

Abstract
Despite having been extensively studied at both the biochemical, haematological and molecular levels, the haemoglobinopathies continue to provide a diagnostic challenge particularly in the multiethnic communities seen in Australia. Early detection and characterisation of the haemoglobinopathies is essential so that appropriate counselling can be provided to couples and families who may be at risk of severe haematological consequences. Although DNA diagnostics have made a major impact on our understanding and detection of the haemoglobinopathies, DNA mutation testing should never be considered a short cut or the test of first choice in the workup of a haemoglobinopathy. A careful three tier approach involving: (1) Full blood count (2) Special haematological tests, followed by (3) DNA mutation analysis, provides the most effective way in which to detect primary gene mutations as well as gene-gene interactions that can influence the overall phenotype. Just as important as the laboratory investigations is the family work up. Often, the first and most helpful clue to gene gene interactions comes from the family study. In Australia, there are many different forms of α and β thalassaemia. Increasingly, different Hb Variants are being detected, and their effects per se, or in combination with the thalassaemias, provide additional diagnostic challenges.
Overview
Haemoglobinopathies are inherited disorders of globin, the protein component of haemoglobin (Hb). Mutations in genes coding for the globin proteins that alter protein output produce the thalassaemia syndromes. Mutations in the globin genes that lead to abnormal proteins are called variant Hbs.1 Haemoglobinopathies are the commonest genetic defect worldwide with an estimated 269 million carriers.2 Certain populations are particularly at risk of having a haemoglobinopathy, for example, in South East Asia, there are 90 million carriers, about 85 million in sub-Saharan Africa and 48 million in the West Pacific region.2
The thalassaemia syndromes and some of the Hb Variants are inherited as autosomal recessive conditions. Very rarely, β thalassaemia demonstrates an autosomal dominant inheritance pattern. Some Hb Variants also have an autosomal dominant inheritance pattern, while others can occur spontaneously.
There are many genes coding for the globins. They are found on chromosome 11 (β globin cluster) or chromosome 16 (α globin cluster) (Figure 1). For the purpose of this review, the important globin genes in humans are the 2 β globin genes and the 4 α globin genes. Despite different numbers of α and β globin genes, the net α/β globin protein output from these genes is balanced to give a ratio of 1.1
Figure 1.
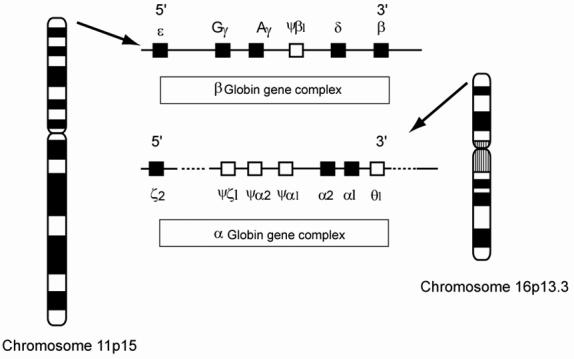
The ancestral globin gene duplicated into the α and β globin genes about 500 million years ago.3 Subsequently, the α and β globin gene clusters have evolved by various duplication events and are now represented by two clusters containing active genes (▪) and inactive or pseudogene genes (□). Another feature of the globin genes is the regulated developmental expression which occurs in a 5′ to 3′ direction.
Classification and Nomenclature
Haemoglobinopathies are primarily grouped into thalassaemia syndromes or variant Hbs (Figure 2). Thalassaemia syndromes are sub-classified based on the gene involved. α and β thalassaemias are further sub-divided into α+, β+ or αo, βo depending on whether some (+) or no (o) globin protein is produced as a result of the causative mutation.
Figure 2.
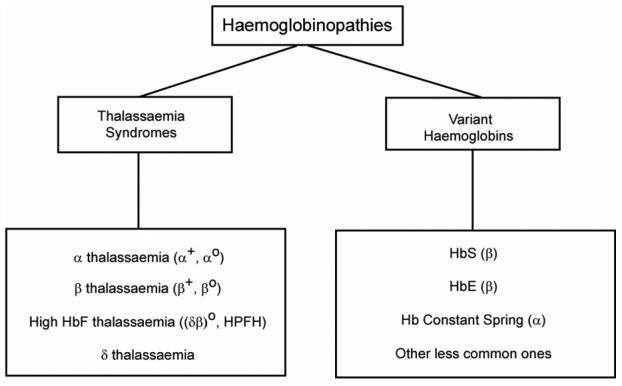
A simple classification of the haemoglobinopathies is based on: (1) An α/β globin protein ratio that is imbalanced i.e. the thalassaemia syndromes. In these conditions, the imbalance is reflected in a low MCV and low MCH. (2) A normal α/β globin ratio but the globin chains produced are abnormal in structure i.e. most variant Hbs have a normal MCV and MCH. The one exception is HbE because of mRNA instability associated with the β globin gene mutation and so HbE is usually found to have a low MCV and MCH.
