

Abstract
Bacteria have developed complex strategies to detoxify and repair damage caused by reactive oxygen species. These compounds, produced during bacterial aerobic respiration as well as by the host immune system cells as a defense mechanism against the pathogenic microorganisms, have the ability to damage nucleic acids, proteins and phospholipid membranes. Here we describe the crystal structure of Pseudomonas aeruginosa Ohr, a member of a recently discovered family of organic hydroperoxide resistance proteins. Ohr is a tightly folded homodimer, with a novel α/β fold, and contains two active sites located at the monomer interface on opposite sides of the molecule. Using in vitro assays, we demonstrate that Ohr functions directly as a hydroperoxide reductase, converting both inorganic and organic hydroperoxides to less toxic metabolites. Site-directed mutagenesis confirms that the two conserved cysteines in each active site are essential for catalytic activity. We propose that the Ohr catalytic mechanism is similar to that of the structurally unrelated peroxiredoxins, directly utilizing highly reactive cysteine thiol groups to elicit hydroperoxide reduction.
Keywords: bacterial resistance/organic hydroperoxides/oxidative stress/peroxiredoxin/Pseudomonas aeruginosa
Introduction
Pseudomonas aeruginosa, a versatile and ubiquitous Gram-negative bacterium, is one of the major causes of opportunistic human infections, such as bacteremia in burn victims, urinary tract infections in catheterized patients and pneumonia. It is also a significant cause of morbidity and mortality in cystic fibrosis patients (Stover et al., 2000). Pathogenic bacteria, including P.aeruginosa, that contact the human host at the mucosal surface are exposed to a variety of reactive oxygen and nitrogen species, including superoxide, hydroxyl radical, hydrogen peroxide (H2O2), peroxynitrite and organic hydroperoxides (OHPs) (Skaar et al., 2002). The latter are generated as by-products of bacterial aerobic metabolism (Gonzalez-Flecha and Dimple, 1997), and as reaction intermediates during host defense responses following the release of the lysosomal contents within inflammatory cells and the neutrophil oxidative burst (Atichartpongkul et al., 2001; Shea et al., 2002). OHPs can damage cellular macromolecules via their ability to generate free organic radicals, which can react with nucleic acids, proteins and lipids, causing DNA mutations, inactivation of enzymes and oxidation of phospholipid membranes (Niki, 1992; Mongkolsuk et al., 1998). Consequently, OHP detoxification is essential for bacterial survival and proliferation in the infected host, and genes involved in protection against organic peroxide toxicity play important roles in host– pathogen interactions (Mongkolsuk et al., 1998).
Microorganisms have evolved several defense systems to detoxify reactive oxygen species (Storz and Zheng, 2000). Examples include the P.aeruginosa exopolysaccharide alginate and the phenolic glycolipid of mycobacteria, which are both effective scavengers of oxidants (Neill and Klebanoff, 1988; Simpson et al., 1989). Catalases, glutathione peroxidases/glutathione reductases and peroxiredoxins represent enzymatic hydroperoxide detoxification systems (Haas and Goebel, 1992). Peroxiredoxins are a family of peroxidases that are present in organisms from all kingdoms, and that exist in multiple isoforms in most eukaryotic cells (Rhee et al., 2001). For example, AhpR, a Salmonella typhimurium peroxiredoxin, plays a role in protecting the bacterium against OHP-induced mutagenesis of DNA by detoxifying thymine hydroperoxide, 5-hydroperoxy-6-hydroxy-dihydrothymine and linoleic acid hydroperoxide into the corresponding less noxious alcohols (Jacobson et al., 1989). Bacteria also possess enzymes, such as the peptide methionine sulfoxide reductase (MsrA/B) and the adenine glycosylase (mutY), which repair oxidatively damaged proteins and DNA, respectively (Bernards et al., 2002; Skaar et al., 2002).
A recently described family predicted to have the ability to detoxify OHPs encompasses the organic hydroperoxide resistance (Ohr) and the osmotically inducible (OsmC) proteins (Mongkolsuk et al., 1998; Atichartpongkul et al., 2001). Pseudomonas aeruginosa Ohr is essential for optimal resistance against oxidative stress generated by exposure to cumene hydroperoxide (CHP) and tert-butylhydroperoxide (t-BHP), and contributes to the decomposition of OHPs in bacterial culture (Mongkolsuk et al., 1998; Ochsner et al., 2001). In Enterococcus faecalis, a bacterium responsible for endocarditis and meningitis, Ohr protects against t-BHP and ethanol stress (Rince et al., 2001). In the pulmonary pathogen Actinobacillus pleuropneumoniae, ohr expression is specifically induced by exposure to CHP both in broth cultures and in an in vivo model of porcine lung infection (Shea and Mulks, 2002). The bacterial phytopathogen Xanthomonas campestris, lacking functional Ohr, is hypersensitive to t-BHP, but its resistance can be restored to wild-type levels by the expression of the AhpC–AhpF peroxiredoxin system (Mongkolsuk et al., 1998). Additionally, ohr expression plasmid complements the OHP-hypersensitive phenotype of an Escherichia coli ahpC-ahpF double deletion mutant (Mongkolsuk et al., 1998). Escherichia coli lacking OsmC is also hypersensitive to H2O2 and t-BHP, indicating that OsmC is also involved, either directly or indirectly, in defense against oxidative stress (Conter et al., 2001). Taken together, these findings suggest overlapping or complementary functions between the Ohr/OsmC and the peroxiredoxin protein families.
In Bacillus subtilis, Ohr protein levels are regulated by the OHP sensor protein OhrR (Fuangthong and Helmann, 2002). Prior to OHP exposure, OhrR represses ohr transcription by cooperative binding to two inverted repeat elements of the ohr promoter. OHPs can reversibly oxidize OhrR at a single cysteine residue to a cysteine–sulfenic acid (-SOH) derivative, temporarily blocking its ability to bind DNA, and therefore allowing for ohr transcription and peroxide detoxification by the newly accumulated Ohr.
Ohr and OsmC family members are present in both Gram-positive and Gram-negative bacteria and share no significant sequence homology to other prokaryotic or eukaryotic proteins. The sequence identity within each of the Ohr and OsmC subfamilies is between 40 and 70%, while between the subfamilies it is ∼20% (Figure 1A). The more conserved C-terminal region (residues 40–143) contains two invariant cysteine residues that have been suggested to play a role in OHP detoxification (Mongkolsuk et al., 1998). The residues around the second cysteine (Cys124 in P.aeruginosa Ohr) are conserved within members of the Ohr and OsmC subfamilies, but are different between the two subfamilies (Atichartpongkul et al., 2001). In Ohr, Cys124 is a part of a Val-Cys-Pro sequence motif, which is also found around the catalytically active cysteine residues in many peroxiredoxins, suggesting that these cysteines may play a similar role during catalysis.

