

Abstract
Protein disulfide isomerase (PDI) is a multifunctional protein catalysing the formation of disulfide bonds, acting as a molecular chaperone and being a component of the enzymes prolyl 4-hydroxylase (P4H) and microsomal triglyceride transfer protein. The role of PDI as a molecular chaperone or polypeptide-binding protein is mediated primarily through an interaction of substrates with its b′ domain. It has been suggested that this binding is regulated by the redox state of PDI, with association requiring the presence of glutathione, and dissociation the presence of glutathione disulfide. To determine whether this is the case, we investigated the ability of PDI to bind to a folding polypeptide chain within a functionally intact endoplasmic reticulum and to be dissociated from the α-subunit of P4H in vitro in the presence of reducing or oxidizing agents. Our results clearly demonstrate that binding of PDI to these polypeptides is not regulated by its redox state. We also demonstrate that the dissociation of PDI from substrates observed in the presence of glutathione disulfide can be explained by competition for the peptide-binding site on PDI.
Keywords: glutathione/molecular chaperone/procollagen/prolyl 4-hydroxylase/protein disulfide isomerase
Introduction
Protein disulfide isomerase (PDI) has two inter-related activities, the ability to catalyse the formation, reduction and isomerization of disulfide bonds and the ability to bind to polypeptide chains (Freedman et al., 1994). The latter activity enables PDI to function as a molecular chaperone assisting the folding of polypeptides. Here a transient association of PDI with polypeptides during their folding prevents non-productive interactions occurring, thereby increasing the yield of correctly folded proteins (Cai et al., 1994; Puig and Gilbert, 1994). PDI also forms more permanent associations with specific proteins to become a subunit of prolyl 4-hydroxylase (P4H) and microsomal triglyceride transfer protein (Pihlajaniemi et al., 1987; Wetterau et al., 1990). The function of PDI as a component of these enzymes appears to be to maintain the catalytic subunits in a soluble form rather than participating directly in catalysis (Wetterau et al., 1991; John et al., 1993). Both of these polypeptide-binding activities are mediated primarily by the b′ domain of PDI, suggesting that there is a common polypeptide-binding site within PDI (Klappa et al., 1998; Pirneskoski et al., 2001). The binding of PDI to polypeptide chains is also likely to enhance the enzymatic activity of PDI during the catalysis of disulfide bond isomerization (Winter et al., 2002). Therefore, a clearer picture of how PDI binds to polypeptides and how this binding is regulated is crucial for our understanding of the mechanism of action of PDI as an enzyme and as a molecular chaperone.
An elegant explanation for the regulation of the polypeptide-binding activity of PDI has been proposed recently. Tsai et al. (2001) demonstrated that PDI acts as a redox-dependent chaperone to unfold the cholera toxin A1 fragment, thereby facilitating retrograde transport of the protein from the endoplasmic reticulum (ER) into the cytosol. The authors conclude that the binding of PDI to the A1 subunit is regulated by the redox state of PDI; when PDI is reduced it binds to the substrate and when oxidized it is released. Whether the same principle of a redox-dependent regulation of binding can be applied to the binding of PDI to other substrates is not clear. Previous work carried out with purified proteins suggests that binding is independent of the redox state of PDI. Hence, the addition of reducing agents has no effect on the molecular chaperone activity of PDI during the folding of substrates with no disulfide bonds (Cai et al., 1994). Also, mutation (Vuori et al., 1992) or alkylation (Quan et al., 1995; Yao et al., 1997) of the active site of PDI does not prevent either chaperone activity or the ability of PDI to assemble into an active P4H complex. It is therefore still an open question as to whether the polypeptide-binding activity is regulated by the redox state of PDI.