The high HbF producing thalassaemias are called (δβ)o or HPFH (hereditary persistence of foetal Hb). These relatively rare disorders are primarily associated with defects involving the foetal or γ genes and arise because the switch from HbF (foetal Hb) to HbA (adult Hb) that normally occurs at about 6 months of life is delayed or incomplete. The high HbF thalassaemias do not produce disease phenotypes. However, they are important because they can reduce the severity of β thalassaemias and some Hb Variants such as HbS.
Apart from the earliest described variant Hbs which have an alphabetical number e.g. HbC, HbD, HbE etc., variant Hbs are usually called after the place in which they were discovered. Hence, Hb Constant Spring (a suburb in Jamaica), Hb Camperdown and Hb Westmead (found in the Royal Prince Alfred Hospital in Camperdown or the Westmead Hospital in Sydney etc.). The important variant Hbs in the Australian community include HbE (β CD26 glu>lys), HbS (β CD6 glu>val) and Hb Constant Spring (α CD142 termination TAA>CAA). Another variant associated with sickling and increasingly being detected in Australia is HbC (β CD6 glu>lys). Although a small number of Hb Variants are mentioned in this review, it should be noted that there are over 1000 mutations associated with the haemoglobinopathies, most of which will produce variants.4
Family Studies
In view of the molecular heterogeneity found in the haemoglobinopathies, it is important to characterise each particular underlying abnormality so that appropriate advice can be given about the health implications to individuals, offspring and other family members. Since the haemoglobinopathies are usually inherited as autosomal recessive traits, there is a 1 in 4 (25%) risk of inheriting a potentially severe disorder in children of parents who are carriers of haemoglobinopathies. The severity of the haemoglobinopathy would depend on the type of disorder inherited by the children as well as a number of other factors.5 These include genetic factors such as the ability to produce excessive HbF, and co-inheritance of α thalassaemia (which would make β thalassaemia less severe) or co-inheritance of triplicated α genes (making β thalassaemia more severe). More discussion about these genetic factors is given under complex haemoglobinopathies below. Non-genetic factors that can influence the haemoglobinopathy phenotype include access to and compliance with treatments such as blood transfusion and iron chelation.
The molecular heterogeneity makes accurate prediction of the clinical phenotype difficult since it is possible for various gene-gene interactions to occur.1 This fact means it is essential to undertake family studies in difficult cases. It is only after a pedigree is drawn and the various haematological changes attributed to different family members that it becomes possible to identify these interactions, and then confirm them through DNA testing. See below for further discussion on complex phenotypes.
Risk Populations
The distribution and high frequency of thalassaemia and some Hb variants such as sickle cell Hb (HbS), HbC and HbE reflects the fact that these genetic abnormalities provide a selective advantage in a region that is endemic for P. falciparum malaria. This is because red blood cells, particularly those with thalassaemia, are poor environments for the malaria parasite and so carriers for these haemoglobinopathies are less likely to develop severe malaria and die from it. From an evolutionary perspective, carriers do not have a “disease” but actually have a more appropriate genetic make up for their particular environment. Those with severe haemoglobinopathy will die, but from an evolutionary viewpoint this is an acceptable price to pay for the protection afforded to the more commonly occurring carriers.6
An exception to the association between haemoglobinopathies and malaria is found in Polynesia with over 10% of Maoris and Cook Islanders having α+ thalassaemia. Since malaria is not endemic to this region another explanation is needed. In the case of the Polynesians, this reflects their origin from East Asia with the acquisition of α+ thalassaemia as they migrated through Melanesia. The high frequencies were subsequently reached through genetic drift and founder effects.7
In Australia, the traditional risk populations for thalassaemia were those with origins from the Mediterranean, particularly Greek and Italian forebears. Today, the very diverse ethnic backgrounds in Australia (1 in 4 Australians are now born overseas) provide a very interesting challenge in terms of detection and then mutation testing for the haemoglobinopathies. Risk populations in this country include: South East Asians, Chinese, Middle Eastern, Mediterranean, Black African and various European communities. Our neighbours in the Pacific including some Melanesians and Polynesians are also at risk of having thalassaemia.
Diagnostic Workup
The detection and characterisation of a haemoglobinopathy involves a 3 tier work up. (1) Full blood count (2) Special haematological tests and (3) DNA testing.
Full Blood Count
The key to successful detection and characterisation of the haemoglobinopathies, particularly the thalassaemias, is the initial haematological data. The clue for a thalassaemia comes with a low MCV (mean corpuscular volume) or MCH (mean corpuscular haemoglobin). Although iron deficiency is the other explanation for a low MCV or MCH, it is likely that this finding will point to thalassaemia in regions of Australia with at risk ethnic populations. The first step after the initial abnormal blood count as described above, is to exclude iron deficiency and if present, to treat it. The blood count is then repeated, and if the MCV/MCH remains low, a thalassaemia is most likely.