Fig. 1. (A) Structure-based sequence alignment of representative Ohr proteins prepared with CLUSTAL_X (Thompson et al., 1997). Identical residues are depicted on a blue background, conservative substitutions are in yellow and the catalytically active cysteines in red. The three P.aeruginosa leucine residues (Leu39, Leu65 and Leu108) replaced by methionines are indicated in green. The Ohr sequences are as follows: Pa, Pseudomonas aeruginosa; Li, Listeria innocua; Sa, Staphylococcus aureus; Rs, Ralstonia solanacearum; Xp, Xanthomonas campestris pv. phaseoli; Xf, Xylella fastidiosa; Dr, Deinococcus radiodurans; Ap, Actinobacillus pleuropneumoniae; Vc, Vibrio cholerae; Mg, Mycoplasma genitalium; Mp, Mycoplasma pneumoniae. Escherichia coli (Ec) OsmC sequence is aligned below the Ohr sequences. Both identical and conserved amino acids within the OsmC subfamily are marked in purple. The asterisk corresponds to a four amino acid (Ala-Ile-Thr-Lys) insertion in the OsmC sequence. (B) Topology diagram of the Ohr monomer. β-strands (S1–S6) are depicted in green; 310 helix (H1) and α-helices (H2–H4) are in red.
The molecular mechanism that the Ohr/OsmC proteins utilize to protect bacteria has been a subject of speculation. In addition to the suggestion that they might facilitate OHP reduction either directly or indirectly, it was also proposed that they may act as transmembrane transporters extruding OHPs outside of the bacterial cell. The latter hypothesis was based on the presence of a long stretch of hydrophobic amino acids, consistent with the existence of a transmembrane region (Mongkolsuk et al., 1998). To understand better the biological function of these proteins, we crystallized and determined the structure of P.aeruginosa Ohr, revealing a structurally novel peroxidase family. We further demonstrate that Ohr possesses hydroperoxide reductase activity with a preference for organic substrates. Based on the structural, biochemical and mutagenesis data, we propose a model for the Ohr catalytic mechanism.
Results
Structure determination
Pseudomonas aeruginosa Ohr was expressed in E.coli and crystallized by hanging-drop vapor diffusion. The protein contains only one methionine residue and is, therefore, not suitable for selenomethionine-based multiwavelength anomalous dispersion (MAD) experiments (Hendrickson, 1991). An Ohr variant was therefore engineered, containing three additional methionines. Using information provided by the sequence alignment of Ohr proteins from different bacteria, a site-directed mutagenesis strategy was employed to replace three leucine residues (Leu39, Leu65 and Leu108) with methionines, which are naturally present in these positions in some homologs (Figure 1A). The structure of the selenomethionine-modified mutant Ohr, which is biochemically indistinguishable from the wild-type enzyme, was determined using the MAD method at 2.0 Å resolution (Table I). The model contains all 142 residues and is refined to an R-factor of 22%, with restrained temperature factors and good stereochemistry.
Table I. Summary of crystallographic analysis.
| |
Inflection |
Peak |
Remote |
| Wavelength (Å) | 0.979 | 0.979 | 0.961 |
| Resolution (Å) | 20–2.0 | 20–2.0 | 20–2.0 |
| Completeness (%) | 97.9 | 97.6 | 97.1 |
| Anomalous completeness (%) | 97.4 | 97.2 | 96.9 |
| Redundancy (fold) | 2.1 | 2.1 | 1.9 |
| I/σI | 15.6 | 15.1 | 15.1 |
| Rmergea | 5.3 | 4.5 | 5.6 |
| F.O.M. (SOLVE) | 0.60 | ||
| F.O.M. (RESOLVE) | 0.76 | ||
| Space group | P21 | ||
| Cell dimensions (Å) | a = 39.95 b = 77.56 c = 43.12 | ||
| |
α = 90.0 β = 116.3 γ = 90.0 |
|
|
| Refinement |
|
|
|
| Resolution (Å) | 8.0–2.0 | ||
| Reflections | |||
| Working | 26 831 | ||
| Test | 2848 | ||
| Non-hydrogen atoms | 2220 | ||
| No. of waters | 170 | ||
| Rcrys/Rfree | 22%/25% | ||
| R.m.s.dsb | |||
| Bonds (Å) | 0.005 | ||
| Angles (°) | 1.31 |
aRmerge = ∑|I – <I>|/∑I, where I is the observed intensity and <I> is the average intensity obtained from multiple observations of symmetry-related reflections.
bR.m.s.ds in bond lengths and angles are the respective r.m.s.ds from ideal values.
Overall structure
The biologically active Ohr is a homodimer and its structure is illustrated in Figure 2A–C. The protein has overall dimensions of ∼35 × 25 × 25 Å and a symmetrical, oval shape, with the two monomers tightly wrapped around each other in a head-to-tail orientation to generate a compact quaternary structure. The monomers are structurally identical, except for the crystallographic packing at a flexible surface loop. The two conserved cysteines of each molecule are located in the cleft regions at the dimer interface, indicating the positions of the two active sites. The interactions between the monomers are dominated by van der Waals helix–helix packing interactions of two long helices at the center of the hydrophobic core of the dimeric enzyme. Two large antiparallel β-sheets wrap around the central helices to form a barrel-like structure, which is completed by the four shorter α-helices to fully surround the central helical core. Each β-sheet is composed of six strands, three of which derive from one monomer and three from the other. Since the two-helix bundle at the center of the hydrophobic core, as well as the surrounding β-sheets, are generated by combining elements of both monomers, it is clear that the two polypeptide chains have to fold together to form active Ohr, and that each monomer would individually be unstable. The solvent-exposed sur face area of each monomer is ∼9200 Å2 and that of the Ohr dimer is ∼12 000 Å2. The interface between the monomers buries a total of 3200 Å2 of solvent-accessible surface per molecule, or ∼35% of its total surface.
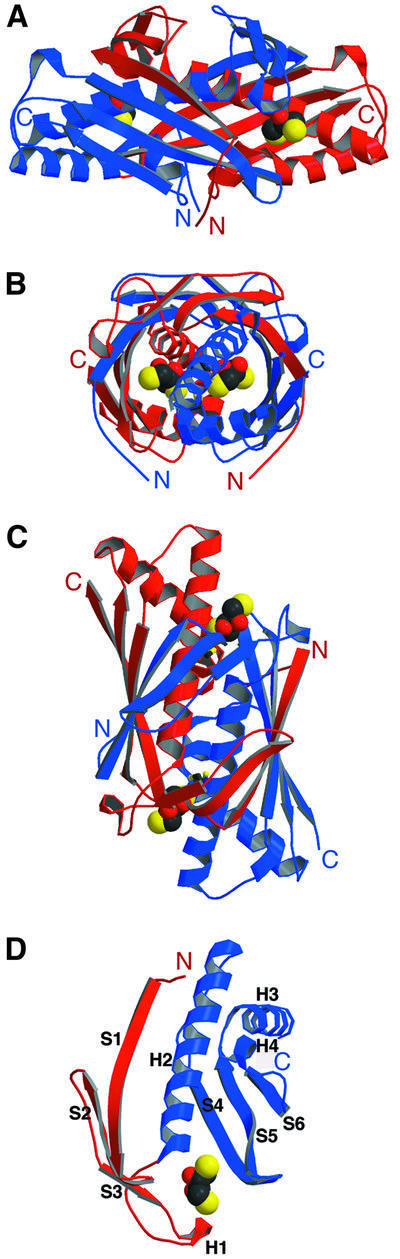
Fig. 2. (A–C) Structure of the Ohr dimer. One monomer is in red, the other is in blue. Three 90° views of the Ohr dimer bound to DTT. (D) Structure of the Ohr monomer depicting the two domains (the N-terminal subdomain is shown in red and the C-terminal subdomain is shown in blue) and a DTT molecule. DTT is shown in CPK format, with oxygen atoms in red, carbon atoms in black and sulfur atoms in yellow.