To address this point, we have characterized the binding of PDI to two different substrates. The first of these is the C-propeptide of procollagen that forms a transient association with PDI during the assembly of procollagen. The fibrillar procollagen molecule is a trimer consisting of three distinct regions, two trimeric regions (the N- and C-propeptides) that are separated by a long triple helix-forming region (Prockop and Kivirriko, 1995). Each region within the procollagen chain interacts with a distinct set of ER resident proteins during folding and assembly, thus the triple helix-forming region interacts with P4H (Chessler and Byers, 1992; Walmsley et al., 1999) and Hsp47 (Tasab et al., 2000; Koide et al., 2002), whereas the C-propeptide interacts with BiP (Chessler and Byers, 1993) or PDI (Wilson et al., 1998). PDI interacts specifically with C-propeptides prior to their trimerization, an interaction that retains unassembled chains within the ER, directly preventing secretion until the correct native structure is reached (Bottomley et al., 2001). We have shown previously that the interaction of PDI with the C-propeptide can be studied using an experimental system that reproduces the biosynthesis, folding and assembly of the C-propeptides within a functionally intact ER, allowing us to assess the regulation of the binding in a physiologically relevant environment (Wilson et al., 1998).
In contrast to the C-propeptide, the α-subunit of P4H forms a more prolonged association with PDI, giving rise to an α2β2 tetramer, the β-subunit being PDI. P4H is an enzyme localized to the ER that catalyses the co- and post-translational hydroxylation of proline residues in X-Pro-Gly sequences of collagens and proteins with collagen-like amino acid sequences (reviewed by Kivirikko et al., 1992). The enzyme is assembled in vivo from newly synthesized α-subunits associating with an endogenous pool of PDI (John et al., 1993). It has been shown previously that the α-subunit will assemble with a PDI mutant in which both pairs of cysteine residues within the active site are mutated, demonstrating that the interaction of these proteins is not reliant upon the enzymatic activity of PDI (Vuori et al., 1992). The stability of the complex allows its purification, enabling the redox-dependent association of the α-subunit with PDI to be assessed in vitro. It has already been shown that the enzyme is sensitive to reducing agents that cause the tetramer to dissociate both in vitro and in vivo (Nietfeld et al., 1981; John and Bulleid, 1996). It remains to be established whether this dissociation is due to a change in the redox state of PDI or whether it is due to a reduction of stabilizing intra-chain disulfide bonds within the α-subunit.
Hence, here we have investigated the interaction of PDI with a substrate with which it forms a transient association during its folding pathway and a substrate with which it forms a more permanent association. Our data demonstrate that binding is not dependent on the redox state of PDI. We were able to reproduce the apparent redox-dependent dissociation of PDI from its substrates, and we show that this is not due to a change in the redox state of PDI but is likely to be due to direct competition for the peptide-binding site. Therefore, our results do not support the view that PDI binding to polypeptide substrates is regulated by its redox state.
Results
Redox dependence of the interaction between PDI and the C-propeptide of procollagen
Previous work from our laboratory has established that PDI forms an association with the C-propeptide of procollagen during its assembly in the ER of mammalian cells. Hence, when the C-propeptide from a homotrimer forming procollagen chains [proα1(III)] is expressed individually, it can fold correctly, assemble into trimers (Bulleid et al., 1997) and be secreted from mammalian cells (Bottomley et al., 2001). In contrast, when the C-propeptide from a procollagen chain that does not self-associate to form trimers [proα2(I)] is expressed individually, the protein folds to a monomer but is not secreted (Bottomley et al., 2001). The reason for this lack of secretion is that PDI interacts specifically with the monomeric C-propeptide chains within the ER, thereby facilitating their retention in the cell (Bottomley et al., 2001). The ability to reproduce the binding of PDI to the C-propeptide using a cell-free translation system allows us to evaluate the redox-dependent regulation of this interaction.
The C-propeptide from proα2(I) used for our studies contains seven cysteine residues, six of which are known to form essential intra-chain disulfide bonds (Lees and Bulleid, 1994). The remaining cysteine residue (third from the N-terminus) potentially can form an inter-chain disulfide bond or remain unpaired. To eliminate any potential effect that this thiol group could have on the interaction of PDI with the C-propeptide, we mutated it to serine. The folding of the resulting mutated C-propeptide (S3) was evaluated by translation in the presence of semi-permeabilized (SP) cells. When the translation products were separated under reducing conditions, the radiolabelled S3 and wild-type C-propeptides migrated with identical mobility (Figure 1, lanes 1 and 2). Under non-reducing conditions, an additional band (arrowhead) is seen with the wild-type C-propeptide (lane 3) with the mobility of a dimer, which is absent in the S3 translation products (lane 4). The monomeric C-propeptides from both translations migrate with an increased mobility under non-reducing conditions, indicating the formation of intra-chain disulfide bonds. The identical mobility of the S3 translation products indicates that the correct set of intra-chain disulfide bonds has formed and therefore the protein has folded correctly. The formation of a dimer by the wild-type C-propeptide but not the S3 mutant indicates that the third cysteine from the N-terminus can form an inter-chain disulfide bond; however, most of the translated protein remains monomeric and consequently contains a free thiol residue.