It should be noted that the MCV can be raised by a number of conditions. In particular, vitamin B12 and folic acid deficiency leads to an elevated MCV. More recently, it has become evident that patients with HIV taking nucleoside analogues also develop an elevated MCV. The latter is thought to occur because of impaired DNA synthesis.9 In this circumstance, the main clue to a thalassemia might be masked because the MCV is falsely normal or even elevated.
There is one important haemoglobinopathy that will be missed if only the MCV or MCH is used for the initial screen. This is the carrier for HbS. Therefore, health professionals or laboratories dealing with populations in which HbS occurs should always include a HbEPG with the request for a full blood count.
As part of the routine full blood count, a blood film can be useful because it might provide a clue to the presence of sickle cell disease (HbS) or an unstable Hb. Changes such as stippling and target cells in the blood film are not definitively associated with a haemoglobinopathy but would be helpful ancillary findings if, in the case of thalassaemia, the MCV or MCH was also low.
TIPS
(1) Some prefer the MCH to the MCV as the key red blood cell parameter since the MCV can change if there is a delay before the blood is processed for testing. Depending on the instrumentation used, there are subtle inter-laboratory differences in the normal ranges for the MCV and these need to be remembered (Table 1). The wider ranges for normal MCV values in Table 1 provide additional reasons for preferring the MCH to the MCV.
Table 1.
Normal ranges for haematological parameters used to detect and characterise haemoglobinopathies in 6 different NSW laboratories.
| NSW Laboratory Number | MCV | MCH pg | HbA2 % |
|---|---|---|---|
| 1 | 76–96 | 27–32 | 1.8–3.5 |
| 2 | 77–98 | 27–32 | 1.5–3.4 |
| 3 | 82–100 | 26–34 | 1.0–3.4 |
| 4 | 80–100 | 26.5–33 | 2.0–3.5 |
| 5 | 80–100 | 27–32 | 1.5–3.5 |
| 6 | 82–98 | 27–32 | 1.8–3.4 |
(2) The RDW (red cell distribution width) measures the coefficient of variation about the MCV. It tends to be higher in iron deficiency but not in the thalassaemias and so can help in pointing to which is more likely. However, particularly during pregnancy, it is not unusual to find both could be present.
(3) Some haemoglobinopathies, particularly HbS, will have a normal MCV and normal MCH and so will be missed if the full blood count is used as a screening test.
(4) Since the foetal to adult β globin switch is not usually complete until about 6 months of life, it is difficult detecting β thalassaemia in the neonate based on the full blood count. However, a paediatric haematologist experienced in looking at blood films and dealing with thalassaemia might make a reasonable guess based on the haematologic parameters and red blood cell changes in the blood film whether these changes are suggestive of thalassaemia.
(5) In risk pregnancies not monitored by prenatal diagnosis (a late diagnosis or the couple is not interested in prenatal testing), it is important to keep some of the cord blood for DNA testing to be undertaken.
Once a haemoglobinopathy is suspected on the basis of a family history, or an abnormal full blood count, a number of additional special haematological tests are ordered. These provide the second tier of knowledge i.e. confirmation of a haemoglobinopathy and the likely type.
Iron Studies
Although it has been suggested that iron deficiency is uncommon in Australian communities with a risk for thalassaemia, it is essential that iron deficiency is always excluded before any further tests are undertaken in haemoglobinopathy workups. Therefore, ferritin levels (and if necessary serum iron, iron binding capacity and percentage saturation) are sought. This is recommended because at times, particularly during pregnancy, it is possible that iron stores will be low or, in the presence of iron deficiency, it is possible that the MCV or MCH are influenced by the iron deficiency. It is also occasionally seen that the HbA2 level can be falsely lowered by iron deficiency. If iron deficiency is present, it is essential to correct this, and then repeat the full blood count and all other investigations.
Special Haematological Tests
Once a haemoglobinopathy is suspected, the next tier of investigation requires a number of special haematological tests. These are often described as a “thalassaemia screen”. However, a better term in the context of the modern multiethnic community in Australia is a “haemoglobinopathy screen”. The tests are listed in Table 2. It is appropriate to order all tests simultaneously since they take time to do, and often information can be obtained from tests that might not have been seen to be relevant when first starting off the investigation of a haemoglobinopathy. Some of the tests are technically demanding and so the person ordering the tests should have some knowledge of the laboratory’s technical skills as well as experience in interpreting results.
Table 2.