The Ohr monomer is a two-domain molecule, with an α/β fold, composed of ∼40% α-helices and 40% β-sheet (Figures 1B and 2D). The larger C-terminal subdomain (Figure 2D, blue) contains an antiparallel β-sheet composed of strands S4, S5 and S6, which pack against three α-helices. The longest helix, H2, interacts with the N-terminus of strand S4 and the C-termini of strands S5 and S6, while helix H3 interacts with the N-terminus of S6. Helices H2 and H3 also pack against each other, completing the hydrophobic core of the monomer. The small N-terminal subdomain (Figure 2D, red) consists of a three-stranded antiparallel β-sheet (strands S1, S2 and S3) and is spatially isolated from the C-terminal subdomain. In the Ohr dimer, this subdomain interacts with the large C-terminal subdomain of another monomer to form a six-stranded antiparallel β-sheet, which folds around the joint α-helical core. A flexible 13-amino-acid linker containing a small, solvent-exposed 310 helix (H1) separates the two subdomains.
The structure of Ohr represents the first structure of a member of the Ohr/OsmC family of resistance proteins. A comparison with the contents of the Protein Data Bank (PDB) database (Sussman et al., 1998) using DALI (Holm and Sander, 1993) reveals that the Ohr structure is different from that of other known ones. The Ohr C-teminal subdomain bears some structural homology to the C-terminal domain of the E.coli ERA and the N-terminal domain of the Streptococcus pneumoniae SP14.3. The common structural motif contains 80–90 amino acids, which can be aligned between the three proteins, with a r.m.s.d. of 3.5–4.0 Å. ERA is an essential bacterial GTPase that has a regulatory role in cell cycle control, coupling cell growth rate with cytokinesis (Chen et al., 1999). The SP14.3 protein, on the other hand, is essential for the growth of S.pneumoniae, but its exact function is still unknown (Yu et al., 2001). Since the three proteins show no sequence homology and appear to have different biological functions, the common α/β motif most probably represents a stable structural scaffold, which is utilized as an architectural module in building different enzymes.
Structure of the active site
The two active-site pockets containing the invariant cysteines are located on opposite faces of the protein, and the structure suggests that they function independently. The conserved Cys60 and Cys124 in each active site come from the same monomer, with Cys60 located in the middle part of helix H2 and Cys124 at the C-terminus of helix H3. The entrance to each pocket is guarded by hydrophobic side chains contributed by both mono mers (Figure 3A, green), including Leu7, Ala64, Phe67, Phe95*, Ile88* and Val123 (the asterisk indicates residue from the second monomer). Cys124 is located at the bottom of the active-site cavity, surrounded by the conserved Tyr57, Phe61, Pro125, Ser127, Tyr126 and Cys60. The side chain of Cys60 is solvent exposed and is located proximal to the entrance to the active site, in front of Cys124 (the distance between S atoms is 3.6 Å). Cys60 is surrounded by the conserved Pro49*, Pro125, Tyr126, and the invariant Arg18* and Cys124. Arg18* (Nη1) hydrogen-bonds with Cys60 (S) (3.4 Å) and also forms a salt bridge with Glu50*.

Fig. 3. (A) Molecular surface rendering of the Ohr dimer. The catalytically active Cys60 (shown in yellow) lies at the bottom of the active-site pocket, which is outlined with hydrophobic residues (shown in green). (B) Stereo view of the Ohr active site. Hydrogen bonds are depicted by light blue, dashed lines. Monomer A (shown in green) contributes Cys60 and Cys124 residues (also in green) and monomer B (shown in purple) contributes Arg18 and Glu50 residues (also in purple) to the active site. The DTT molecule (orange) is depicted in a ball-and-stick format. Nitrogen atoms are dark blue, oxygen atoms are red and sulfur atoms are yellow. (C) Representative region of the density-modified experimental electron density map showing the active-site pocket in the Ohr structure (the refined model) contoured at 1.5σ.
The structure of reduced Ohr was solved using crystals grown in the presence of the reducing agent DTT and each active site contains well-defined electron density corresponding to a bound DTT molecule (Figure 3C). One of the DTT sulfur atoms is positioned 3.0 Å away from the sulfur atom of Cys60 and 4.5 Å away from the sulfur atom of Cys124. In addition, the (Nε) atom of Arg18* makes a strong hydrogen bond with a DTT oxygen atom at a distance of 2.9 Å, stabilizing the DTT molecule in a position favoring the reduction of Ohr.
Peroxidase activity
Recombinant Ohr was tested for peroxidase activity towards both inorganic (H2O2) and organic hydroperoxides (CHP and t-BHP). Ohr showed no detectable enzymatic activity in the absence of reducing agents and since the endogenous Ohr reductant is unknown, we tested several small organic molecules, including DTT, NADH, NADPH and glutathione, as potential electron donors. DTT, which has no activity on its own, was the only molecule capable of supporting Ohr-mediated peroxidase activity. We used a colorimetric Ferrous Oxidation in Xylenol (FOX) assay (Wolff, 1994), where H2O2, CHP and t-BHP were individually incubated with Ohr in the presence of DTT, and the decrease in hydroperoxide concentrations was monitored spectrophotometrically. The results show that H2O2, CHP and t-BHP can all serve as Ohr substrates, but the enzyme’s ability to turn them over varies substantially. Compared with CHP and t-BHP, H2O2 is a very poor Ohr substrate. Indeed 10 µM Ohr can remove ∼12 µM H2O2 in a minute (Figure 4A), while only 3 µM Ohr can metabolize >1900 µM CHP during the same time period (Figure 4C). Similarly, it takes >600 s for 1 µM Ohr to remove 15 µM H2O2 (Figure 4B), but only 1–2 s to metabolize an equivalent amount of CHP (Figure 4D). Our Ohr steady-state kinetic assay data are consistent with an apparent Km value for H2O2 in the 200–800 µM range, and an approximately two order of magnitude lower Km for CHP. The fact that H2O2 is metabolized inefficiently is not due to enzyme inactivation by H2O2, because if an equivalent concentration of H2O2 is included with CHP in an HPLC-based assay (Figure 5F), Ohr’s activity towards CHP is not altered.

Fig. 4. FOX assays demonstrating that Ohr metabolizes hydrogen peroxide (HP) (A and B) and cumene hydroperoxide (CHP) (C and D) in a protein concentration- and time-dependent manner in the presence of DTT. Note the significant quantitative differences in metabolism rates between inorganic H2O2 and organic CHP. Each point represents a mean measurement value from three separate experiments and the bars represent standard errors.