Fig. 1. Synthesis of α2(I) C-propeptides in the presence of SP cells. RNA coding for either the wild-type or S3 C-propeptide was translated in a rabbit reticulocyte lysate in the presence of semi-permeabilized HT1080 cells (SP cells). SP cells were isolated by centrifugation. (A) Products of translation were immunoprecipitated using anti-HA antibody, separated by SDS–PAGE on a 10% polyacrylamide gel under reducing (lanes 1 and 2) or non-reducing conditions (lanes 3 and 4) and visualized by autoradiography. (B) Products of translation were left untreated (lanes 1 and 5) or chemically cross-linked with BMH (lanes 2–4 and 6–8), and immunoprecipitated with anti-HA, PDI or myc antibody. Precipitated samples were separated by SDS–PAGE on a 10% poly acrylamide gel under reducing conditions and visualized by autoradiography.
The interaction of the C-propeptide with PDI can be stabilized after translation by cross-linking with the thiol-specific reagent bismaleimidohexane (BMH) (Bottomley et al., 2001). As can be seen, the wild-type C-propeptide forms several cross-linked products after cross-linking with BMH (Figure 1B, lane 2). The C-propeptide used in these studies contains a haemagglutinin (HA) epitope tag to aid immunoprecipitation (Bottomley et al., 2001). Two of these cross-linked products were immunoprecipitated with an antibody raised against PDI, demonstrating that PDI associates with the C-propeptide (lane 3). The appearance of two cross-linked products immunoprecipitated with the PDI antibody could represent cross-links between different regions of PDI, cross-links to dimers or cross-links to a third as yet unidentified protein. Under identical conditions, the S3 C-propeptide formed only minor (barely visible) cross-linked products (lane 6), none of which were immunoprecipitated with the PDI antibody. For both C-propeptides, some non-cross-linked material was also immunoprecipitated, suggesting that some of the C-propeptide could be co-immunoprecipitated with the antibody to PDI. Hence, mutation of the free cysteine abolished BMH-dependent cross-linking of PDI to the C-propeptide. These results demonstrate that PDI must contain a free thiol when present in a complex with the C-propeptide and that the free thiol in the C-propeptide to which PDI is cross-linked is the third cysteine from the N-terminus. The mutated C-propeptide can fold correctly, but removal of the free cysteine residue eliminates potential non-specific effects that would complicate any study of the redox-dependent interaction of PDI with the C-propeptide.
To assay for an interaction between PDI and the S3 C-propeptide, we used a non-thiol-specific cross-linking agent to stabilize the complex. After cross-linking with the amine-specific agent dithiobis[succinimidyl propionate] (DSP), translation products were separated under reducing conditions; any DSP-cross-linked products are cleaved under these conditions. Both the wild-type and the S3 mutant C-propeptide formed a cross-linked product that was immunoprecipitated with the PDI antibody (Figure 2, upper and lower panel, lane 4). The specificity of this interaction was demonstrated by a lack of cross-linked product immunoprecipitated by a non-specific antibody (lane 6) and an absence of a PDI cross-link to the homo trimer forming C-propeptide α1(III)CP (middle panel).

Fig. 2. The α2(I) C-propeptides can be cross-linked to PDI in a non-thiol-dependent manner. RNA coding for the α2(I), the α1(III) and the S3 C-propeptides were translated in a rabbit reticulocyte lysate in the presence of SP cells. SP cells were isolated by centrifugation and translation products were left untreated (lanes 1, 3 and 5) or chemically cross-linked with DSP (lanes 2, 4 and 6). Cross-linked samples were immunoprecipitated using the indicated antibodies before separation by SDS–PAGE on a 10% polyacrylamide gel, and were visualized by autoradiography.