Special haematology tests requested once a haemoglobinopathy is suspected based on family history and/or full blood count. Often these tests are ordered by asking for a “thalassaemia or haemoglobinopathy screen”.
| Test | What does it measure or detecta | What does it mean |
|---|---|---|
| HbEPG | Electrophoresis of globin proteins. Different techniques possible from gel or membrane based kits to HPLC. Abnormal bands apart from the usual HbA, HbF and HbA2 peaks can be detected. | (1) Gives some idea of the HbA2 level but more importantly (2) identifies if there are any variant Hbs – particularly Hbs such as HbE and HbS in the Australian context. |
| HbA2 | Globin electrophoresis and quantitation of the HbA2 peak. Different techniques used from membrane or column based kits to the more universally suited HPLC are in use. | A raised HbA2 is the key parameter indicating the presence of β thalassaemia.b It is said that variant Hbs can raise the HbA2 but this must be a rare event. More of an issue is the borderline normal-raised HbA2 because this might indicate silent β thalassaemia. A low HbA2 is also important to note as this might indicate δ thalassaemia (discussed further in Table 4). |
| HbF | Globin electrophoresis and quantitation with different methods available for the latter. | A slightly raised HbF to 2–3% (normal is <1% in an adult) might indicate heterocellular HPFH or may be a subtle pointer to an underlying silent β thalassaemia. HbF levels 5% and above are more likely to be due to δβ thalassaemia or HPFH (heterocellular or pancellular). In the case of δβ thalassaemia or deletional HPFH one would expect the HbA2 level to be low. |
| Kleihauer | Red blood cells are stained to detect HbF. This test is used to distinguish heterocellular from pancellular HPFH. | Not a particularly useful test for distinguishing the types of HPFH because these are very rare and most laboratories are not sure how to interpret the results. The only practical value for a Kleihauer stain might be in foetal blood sampling to confirm that maternal blood has not contaminated a foetal sample (the latter would be homogeneously stained for HbF). Any cells not staining for HbF would represent maternal blood. |
| HbH inclusions | Red blood cells are stained to detect HbH inclusions (aggregates of β globin protein) | Definitive test to confirm the presence of α thalassaemia. Of the tests described, this is the one most likely to cause problems because of a false negative result. Requires patience and skill to find the HbH inclusions and even with a 2 gene deletion α thalassaemia, only 1–2 such inclusions might be found after a search lasting many minutes. Therefore, HbH inclusions are easy to miss if the laboratory is inexperienced or the individual looking down the microscope does not spend enough time searching for these inclusions. |
| Sickle solubility and instability tests | Various tests ranging from biochemical to immunoassay are used to detect HbS and unstable variant Hbs | HbS disease as well as interactions of HbS with β thalassaemia are increasingly being detected in many Australian cities. Therefore, efficient and accurate tests for sickling (sickle solubility, HbEPG) are important components of the haemoglobinopathy workup. |
See reference 8 for more information on the various techniques listed in this table.
There are other reasons why the HbA2 might be elevated. These are very uncommon and include: some unstable Hbs, hyperthyroidism, and megaloblastic anaemia. However, in most cases it should be possible to distinguish a thalassaemia from the above.
Other special haematological tests are possible, particularly when investigating the more uncommon variant Hbs. These include tests for oxygen affinity, haemoglobin stability and detection of methaemoglobin. Mass spectrometry has been used to characterise various variant Hbs.10 The latter approach might be very valuable for population screening, but for detection involving individual cases, DNA based approaches remain the methods of choice.
DNA tests, discussed in more detail below, are only as good as the special haematology tests described in Table 2. This is because the DNA laboratory needs to know whether the α or β globin genes or both need to be characterised. Since DNA technology in haemoglobinopathy testing is based predominantly on PCR (polymerase chain reaction) and Southern blotting is less readily available, the DNA laboratory is also faced with deciding whether it is looking for point mutations or deletions. The latter are particularly relevant since PCR will miss deletions unless the appropriate Gap-PCR strategy is followed (see below). Another common source of error with the potential to mislead the DNA laboratory is the failure to find HbH inclusion bodies. Not infrequently, a DNA laboratory works on the β globin gene because HbH inclusion bodies had not been detected. After considerable effort, including DNA sequencing, no mutation is found. At this stage, the DNA laboratory usually goes back to the haematology laboratory to ask for a review of the HbH inclusions, and these are now detected.
Another useful specialised test helpful in confirming a thalassaemia and whether it is due to an α or β globin gene problem is the α/β globin protein ratio. This requires the incubation of red blood cells with a radioactive tracer such as 3H-leucine. The peaks representing α and β globin proteins are then quantitated to provide an α/β ratio which should equal 1. A ratio > 1 indicates β thalassaemia while a ratio <1 is caused by α thalassaemia. Although a useful test in some cases, the α/β ratio is no longer routinely available. This has occurred because DNA testing has essentially become the strategy of choice for testing and so few α/β ratios are now requested that laboratories do not have sufficient experience. The requirement for fresh radioactive material is another disincentive for setting up this assay. It should also be noted that the α/β ratio may not be particularly helpful if gene-gene interactions are occurring, for example, both α and β thalassaemia are co-inherited.