Fig. 5. The effect of substitution of Cys60 or Cys124 with serines and of Arg18 with glutamine on the initial metabolism rates of (A) cumene hydroperoxide (CHP), (B) t-BHP and (C) hydrogen peroxide (HP), as measured using FOX assays. Each measurement represents a mean value of three separate experiments and the bars represent standard errors. (D) Protective effect of wild-type and mutant Ohr proteins against oxidative inactivation of glutamine synthetase (GS) by DTT–FeCl3–O2. Each assay was performed in duplicate, with each point representing a mean value of six separate measurements. (E) DTT oxidation as a function of concentration of CHP in the presence of 5 µM Ohr. Inset: DTT oxidation as a function of Ohr concentration in the presence of 6 mM CHP. Each assay was performed twice, with each point representing a mean value of four separate measurements. (F) Product formation from cumene hydroperoxide as analyzed by HPLC. The substrate reaction mixture prior to addition of Ohr (left panel) and after incubation with Ohr (right panel). Note the disappearance of the CHP substrate peak and the increase in the cumyl alcohol (CA) product peak area and height following the addition of Ohr.
Next, we examined the individual roles of the conserved cysteines in Ohr’s enzymatic mechanism. Ohr variants containing substitutions of these residues with serines were generated via site-directed mutagenesis and evaluated for their ability to directly metabolize peroxides. The initial hydroperoxide metabolism rates of the mutants were either compromised or completely abolished as compared with the wild-type enzyme (Figure 5A–C, orange bars). More specifically, the substitution of Cys60 for serine completely inactivates Ohr (blue bars), as does the C60S/C124S double substitution (data not shown). The C124S mutation, on the other hand, leads to a 60–75% loss of activity of the wild-type Ohr with all tested substrates (Figure 5A–C, green bars).
We hypothesize that the high reactivity of the Cys60 thiol group may be due in part to the lowering of its pKa via a hydrogen bond with the Arg18* side chain, as observed in other peroxidases (see Discussion). To evaluate the role of the Cys60–Arg18* interaction in the catalytic mechanism, an Ohr R18Q mutant that replaces the positively charged side chain of arginine with the polar, but uncharged, side chain of glutamine was generated and evaluated for its ability to metabolize peroxide substrates. The initial hydroperoxide metabolism rates of this mutant with H2O2, CHP and t-BHP were substantially compromised, but not abolished (Figure 5A–C, gray bars).
The C60S and C124S mutants were also tested for their ability to protect glutamine synthetase (GS) from inactivation by a mixed-function oxidase system composed of DTT, FeCl3 and O2 (Figure 5D) (Kim et al., 1988). In this assay, a protein that is capable of removing endogenously generated H2O2 will prevent inactivation of the GS enzyme. The DTT/FeCl3/O2 mixture alone (positive control) causes an 80% decrease in GS activity within 30 min, whereas addition of the wild-type Ohr fully protects GS from inactivation. In full agreement with the data from the FOX assays, the C60S mutant is inactive, while the C124S mutant displays weak residual activity (Figure 5A–C). The high, 100 µM Ohr concentration required to fully protect GS from inactivation is most likely due to the fact that the H2O2 is a poor substrate for the enzyme (see Figure 4A and B).
We next evaluated whether the Ohr cysteines form disulfide bonds during enzyme catalysis. An assay described by Iyer and Klee (1973) was employed, in which oxidation of DTT (monitored spectrophotometrically) is used to determine the presence and extent of disulfide bond cleavage in proteins. The results, illustrated on Figure 5E, show that the overall extent of DTT oxidation is a linear function of the initial concentration of the CHP substrate, as the reaction is carried out in the presence of a constant enzyme concentration (Figure 5E). A linear dependence between the extent of DTT oxidation and Ohr concentration was also observed (Figure 5E, inset). These findings document, though indirectly, the formation of disulfide bonds in Ohr during the catalytic cycle.
Finally, HPLC-based methodology was used to identify the products generated by Ohr during the catalytic reaction (Figure 5F). Three peaks are observed when the CHP substrate is analyzed directly as purchased: a CHP peak [retention time (RT) of ∼55 min)], a cumyl alcohol (CA) peak (RT of ∼37.5 min) and a signal from an unidentified compound (RT of 40 min, which corresponds to the RT of acetophenone, a possible CHP contaminant) (Figure 5F, left panel). Following incubation with Ohr and an injection of the same volume of the reaction mixture, the CHP peak disappears, with a concomitant increase in the area of the CA peak, indicating a full conversion of the CHP substrate into a CA product (Figure 5F, right panel). An independent set of identical reactions evaluated with the colorimetric FOX assay confirms the full disappearance of CHP from the reaction mixture (data not shown).
Discussion
Structural comparison to other peroxidases
In the present report, we describe the first crystal structure of a member of the recently identified Ohr/OsmC family of bacterial resistance proteins and provide evidence that Ohr functions as a peroxidase. The dimeric enzyme has a novel oval-shaped structure and lacks significant structural similarity to other known peroxidases, including OHP reductases such as peroxiredoxins (Choi et al., 1998), glutathione peroxidases (Epp et al., 1983) and glutathione S-transferases (Dirr et al., 1994). These multidomain enzymes share a common structural core comprised of an ∼80 amino acid thioredoxin fold, dominated by a four-stranded β-sheet and three α-helices. This domain provides a structural framework upon which the rest of the protein is constructed, and incorporates the structurally equivalent, enzymatically active cysteine (in peroxiredoxins), selenocysteine (in glutathione peroxidases) and tyrosine (in glutathione S-transferase) (Martin, 1995). Although Ohr lacks a thioredoxin fold, it somewhat resembles thioredoxin in the overall arrangement of its secondary structural elements around the active site. More specifically, when the Ohr S4–S5–S6 β-sheet is superimposed on the β-sheet of human thioredoxin, the Ohr α-helices H3 and H4 are positioned similarly to the thioredoxin helices α4 and α2. The long Ohr α-helix H2, on the other hand, lacks a structural counterpart in the thioredoxin. Despite the overall similarity, the active-site cysteines are located in different positions in this three-strand–two-helix arrangement in each protein.
Substrate specificity
The in vitro assays demonstrate that Ohr preferentially metabolizes organic hydroperoxides (t-BHP and CHP) over inorganic H2O2. These findings are consistent with the results of experiments in bacterial cell cultures, which show that deletion/inactivation ohr mutants are hypersensitive to OHPs, but not to H2O2 (Mongkolsuk et al., 1998; Ochsner et al., 2001; Rince et al., 2001; Shea and Mulks, 2002), and that ohr is highly induced by bacterial exposure to organic hydroperoxides, but only weakly by exposure to H2O2 (Mongkolsuk et al., 1998).
