;408:131–136. doi: 10.1016/s0003-9861(02)00535-0. [DOI] [PubMed] [Google Scholar]
- 16.Morrow J, Segall M, Lund-Katz S, Phillips M, Knapp M, Rupp B, Weigraber K. Biochemistry. 2000;39:11657–11666. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 17.Gomis-Ruth F, Sola M, Acebo P, Parraga A, Guasch A, Eritja R, Gonzalez A, Espinosa M, del Solar G, Coll M. EMBO J. 1998;17:7404–7415. doi: 10.1093/emboj/17.24.7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki M. Protein Eng. 1995;8:1–4. doi: 10.1093/protein/8.1.1. [DOI] [PubMed] [Google Scholar]
- 19.Milla M, Sauer R. Biochemistry. 1995;34:3344–3351. doi: 10.1021/bi00010a025. [DOI] [PubMed] [Google Scholar]
- 20.Milla M, Brown B, Sauer R. Struct. Biol. 1994;1:518–523. doi: 10.1038/nsb0894-518. [DOI] [PubMed] [Google Scholar]
- 21.Brown B, Sauer R. Proc. Natl. Acad. Sci. USA. 1999;96:1983–1988. doi: 10.1073/pnas.96.5.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen L-M, Maloy S. J Bacteriol. 1991;173:783–790. doi: 10.1128/jb.173.2.783-790.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vílchez S, Manzanera M, Ramos J. Appl. Environ. Microbiol. 2000;66:5221–5225. doi: 10.1128/aem.66.12.5221-5225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Straub PF, Reynolds PHS, Althomsons S, Mett V, Zhu Y, Shearer G, Kohl DH. Appl. Environ. Microbiol. 1996;62:221–229. doi: 10.1128/aem.62.1.221-229.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soto MJ, Jiménez-Zurdo JI, van Dillewijn P, Toro N. J. Bacteriol. 2000;182:1935–1941. doi: 10.1128/jb.182.7.1935-1941.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golovanov A, Barillà D, Golovanova M, Hayes F, Lian L-Y. Mol. Microbiol. 2003;50:1141–1153. doi: 10.1046/j.1365-2958.2003.03750.x. [DOI] [PubMed] [Google Scholar]
- 27.Somers WS, Phillips SEV. Nature. 1992;359:387–393. doi: 10.1038/359387a0. [DOI] [PubMed] [Google Scholar]
- 28.Raumann BE, Rould MA, Pabo CO, Sauer RT. Nature. 1994;367:754–757. doi: 10.1038/367754a0. [DOI] [PubMed] [Google Scholar]
- 29.Berggrun A, Sauer R. Proc. Natl. Acad. Sci. USA. 2001;98:2301–2305. doi: 10.1073/pnas.041612198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chivers P, Sauer R. Protein Sci. 1999;8:2494–2500. doi: 10.1110/ps.8.11.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nooren IMA, Kaptein R, Sauer RT, Boelens R. Nat. Struct. Biol. 1999;6:755–759. doi: 10.1038/11531. [DOI] [PubMed] [Google Scholar]
- 32.Rafferty JB, Somers WS, Saint-Girons I, Phillips SEV. Nature. 1989;341:705–710. doi: 10.1038/341705a0. [DOI] [PubMed] [Google Scholar]
- 33.Oberer M, Zangger K, Prytulla S, Keller W. Biochem. J. 2002;361:41–47. doi: 10.1042/0264-6021:3610041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts RC, Spangler C, Helinski DR. J. Biol. Chem. 1993;268:27109–27117. [PubMed] [Google Scholar]
- 35.Chivers P, Sauer RT. J. Biol. Chem. 2000;275:19735–19741. doi: 10.1074/jbc.M002232200. [DOI] [PubMed] [Google Scholar]
- 36.Schreiter E, Sintchak M, Guo Y, Chivers P, Sauer R, Drennan C. Nat. Struct. Biol. 2003;10:794–799. doi: 10.1038/nsb985. [DOI] [PubMed] [Google Scholar]
- 37.Ling M, Allen SW, Wood JM. J. Mol. Biol. 1994;245:950–956. doi: 10.1006/jmbi.1994.1696. [DOI] [PubMed] [Google Scholar]
- 38.Ostrovsky PC, Maloy S. Genes Dev. 1995;9:2034–2041. doi: 10.1101/gad.9.16.2034. [DOI] [PubMed] [Google Scholar]
- 39.Muro-Pastor AM, Ostrovsky P, Maloy S. J. Bacteriol. 1997;179:2788–2791. doi: 10.1128/jb.179.8.2788-2791.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]