TIPS
(1) Failure to find HbH inclusions when they are present is an important error that produces a lot of unnecessary DNA testing. In these circumstances, the DNA testing laboratory may be unsure of whether to test for α or β thalassaemia. Prior to ordering expensive and time consuming DNA tests, it is often beneficial to ask the haematology laboratory to check again for HbH inclusions.
(2) Another dilemma for the DNA laboratory is the equivocally-raised HbA2 result because it means that normal HbA2 β thalassaemia cannot be excluded. In this circumstance, the HbA2 test should be repeated.
(3) It is important to refer your samples to a haematology laboratory that is experienced in thalassaemia and variant Hb work, and you are confident that sufficient time will be allowed for the haemoglobinopathy screen to be undertaken optimally.
DNA Tests
The 3rd diagnostic tier involves DNA testing. This is requested in two circumstances: (1) A haemoglobinopathy cannot be confirmed by the special haematological tests. At times it is possible to suspect a haemoglobinopathy but the haematology tests (as well as the family studies) cannot determine which gene is likely to be involved. (2) The underlying mutation is being sought in a confirmed haemoglobinopathy. This is usually required as part of a prenatal workup. A detailed description of the various approaches used to detect DNA mutations is beyond the scope of this review. Readers interested in more technical aspects of DNA mutation testing should read the monograph edited by Cotton, Edkins and Forrest.11
In general, the α thalassaemias are caused by gene deletions although increasingly non-deletional forms of α thalassaemia are being sought in difficult cases. In contrast, the β thalassaemias and Hb Variants are, in most cases, the result of point mutations. Thus, DNA mutation strategies need to be developed with some knowledge of the likely underlying defect that will need to be detected.
Mutation analysis
Most laboratories providing a DNA testing service for thalassaemia do so through the identification of common mutations present in the population serviced by the laboratory, or on the basis of the underlying haematological phenotypes (Table 3). There are many different DNA mutation detection methods in use but, with very few exceptions, they are based on PCR. An interesting paper by Chan and colleagues describes the development of a thalassaemia array for the simultaneous analysis of 15 non-deletional α gene defects and 23 β thalassaemia mutations likely to be present in a Hong Kong population.12 These 38 defects involved single base mutations and so could be simultaneously detected by printing the relevant oligonucleotides onto glass slides. This approach will be increasingly utilised, particularly as the cost of microarrays comes down, and will prove particularly valuable for population screening.
Table 3.
Strategies used in the author’s laboratory to screen for likely mutations leading to α or β thalassaemia. Abnormal results are then confirmed using a second and different mutation detection method.
| Assay | Range of Mutations Detecteda | Comments |
|---|---|---|
| 5 Plex α thalassaemia deletional screen | αo defects = --/MED; --/SEA and 20.5 kb deletion | Gap-PCR strategy for an initial screen to detect some of the common forms of deletional α thalassaemia. |
| α+ defects = 3.7, 4.2 | ||
| 6 Plex α thalassaemia non-deletional screen | Initiation codon ATG>A-G, CD30 (ΔGAG), CD35 (T>C), Hb Adana (CD59 G>A), Hb Quong Sze (CD125 T>C) and Hb Constant Spring (termination codon TAA>CAA). | ARMS based screening for some of the non deletional α thalassaemia defects. |
| Mediterranean 5 Plex β thalassaemia screen | Detects the following point mutations: IVS1-1 (G>A); IVS1-6 (T>C), IVS1- 110 (G>A), IVS2-1 (G>A) and CD39 (C>T). | ARMS based screening for some of the more common non deletional β thalassaemia defects. |
| Chinese and Indian Asian 7 Plex β thalassaemia screen | Detects the following mutations: IVS1-1 (G>T), IVS1-5 (G>C), −28 (A>G), CD17 (A>T), CD41-42 (− TCTT), CD8/9 (+G), CD30 (G>C) | ARMS based screening for some of the more common non deletional β thalassaemia defects. |
| Silent β thalassaemia (or normal HbA2 β thalassaemia) | β globin gene mutations associated with this phenotype include: −101 (C>T), −92 (C>T), CAP+1 (A>C), +10 (−T), +33 (C>G), CD27 (G>T), +1480 (C>G), and the polyA tail mutation AATAAA > AATAGA. | In the difficult situation of trying to characterise silent β thalassaemia, the various mutations spread across the β globin gene were individually characterised. Now, it is easier to sequence the β globin gene in this phenotype. |
| Corfu type δβ thalassaemia also produces this phenotype |
MED – Mediterranean; SEA – South East Asian; ARMS – amplification refractory mutation system.