The difference in the apparent affinity of H2O2 and CHP for Ohr can be compared with the Km values determined for other peroxidases with the same substrates. The Mycobacterium tuberculosis peroxiredoxin AhpC, for example, has an apparent Km of ∼1500 µM for H2O2 and ∼500 µM for CHP (Hillas et al., 2000). Similarly, the M.tuberculosis peroxidase AhpD has an apparent Km of ∼1500 µM for H2O2 and ∼52 µM for CHP (Hillas et al., 2000), and the yeast glutathione peroxidase PHGpx2 has a Km value of 2.2 mM for H2O2 and 310 µM for CHP (Collinson et al., 2002). On the other hand, some peroxiredoxins, including the E.coli p20 (Cha et al., 1995) and the Entamoeba histolytica Eh29 (Bruchhaus et al., 1997), possess lower Km values for H2O2: 60 and 100 µM, respectively. Moreover, in contrast to Ohr, the M.tuberculosis catalase–peroxidase, for example, possesses higher apparent affinity for H2O2 than for organic hydroperoxide (Nagy et al., 1997).
The Ohr structure reveals that the preferential metabolism of hydrophobic hydroperoxides over inorganic ones is probably due to the fact that the entrance to each active site is surrounded mostly by hydrophobic side chains (Figure 3A, see also Results, Structure of the active site). The inside of the active-site pocket, on the other hand, contains many polar, uncharged side chains around the catalytic Cys60. This is in contrast to the active site of the homodimeric 1-Cys peroxiredoxin hORF6, whose cata lytically active cysteine is also solvent exposed, but is located at the bottom of a very narrow and positively charged pocket (Choi et al., 1998). It is interesting that the size of the active-site pocket in hORF6 decreases upon dimerization, which may be an important determinant in the regulation of enzyme activity and substrate specificity. This is not the case with the tightly folded Ohr homodimer, which is unlikely to change either its oligomerization state or the structure of its active-site cavity under physiological conditions. It should be noted that both the Ohr structure and the reported peroxiredoxin structures are determined in the absence of a physiological substrate. Furthermore, in most cases, the cognate endogenous substrates are as yet unknown. It is, therefore, difficult to fully rationalize the substrate specificity of these enzymes based solely on the structural characteristics of their active sites.
Catalytic cysteines
Several lines of evidence suggest that the two P.aeruginosa Ohr cysteines are directly involved in the metabolism of hydroperoxides. First, they are invariant in all known Ohr and OsmC proteins. Secondly, the cysteines are located very close to each other in the crystal structure (3.6 Å), ideally positioned for the formation (within each monomer’s active site) of an intramolecular disulfide bond. Thirdly, a disulfide-reducing agent, such as DTT, is required for catalysis, and in the Ohr crystal structure a DTT molecule is present next to the Cys60 thiol group (Figure 3B and C). To clarify the role of these residues, we performed mutagenesis experiments, which document that Cys60 is indispensable for Ohr enzymatic activity, and that Cys124, while not absolutely required, is necessary for the maintenance of a high peroxidase reaction rate of the enzyme (Figure 5A–C). In addition, data from experiments measuring the extent of DTT oxidation during peroxide catalysis (Figure 5E) are consistent with a transient formation of a C60-S–S-C124 disulfide bond.
We propose that Ohr metabolizes OHPs by directly using cysteine thiol groups as the catalytic centers in an analogous fashion to the well-characterized peroxiredoxins. Unlike other reactive-oxygen-species-detoxifying enzymes (such as catalases, superoxide dismutases or glutathione peroxidases), peroxiredoxins do not rely upon any prosthetic groups or cofactors for their enzymatic activity, but instead utilize a highly reactive cysteine as the primary catalytic group (Poole, 1996). Most peroxiredoxins contain two conserved cysteine residues that are directly involved in peroxide catalysis (2-Cys peroxi redoxins). During the reaction, the OHP molecule initially oxidizes the attacking N-terminal cysteine, which then condenses with the reduced C-terminal cysteine to form a disulfide bond with the simultaneous release of a water molecule and the corresponding alcohol product. The disulfide bond can finally be reduced by thioredoxin, glutathione, trypanothione or a thioredoxin-like protein (e.g. AhpF), regenerating the peroxiredoxin back to its catalytically active form. Some peroxiredoxins contain only one conserved cysteine, which is both necessary and sufficient for full enzymatic activity (1-Cys peroxiredoxins), but the identities of their endogenous reductants are still unknown.
Proposed enzymatic mechanism
Based on the collective evidence presented here, we suggest the following enzymatic mechanism for Ohr. First, Cys60 reacts with a peroxide molecule and in the process becomes transiently oxidized to a cysteine–sulfenic acid (-SOH) intermediate. The OHP is simultaneously reduced to its corresponding alcohol. Secondly, the Cys60-SOH group quickly condenses with the reduced Cys124 thiol group (-SH), forming an intramolecular disulfide bond and releasing a molecule of water (Cys60-S-OH + HS-Cys124 → Cys60-S–S-Cys124 + H2O). Thirdly, the oxidized Ohr is regenerated back to its enzymatically active, reduced state using an as yet unidentified reductant.
A possible pathway that could function to regenerate oxidized Ohr back to its reduced state in vivo may include the thioredoxin/thioredoxin reductase/NADPH system, which is utilized by many peroxiredoxin isoforms (Powis and Montfort, 2001). Alternatively, the other major microbial intracellular disulfide-reducing system may be involved, which is composed of glutaredoxin, glutathione reductase and glutathione. It is also possible that Ohr is directly reduced in vivo by a low-molecular-weight organic molecule or, alternatively, by an enzyme possessing thioredoxin-like activity, in a similar fashion to the S.typhimurium and M.tuberculosis AhpC peroxiredoxins, which are reduced by the AhpF and AhpD proteins, respectively (Poole, 1996; Bryk et al., 2002).
The initially attacking Cys60 is more reactive than Cys124 due to its interaction with the conserved Arg18* (Figure 3B and C). The hydrogen bond between the side chains of the two residues has a major role in lowering the pKa of the Cys60 thiol, polarizing and stabilizing it in the ionized (-S–) state, which is more nucleophilic and therefore more reactive than the unionized (-SH) form. This observation is further supported by structure-based mutagenesis experiments, demonstrating that the catalytic activity of a R18Q mutant is severely compromised (Figure 5A–C, gray bars). The fact that the R18Q mutant retains some enzymatic activity indicates that other residues are also responsible for creating the favorable microenvironment necessary for sustaining the high reactivity of the Cys60 γ-thiolate. Similar interactions, where the increased reactivity of an attacking cysteine results from its hydrogen-bonding to histidine and/or arginine amino acids, are observed in the active sites of other enzymes (Lewis et al., 1981), including peroxiredoxins (Alphey et al., 2000; Declercq et al., 2001).