In replacing gene mapping, PCR has revolutionised the turnaround time possible for DNA testing, as well as expanding the repertoire of tests, particularly in terms of identifying single point changes, which overall are the most common mutations in the haemoglobinopathy. However, the inability of many DNA laboratories to do a Southern blot and the complete reliance on PCR comes at a cost if unknown or rare deletions are present. While PCR detects deletions if a technique such as gap-PCR is used, it is necessary when doing gap-PCR to know breakpoints, and this information is not always available.13 In these circumstances, PCR amplifies the remaining (normal) allele, and so the DNA laboratory completes a lot of work but to no avail since it is based on amplification of the normal allele i.e. the DNA region being characterised is actually hemizygous.
When dealing with deletional disorders such as α thalassaemia (and to a much lesser extent the β thalassaemias), it would be worthwhile having mutation testing strategies based on Q-PCR because these will allow the loss of an allele or a locus to be detected. Although some Q-PCR based approaches have been described in the deletional haemoglobinopathies, they are not techniques that have attracted much interest, perhaps because they are likely to be needed only occasionally. A nice example of Q-PCR based on TaqMan probe technology has been described to detect a common deletional form of αo thalassaemia associated with Hb Bart’s hydrops foetal is in Chinese communities.14 Another potentially interesting approach to detecting copy number has recently proven useful in mutation testing for genomic deletions and duplications in the BRCA1 gene. This method is called MPLA (multiplex ligation-dependent probe amplification) and relies on a ligation based assay and PCR.15,16 It is reported that up to 40 target sequences (or exons) can be quantitated in the one multiplex reaction with MPLA.
DNA Scanning
A strategy often used when many different underlying mutations are possible involves the screening (a better term is scanning since screening can be confused with other DNA uses such as population screening) of DNA segments for nucleotide changes using techniques such as SSCP (single stranded conformation polymorphism), and DGGE (denaturing gradient gel electrophoresis).13 Most recently, scanning by DHPLC has become the method of choice. These techniques are based on different melting temperatures for heteroduplexes compared to homoduplexes which are then detected by migration profiles on electrophoresis. DNA scanning allows different DNA segments or exons to be identified as sites for possible mutations. Any changes found must then be confirmed by DNA sequencing. The latter is essential to distinguish mutations from neutral polymorphisms.
DNA scanning is useful when looking for mutations in large genes since it becomes possible to dissect the gene into discrete fragments e.g. 500 bp in size. We have successfully utilised scanning by DHPLC in large genes such as the cardiac β myosin heavy chain when looking for mutations that cause familial hypertrophic cardiomyopathy.17 Scanning would also be feasible in the haemoglobinopathies although with increasing availability and cheaper costs for DNA sequencing most would go straight from direct DNA mutation analysis to DNA sequencing since the globin genes are relatively small in size (about 1.6 kb in size with 3 exons) compared to a large gene such as cardiac β myosin heavy chain with 40 exons over 25 kb of genomic DNA.
DNA Sequencing
Direct DNA sequencing is now routinely used to look for mutations in the β and α globin genes. Generally sequencing is indicated if mutations are not detectable with the preliminary screening approaches described above. Increasingly though, direct DNA sequencing is being used in difficult cases, for example, the finding of normal HbA2 β thalassaemia (also called silent β thalassaemia) is best dealt with by sequencing the β globin gene because there are a number of mutations known to be associated with this phenotype. Occasionally, normal HbA2 β thalassaemia is found in association with the conventional β thalassaemia causing mutations, so overall DNA sequencing is usually preferred.
Some technical advances in DNA sequencing have enhanced this option for DNA mutation detection. They are sequencing using dye primer or dye terminator chemistry and, more recently, the coupling with fluorescently labelled M13 primers to initiate elongation. With this approach the β globin gene can be entirely sequenced (apart from a small segment in IVS2) with four primer sets.13 Both the α1 and α2 globin genes can also be sequenced with four primer sets. Since thalassaemia is a recessive condition, the DNA sequence will show only one mutation superimposed on the normal nucleotide base. Because this can be missed (for technical reasons or perhaps not called by the sequencing software or missed on visual inspection) it is necessary to sequence both forward and reverse strands (Figure 3).
Figure 3.
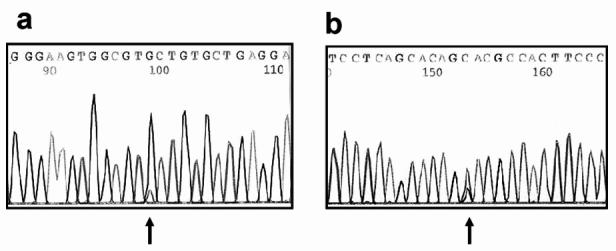
. DNA sequencing trace showing how a heterozygous mutation can be missed on DNA sequencing (a) forward primer, (b) reverse primer. This particular example involves direct DNA sequencing to detect the common A149P mutation in the aldolase B gene (mutations in which cause hereditary fructose intolerance). In both (a) and (b) the DNA sequencing software has miscalled the mutation (indicated by arrows). However, in the reverse primer it is clear on visual inspection that there is a point mutation with both G and C being present. The forward sequence, when it has been magnified, shows a small “blip” representing the mutant C base under the normal G sequence. However, in the actual trace this could easily be missed by a cursory glance at the sequence, or if only the sequence from the forward primer had been used.