In contrast to Cys60, Cys124 is not in close proximity to any histidines or arginines. Furthermore, Cys124 is positioned at the C-terminus of a α-helix (Figures 1A and 3B), which may lead to an increase in its pKa (and therefore to a decreased reactivity) due to an electrostatic interaction with the partially negative helix dipole (Kortemme and Creighton, 1995). Steric factors may also account for the difference in the biochemical behavior of the two cysteine residues. For instance, Cys60 is located closer to the entrance of the active site and is more solvent exposed, thus probably coming first in contact with the substrate. Our model for Ohr’s enzymatic mechanism accounts for the observation that, unlike the completely inactive C60S mutant, C124S still possesses some peroxidase activity in the presence of DTT. Indeed, upon reaction with a peroxide molecule, the Cys60 thiol would be transiently oxidized to a cysteine–sulfenic acid intermediate, which would subsequently be reduced in this case by two molecules of DTT, first to a Cys60-S–S-DTT mixed disulfide and then to the reduced thiol and a molecule of oxidized DTT. An analogous mechanism has been proposed for the selenoprotein glutathione peroxidase, which contains a redox-active selenocysteine and no sulfur or other selenium atoms in proximity (Chaudiere et al., 1984), as well as for the 1-Cys peroxiredoxins, which are also enzymatically active in the presence of DTT (Kang et al., 1998).
Some bacterial genomes, e.g. Pseudomonas putida and Deinococcus radiodurans, possess both ohr and osmC genes, despite the fact that the proteins are structurally and functionally homologous. It is possible that in such cases each protein resides in distinct subcellular locations, which may be beneficial if, for example, one is primarily responsible for detoxification of exogenous OHPs produced by the host immune system, while the other inactivates the peroxide by-products of bacterial metabolism. Since the endogenous substrates for Ohr and OsmC have not yet been characterized, it is also plausible that each enzyme preferentially metabolizes some OHP substrates over others. The biological differences between Ohr and OsmC may also extend to differences in timing of gene induction, or to differences in protein synthesis and/or degradation.
Conclusion
This work builds upon the previously gathered genetic, biochemical and microbiological data by providing the first high-resolution view of the Ohr/OsmC family of bacterial OHP-resistance proteins. The Ohr crystal structure allows us to locate the active-site pockets and to identify the catalytically important residues. The structural, enzymatic and mutagenesis data demonstrate that Ohr detoxifies hydroperoxides by catalyzing their reduction to alcohols, and suggest a catalytic mechanism for this enzyme family. The structural basis for the unusual reactivity of the cysteines and Ohr’s preference for the metabolism of lipophilic peroxide substrates are also elucidated. The results presented will facilitate further research towards determination of the identity of the endogenous substrates as well as the physiological reductant(s) for the Ohr and the OsmC proteins. Finally, our data provide a foundation for a better understanding of the molecular mechanism of bacterial factors involved in protection of microbial pathogens from oxidative stress caused by exposure to peroxides.
Materials and methods
Ohr expression, purification and mutagenesis
Pseudomonas aeruginosa ohr was amplified by Expand High Fidelity PCR System (Boehringer Mannheim) and subcloned into a pET22b vector (Novagen) using E.coli DH5α cells (Promega). Ohr mutants were generated by a two-step PCR site-directed mutagenesis approach. Escherichia coli BL21 (DE3) cells (Novagen) were used for protein expression. The cells were harvested, resuspended in buffer containing 20 mM Tris pH 8.0, 5 mM DTT (buffer A) and lysed. The lysate was centrifuged, soluble proteins were precipitated with ammonium sulfate, resuspended in buffer A, and purified using a NaCl gradient on Q-Sepharose Fast Flow and Mono-Q ion-exchange columns (Pharmacia). Ohr-containing fractions (analyzed by SDS–PAGE) were further purified on a Superdex SD-75 gel filtration column using buffer A plus 200 mM NaCl. Selenomethionine-labeled Ohr triple mutant was expressed in the methionine auxotroph E.coli B834 (DE3) using M9 minimal medium supplemented with selenomethionine (50 mg/l final concentration), and was purified as the wild-type protein. Electrospray ionization mass spectrometry and N-terminal sequencing confirmed the identities of the wild-type and mutant proteins. All chemicals were purchased from Sigma and Fluka, unless stated otherwise. The Ohr mutants were indistinguishable from the wild-type protein, as judged by CD spectroscopy, and behaved in an identical fashion during purification.
Crystallization, X-ray data collection, structure determination and refinement
Purified triple mutant Ohr was concentrated to 20 mg/ml in 5 mM Tris pH 8.0 and 5 mM DTT, and was crystallized at room temperature using hanging-drop vapor diffusion against a reservoir containing 0.2 M Na acetate, 30% PEG5 K MME and 0.1 M Tris pH 8.0. Crystals were flash-frozen in 20% glycerol (v/v) in mother liquor. Diffraction data were collected either at the Brookhaven National Synchrotron Light Source beamline X9A or at CHESS beamlines A1 and F2. Oscillation images were integrated, scaled and merged using DENZO and SCALEPACK (Otwinowski and Minor, 1997). The program SOLVE was used to locate the eight selenium atoms as well as to calculate the initial maps (Terwilliger and Berendzen, 1999). Solvent flattening and non-crystallographic symmetry (NCS) averaging were employed in the program RESOLVE to improve the experimental map (Terwilliger, 2000). The automatic chain tracing procedure of wARP (Perrakis et al., 1999) was used to build 238 out of 284 Ohr residues, while the program O (Jones et al., 1991) was used to complete the tracing and sequence assignment. Refinement proceeded via conventional least squares algorithm with X-PLOR (Brunger, 1993). Stereochemical analysis of the refined models using PROCHECK of the CCP4 package (CCP4, 1994) revealed main-chain and side-chain parameters better than or within the typical range of values for protein structures determined at corresponding resolutions. None of the Ohr residues fell in the disallowed region of the Ramachandran plot. Molecular graphic figures were created with MolScript (Kraulis, 1991), Raster3D (Merritt and Bacon, 1997) and GRASP (Nicholls et al., 1991).
Enzymatic assays
FOX assays (Figures 4 and 5A–C). Metabolism of peroxides by Ohr was measured using the FOX method (Wolff, 1994). Briefly, Ohr was pre-incubated for 1 h on ice with 5 mM DTT. Reactions were typically initiated by the addition of an appropriate amount of Ohr protein (Ohr final concentration 1.5 µM for measurement of Km and Vmax) and carried out at room temperature in 50 µl of 100 mM potassium phosphate pH 7.0, 0.6 mM DTT final concentration (1 mM DTT for determination of the kinetic parameters) and with varying amounts of substrate peroxides (1–1000 nmol). Reactions were terminated by the addition of 950 µl of cold FOX reagent (pH ∼ 1.6). Color development was complete after incubation at room temperature for 1 h. The absorbance was read at 560 nm [using a molar extinction coefficient (ε) of 2.24 × 105 M–1 cm–1], and was compared with the H2O2, CHP and t-BHP standards prepared in the same buffer (100 mM potassium phosphate pH 7.0 with 0.6 or 1.0 mM DTT), but without Ohr. DTT did not interfere with the progress of color development of FOX reactions, and DTT alone (without Ohr) did not measurably alter hydroperoxide concentrations. Lines of best fit were used to generate Figure 4 using Kaleidagraph (Abelbeck Software, Reading, PA). For the determination of steady-state parameters, data were fit to the equation V = Vmax [S]/(Km + [S]) (graphs not shown).
Glutamine synthetase assays (Figure 5D). These assays were performed as described previously (Kim et al., 1988). Briefly, 5 µg of E.coli GS, 10 mM DTT, 3 µM FeCl3 and 100 µM Ohr (wild type or mutant) were incubated for 5, 10, 20 and 30 min at 37°C. Four microliters of this mixture were then transferred to 400 µl of buffer containing 100 mM HEPES pH 7.4, 0.4 mM ADP, 150 mM glutamine, 10 mM potassium arsenate, 20 mM hydroxylamine and 0.4 mM MnCl2, and incubated for 30 min also at 37°C. The reactions were terminated by the addition of 200 µl of a stop mixture (55 g of FeCl3 hexahydrate, 20 g of trichloroacetic acid and 21 ml of concentrated HCl per liter) and the absorbance was read at 540 nm.
DTT oxidation assays (Figure 5E). These assays were performed similarly to the method described in Iyer and Klee (1973). The extent of DTT oxidation catalyzed by Ohr in the presence of peroxide substrate was measured by monitoring the change in absorbance at 310 nm (and using a ε of 110 M–1 cm–1 for oxidized DTT), resulting from the formation of the DTT disulfide. The reactions were carried out at room temperature in 1 ml final volume, and contained 5 µM Ohr (or indicated concentrations in the inset), 100 mM potassium phosphate buffer pH 7.0, 1 mM EDTA, 10 mM freshly prepared DTT and the indicated (or 6 mM for the inset) CHP concentrations. The measurements were corrected for the small background DTT oxidation observed in the presence of CHP, but absence of enzyme. The assay displayed linear oxidized DTT–absorbance correlation in the 0.5–10 mM oxidized DTT range.
HPLC-based assays (Figure 5F). These assays were performed similarly to the method described in Hillas et al. (2000). Briefly, 300 µl of 3.5 mM CHP were mixed with 6.7 ml of 100 mM potassium phosphate buffer pH 7.0 and 1 mM DTT, and the reaction was initiated at room temperature by adding 20 µl of 1 mM Ohr (pre-incubated with 2 mM DTT). The mixture was centrifuged and 5 ml of the filtrate were injected onto a Waters 490E HLPC system equipped with an Axxiom ODS (4.6 × 250 mm) reverse-phase HPLC column. Compounds were separated using 20% acetonitrile and 80% water at a flow rate of 1 ml/min. The detector was set at 260 nm. Control reactions (no Ohr) were performed in the same buffer. Peaks were identified using comparison with authentic standards under the same elution conditions.
Coordinates
Coordinates have been deposited in the PDB under accession code 1N2F.
Acknowledgments
Acknowledgements
We thank Dr Lily Fisher for assistance with CD spectroscopy, Dr Dinshaw Patel’s laboratory members for access to HPLC instrumentation and Dr K.R.Rajashankar for help with data processing. D.B.N. is a PEW Fellow and a Bressler Scholar.
References
- Alphey M., Bond,C., Tetaud,E., Fairlamb,A. and Hunter,W. (2000) The structure of reduced tryparedoxin peroxidase reveals a decamer and insight into reactivity of 2 Cys-peroxiredoxins. J. Mol. Biol., 300, 903–916. [DOI] [PubMed] [Google Scholar]
- Atichartpongkul S., Loprasert,S., Vattanaviboon,P., Whangsuk,W., Helmann,J. and Mongkolsuk,S. (2001) Bacterial Ohr and OsmC paralogues define two protein families with distinct functions and patterns of expression. Microbiology, 147, 1775–1782. [DOI] [PubMed] [Google Scholar]
- Bernards A., Miller,J., Bao,K. and Wong,I. (2002) Flipping duplex DNA inside out: a double base-flipping reaction mechanism by Escherichia coli MutY adenine glycosylase. J. Biol. Chem., 277, 20960–20964. [DOI] [PubMed] [Google Scholar]
- Bruchhaus I., Richter,S. and Tannich,E. (1997) Removal of hydrogen peroxide by the 29 kDa protein of Entamoeba histolytica. Biochem. J., 326, 785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A. (1993) X-PLOR v. 3.1 Manual. Yale University, New Haven, CT.
- Bryk R., Lima,C., Erdjument-Bromage,H., Tempst,P. and Nathan,C. (2002) Metabolic enzymes of mycobacteria linked to antioxidant defense by thioredoxin-like protein. Science, 295, 1073–1077. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 Suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Cha M., Kim,H. and Kim,I. (1995) Thioredoxin-linked ‘thiol peroxidase’ from periplasmic space of Escherichia coli. J. Biol. Chem., 270, 28635–28641. [DOI] [PubMed] [Google Scholar]
- Chaudiere J., Wilhelmsen,E. and Tappel,A. (1984) Mechanism of selenium-glutathione peroxidase and its inhibition by mercaptocarboxylic acids and other mercaptans. J. Biol. Chem., 259, 1043–1050. [PubMed] [Google Scholar]
- Chen X., Court,D. and Ji,X. (1999) Crystal structure of ERA: a GTPase-dependent cell cycle regulator containing an RNA binding motif. Proc. Natl Acad. Sci. USA, 96, 8396–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H., Kang,S., Yang,C., Rhee,S. and Ryu,S. (1998) Crystal structure of a novel human peroxidase enzyme at 2.0 Å resolution. Nat. Struct. Biol., 5, 400–406. [DOI] [PubMed] [Google Scholar]
- Collinson E., Wheeler,G., Garrido,E., Avery,A., Avery,S. and Grant,C. (2002) The yeast glutaredoxins are active as glutathione peroxidases. J. Biol. Chem., 277, 16712–16717. [DOI] [PubMed] [Google Scholar]
- Conter A., Gangneux,C., Suzanne,M. and Gutierrez,C. (2001) Survival of Escherichia coli during long-term starvation: effects of aeration, NaCl and the rpoS and osmC gene products. Res. Microbiol., 152, 17–26. [DOI] [PubMed] [Google Scholar]
- Declercq J.-P., Evrard, Ch., Clippe,A., Vander Stricht,D., Bernard,A. and Knoops,B. (2001) Crystal structure of human peroxiredoxin 5, a novel type of mammalian peroxiredoxin at 1.5 Å resolution. J. Mol. Biol., 311, 751–759. [DOI] [PubMed] [Google Scholar]
- Dirr H., Reinemer,P. and Huber,R. (1994) X-ray crystal structures of cytosolic glutathione S-transferases. Implications for protein architecture, substrate recognition and catalytic function. Eur. J. Biochem., 220, 645–661. [DOI] [PubMed] [Google Scholar]
- Epp O., Ladenstein,R. and Wendel,A. (1983) The refined structure of the selenoenzyme glutathione peroxidase at 0.2-nm resolution. J. Biochem., 133, 51–69. [DOI] [PubMed] [Google Scholar]
- Fuangthong M. and Helmann,J. (2002) The ohrR repressor senses organic hydroperoxides by reversible formation of a cysteine– sulfenic acid derivative. Proc. Natl Acad. Sci. USA, 99, 6690–6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Flecha B. and Dimple,B. (1997) Homeostatic regulation of intracellular hydrogen peroxide concentration in aerobically growing Escherichia coli. J. Bacteriol., 179, 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A. and Goebel,W. (1992) Microbial strategies to prevent oxygen-dependent killing by phagocytes. Free Radic. Res. Commun., 16, 137–157. [DOI] [PubMed] [Google Scholar]