Despite the advances making DNA sequencing cheaper and more accessible, it is still necessary to inspect the sequence visually. This is both tedious and a potential source of error. Increasingly software is being developed to assist in sequence analysis. One program from Applied Biosystems is called SeqScape®. This allows the site of known mutations to be quickly marked in any trace. See Applied Biosystems web site for more information on SeqScape®.18
TIPS
(1) Despite the attraction of going to a definitive test i.e. DNA mutation analysis, and the increasing use of DNA sequencing to characterise difficult cases, it needs to be emphasised that a lot of unnecessary work will be undertaken in a DNA laboratory if adequate and accurate haematologic studies are not available to identify what are likely to be the gene(s) involved as well as the potential for gene to gene interactions. The latter might be found serendipitously by a DNA test, but are more likely to be sought and found if the haematology results coupled with good family studies identify the likelihood that these are present.
(2) Increasingly, DNA laboratories are being asked to undertake mutation analysis without iron deficiency having been excluded. This is not good laboratory practice and will inevitably lead to unnecessary work or even an erroneous result because a HbA2 level was falsely low due to co-existent iron deficiency.
Complex Haemoglobinopathies and Gene - Gene Interactions
An interesting challenge when working with the haemo-globinopathies is the heterogeneity of mutations and gene-gene interactions possible in these disorders. Thus, the finding of an α or β globin gene mutation in one member of a family does not exclude the possibility that the other side of the family may have a completely different mutation affecting the same or another globin gene. Therefore, it is important to always keep this in mind when interpreting results.
Two interesting situations that are not that rare, depending on the ethnic group of the patient, are illustrated in Table 4. In the majority of cases, gene-gene interactions will only be suspected if a careful family study is undertaken. The underlying basis for the gene-gene interaction can then be proven by DNA analysis.
Table 4.
Examples of gene-gene interactions that will not be identified without adequate clinical and laboratory workups.
| Scenario | Possible interactions | Importance of identifying |
|---|---|---|
| South East Asian couple with one partner having heterozygous HbE and the other heterozygous αo thalassaemia. (αα/--). | Since various forms of α thalassaemia are common in this population, it is important to exclude its co-existence in the individual with HbE. | The combination of HbE and α thalassaemia in this couple does not place an increased risk of a severe haemoglobinopathy on their children. However, co-existent α thalassaemia in the individual with heterozygous HbE would be associated with a 25% risk for HbH disease or even Hb Bart’s hydrops foetal is in the offspring. Co-existent α thalassaemia may not easily be detectable on haematologic studies and so DNA testing is needed to exclude this risk. |
| A pregnant couple with a Mediterranean ethnic background requests advice about the risk of β thalassaemia for children. The woman has straightforward β thalassaemia while the male partner has a low MCV and MCH and normal iron stores. All other haematological studies are normal including the HbA2. HbH bodies were not detected. A normal HbEPG excluded HbS. | After iron deficiency is excluded, the male is likely to have thalassaemia and it will be α or normal HbA2 β thalassaemia. This is a dilemma because the haematological studies have not been helpful in identifying which globin gene needs to be investigated at the DNA level. | Without family studies the DNA laboratory will spend a considerable amount of time trying to sort out this problem. However, when the male’s family members were studied i.e. parents and 3 siblings, it became apparent from the haematology results that his phenotype was due to interacting β and δ thalassaemias. This meant that he has β thalassaemia and so this couple is at 1 in 4 risk of having a child with homozygous β thalassaemia. The HbA2 which relies on globin produced by the δ globin gene failed to be elevated because of a mutation in this gene. The mutation was confirmed by DNA sequencing.19 |
Although not strictly a haemoglobinopathy, the triplicated α globin gene (ααα/) may influence the phenotype. This gene rearrangement occurs as a consequence of the inter-chromosomal cross over which produces the α+ thalassaemia defect (-α).20 Per se this rearrangement is associated with normal haematology and would not be detected by the tier 1 and tier 2 tests described earlier. However, the net mRNA output from the triplicated α globin gene is greater than the normal two gene structure. Hence, the triplicated α globin gene can make β thalassaemia more severe as a lack of β coupled with a greater output from the α globin genes would accentuate further the α / β ratio imbalance.
Both the α and β globin gene clusters are regulated by distant control regions known as HS-40 and β LCR respectively. These are located 5–40 kb upstream of the globin gene complexes and comprise DNAase I hypersensitive sites. Very rarely mutations in either the HS-40 or the β LCR have been reported, and their effect is to inhibit the related downstream globin gene complex. In the case of the β LCR this produces a normal HbA2 thalassaemia since down-regulation of the δ globin gene would occur.1 Because these types of mutation are very rare, they are not normally sought by conventional mutation analysis strategies or even DNA sequencing. Hence, irrespective of how much DNA mutation testing is undertaken, failure to find a mutation does not exclude an underlying thalassaemia.
Footnotes
Competing interests: None declared
References
- 1.Weatherall DJ, Clegg JB. The thalassaemia syndromes. (4th ed), Blackwell Scientific; 2001.
- 2.Angastiniotis M, Modell B. Global epidemiology of hemoglobin disorders. Ann N Y Acad Sci. 1998;850:251–69. doi: 10.1111/j.1749-6632.1998.tb10482.x. [DOI] [PubMed] [Google Scholar]
- 3.Jeffreys AJ. Gene evolution. In: Genetic Engineering (2nd ed), Robert Williamson, Academic Press;1981, p 1–48.
- 4.A list of Hb variants and thalassaemias may be found in:http://globin.cse.psu.edu/html/huisman/variants/ (accessed September 30, 2005).
- 5.Rund D, Rachmilewitz E. Beta-Thalassaemia. N Engl J Med. 2005;353:1135–46. doi: 10.1056/NEJMra050436. [DOI] [PubMed] [Google Scholar]
- 6.Weatherall DJ, Miller LH, Baruch DI, et al. Malaria and the red blood cell. In http://www.asheducationbook.org/cgi/reprint/2002/1/35 (accessed September 30, 2005).
- 7.Trent RJ, Mickleson KN, Yakas J, Hertzberg M. Population genetics of the globin genes in Polynesians. Hemoglobin. 1988;12:533–7. doi: 10.3109/03630268808991642. [DOI] [PubMed] [Google Scholar]
- 8.Working party of the general haematology task force of the British committee for standards in haematology. Guideline: the laboratory diagnosis of haemoglobinopathies. Brit J Haemat. 1998;101:783–92. [Google Scholar]
- 9.Howard J, Henthorn JS, Murphy S, Davies SC. Implications of increased haemoglobin A2 values in HIV positive women in the antenatal clinic. J Clin Pathol. 2005;58:556–8. doi: 10.1136/jcp.2004.018697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wild BJ, Bain BJ. Detection and quantitation of normal and variant haemoglobins: an analytical review. Ann Clin Biochem. 2004;41:355–69. doi: 10.1258/0004563041731600. [DOI] [PubMed] [Google Scholar]
- 11.Cotton RGH, Edkins E, Forrest S editors. Mutation detection. A practical approach. IRL Press Oxford; 1998.
- 12.Chan K, Wong MS, Chan TK, Chan V. A thalassaemia array for Southeast Asia. Br J Haematol. 2004;124:232–9. doi: 10.1046/j.1365-2141.2003.04758.x. [DOI] [PubMed] [Google Scholar]
- 13.Clark BE, Thein SL. Molecular diagnosis of haemoglobin disorders. Clin Lab Haematol. 2004;26:159–76. doi: 10.1111/j.1365-2257.2004.00607.x. [DOI] [PubMed] [Google Scholar]
- 14.Chan V, Yip B, Lam YH, Tse HY, Wong HS, Chan TK. Quantitative polymerase chain reaction for the rapid prenatal diagnosis of homozygous alpha-thalassaemia (Hb Barts hydrops foetalis) Br J Haematol. 2001;115:341–6. doi: 10.1046/j.1365-2141.2001.03112.x. [DOI] [PubMed] [Google Scholar]
- 15.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucl Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hogervorst FB, Nederlof PM, Gille JJ, et al. Large genomic deletions and duplications in the BRCA1 gene identified by a novel quantitative method. Cancer Res. 2003;63:1449–53. [PubMed] [Google Scholar]
- 17.Yu B, Sawyer NA, Caramins M, et al. Denaturing high performance liquid chromatography: high throughput mutation screening in familial hypertrophic cardiomyopathy and SNP genotyping in motor neurone disease. J Clin Pathol. 2005;58:479–85. doi: 10.1136/jcp.2004.021642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reference to SeqScape may be found in: http://www.appliedbiosystems.com/ (accessed September 30, 2005).
- 19.Trent RJ, Thein SL. Detection of beta and delta globin gene mutations by PCR and direct DNA sequencing in an individual with normal HbA2 beta thalassemia. Pathology. 1992;24:15–8. doi: 10.3109/00313029209063614. [DOI] [PubMed] [Google Scholar]
- 20.Trent RJ, Higgs DR, Clegg JB, Weatherall DJ. A new triplicated alpha-globin gene rearrangement in man. Br J Haemat. 1981;49:49–52. doi: 10.1111/j.1365-2141.1981.tb07207.x. [DOI] [PubMed] [Google Scholar]