- Hendrickson W. (1991) Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science, 254, 51–58. [DOI] [PubMed] [Google Scholar]
- Hillas P., Soto del Alba,F., Oyarzabal,J., Wilks,A. and Oritz de Montellano,P. (2000) The AhpC and AhpD antioxidant defence system of Mycobacterium tuberculosis. J. Biol. Chem., 275, 18801–18809. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1993) Protein structure comparison by alignment of distant matrices. J. Mol. Biol., 233, 123–138. [DOI] [PubMed] [Google Scholar]
- Iyer K. and Klee,W. (1973) Direct spectrophotometric measurement of the rate of reduction of disulfide bonds. The reactivity of the disulfide bonds of bovine α-lactalbumin. J. Biol. Chem., 248, 707–710. [PubMed] [Google Scholar]
- Jacobson F., Morgan,R., Christman,M. and Ames,B. (1989) An alkyl hydroperoxide reductase from Salmonella typhimurium involved in the defense of DNA against oxidative damage. J. Biol. Chem., 264, 1488–1496. [PubMed] [Google Scholar]
- Jones T., Zou,J., Cowan,S. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kang S., Baines,I. and Goo Rhee,S. (1998) Characterization of mammalian peroxiredoxin that contains one conserved cysteine. J. Biol. Chem., 273, 6303–6311. [DOI] [PubMed] [Google Scholar]
- Kim K., Kim,I., Lee,K., Rhee,S. and Stadtman,E. (1988) The isolation and purification of a specific ‘protector’ protein which inhibits enzyme inactivation by thiol/Fe(III)/O2 mixed-function oxidation system. J. Biol. Chem., 263, 4704–4711. [PubMed] [Google Scholar]
- Kortemme T. and Creighton,T. (1995) Ionization of cysteine residues at the termini of model α-helical peptides. Relevance to unusual thiol pKa values in proteins of the thioredoxin family. J. Mol. Biol., 253, 799–812. [DOI] [PubMed] [Google Scholar]
- Kraulis P. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Lewis S., Johnson,F. and Shafer,J. (1981) Effect of cysteine-25 on the ionization of histidine-159 in papain as determined by proton nuclear magnetic resonance spectroscopy. Biochemistry, 20, 48–51. [DOI] [PubMed] [Google Scholar]
- Martin J. (1995) Thioredoxin—a fold for all reasons. Structure, 3, 245–250. [DOI] [PubMed] [Google Scholar]
- Merritt E. and Bacon,D. (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- Mongkolsuk S., Praituan,W., Loprasert,S., Fuangthong,M. and Chamnongpol,S. (1998) Identification and characterization of a new organic hydroperoxide resistance (ohr) gene with a novel pattern of oxidative stress regulation from Xanthomonas campestris pv. phaseoli. J. Bacteriol., 180, 2636–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy J., Cass,A. and Brown,K. (1997) Purification and characterization of recombinant catalase–peroxidase, which confers isoniazid sensitivity in Mycobacterium tuberculosis. J. Biol. Chem., 272, 31265–31271. [DOI] [PubMed] [Google Scholar]
- Neill M. and Klebanoff,S. (1988) The effect of phenolic glycolipid-1 from Mycobacterium leprae on the antimicrobial activity of human macrophages. J. Exp. Med., 167, 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Niki E. (1992) Peroxides in biological systems. In Ando,W. (ed.), Organic Peroxides. Wiley, New York, NY, pp. 765–787.
- Ochsner U., Hassett,D. and Vasil,M. (2001) Genetic and physiological characterization of ohr, encoding a protein involved in organic hydroperoxide resistance in Pseudomonas aeruginosa. J. Bacteriol., 183, 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Perrakis A., Morris,R. and Lamzin,V. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol., 6, 458–463. [DOI] [PubMed] [Google Scholar]
- Poole L. (1996) Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 2. Cystine disulfides involved in catalysis of peroxide reduction. Biochemistry, 35, 65–75. [DOI] [PubMed] [Google Scholar]
- Powis G. and Montfort,W. (2001) Properties and biological activites of thioredoxins. Annu. Rev. Pharmacol. Toxicol., 41, 261–295. [DOI] [PubMed] [Google Scholar]
- Rhee S., Kang,S., Chang,T., Jeong,W. and Kim,K. (2001) Peroxiredoxin, a novel family of peroxidases. IUBMB Life, 52, 35–41. [DOI] [PubMed] [Google Scholar]
- Rince A., Giard,J., Pichereau,V., Flahaut,S. and Auffray,Y. (2001) Identification and characterization of gsp65, an organic hydroperoxide resistance (ohr) gene encoding a general stress protein in Enterococcus faecalis. J. Bacteriol., 183, 1482–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea R. and Mulks,M. (2002) Ohr, encoding an organic hydroperoxide reductase, is an in vivo-induced gene in Actinobacillus pleuropneumoniae. Infect. Immun., 70, 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J., Smith,S. and Dean,R. (1989) Scavenging by alginate of free radicals released by macrophages. Free Radic. Biol. Med., 6, 347–358. [DOI] [PubMed] [Google Scholar]
- Skaar E., Tobiason,D., Quick,J., Judd,R., Weissbach,H., Etienne,F., Brot,N. and Seifert,S. (2002) The outer membrane localization of the Neisseria gonorrhoeae MsrA/B is involved in survival against reactive oxygen species. Proc. Natl Acad. Sci. USA, 99, 10108–10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G. and Zheng,M. (2000) Oxidative stress. In Storz,G. and Hengge-Aronis,R. (eds), Bacterial Stress Responses. ASM Press, Washington, DC, pp. 47–59.
- Stover C. et al. (2000) Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature, 406, 959–964. [DOI] [PubMed] [Google Scholar]
- Sussman J., Jiang,J., Manning,N., Prilusky,J., Ritter,O. and Abola,E. (1998) Protein Data Bank (PDB): database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. D, 54, 1078–1084. [DOI] [PubMed] [Google Scholar]
- Terwilliger T. (2000) Maximum likelihood density modification. Acta Crystallogr. D, 56, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T. and Berendzen,J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D, 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J., Gibson,T., Plewniak,F., Jeanmougin,F. and Higgins,D. (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff S. (1994) Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol., 233, 182–189. [Google Scholar]
- Yu L., Gunasekera,A., Mack,J., Olejniczak,E., Chovan,L., Ruan,X., Towne,D., Lerner,C. and Fesik,S. (2001) Solution structure and function of a conserved protein SP14.3 encoded by an essential Streptococcus pneumoniae gene. J. Mol. Biol., 311, 593–604. [DOI] [PubMed] [Google Scholar]