Having modified our experimental approach to ensure that any alteration in PDI binding is due to a change in the redox state of PDI, we next evaluated the redox state of PDI in the cell before and after treatment with reducing and oxidizing agents. We used two approaches to determine the redox state of PDI; the first involved adding acid directly to intact cells to quench any disulfide exchange, followed by treatment with acetamidomaleimidylstilbene-disulphonic acid (AMS), an alkylating agent that causes a proportional decrease in electrophoretic mobility upon alkylation of free thiol groups. When cells were pre-treated with the reducing agent dithiothreitol (DTT) at a concentration of 10 mM prior to acid quench, a shift in mobility was observed following AMS alkylation (Figure 3A, lane 4). In contrast, when cells were treated with the oxidizing agent dipyridyl sulfide (DPS) at a concentration of 1 mM prior to acid quench, only a slight increase in mobility was observed following AMS alkylation (lane 6). When cells are not treated with either reducing or oxidizing agent, an intermediate level of alkylation occurs that is neither as extensive as after reduction nor as slight as after oxidation (lane 2). These results demonstrate that PDI can be reduced by the addition of DTT, giving rise to the presence of free thiol groups that can then be alkylated by AMS. It is possible that free thiol groups are also present in PDI even after treatment with DPS, giving rise to the slight decrease in mobility after alkylation. At steady state, PDI would appear to be partially oxidized though it is difficult to verify the exact redox state of PDI using this approach due to the relatively small shifts in mobility following AMS treatment.
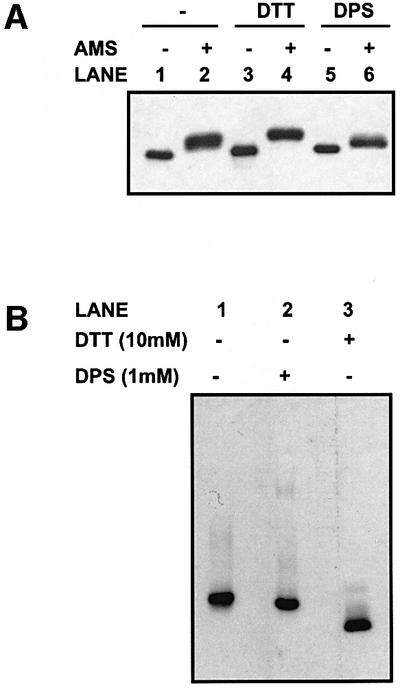
Fig. 3. Determination of the redox state of PDI. (A) HT1080 cells were incubated for 20 min at room temperature alone (lanes 1 and 2), with DTT (lanes 3 and 4) or with DPS (lanes 5 and 6) before lysis in TCA. Free protein thiols were modified with AMS (lanes 2, 4 and 6) or not (lanes 1, 3 and 5), as described in Materials and methods. Proteins were resolved by SDS–PAGE on a 10% polyacrylamide gel, and PDI was detected by western blotting. (B) The HT1080 cell lysate was incubated for 20 min at room temperature alone (lane 1), with DPS (lane 2) or with DTT (lane 3). Samples were treated with NEM and separated by native gel electrophoresis through a 7.5% polyacrylamide gel. Proteins were transferred to nitrocellulose and PDI was detected by western blotting.
As an alternative approach, we evaluated the mobility of PDI after native gel electrophoresis. Cells were either untreated or treated with DTT or DPS prior to N-ethylmaleimide (NEM) alkylation and cell lysis. There was a clear difference in mobility of PDI isolated from cells treated with DTT in comparison with that isolated from cells treated with DPS (Figure 3B, lanes 2 and 3). Thus, the reduction of a disulfide bond or bonds within PDI causes a change in protein conformation resulting in a difference in mobility through a polyacrylamide gel run under native conditions. PDI isolated from cells that had not been treated co-migrated with oxidized PDI, confirming the AMS result that PDI is at least partially oxidized at steady state. It still remains to be established whether the disulfide bond(s) sensitive to reduction that cause this change in conformation is within the active site. What is clear is that treatment of cells with 10 mM DTT causes a reduction of PDI, whereas treatment with DPS at 1 mM maintains PDI in an oxidized form, and that both these forms can be resolved by native gel electrophoresis.
To determine the effect of the redox state of PDI on binding to the C-propeptide, we translated S3-mutated C-propeptide in the presence of SP cells, treated the translation products with either DTT or DPS and then cross-linked with DSP. The concentration of DTT used in these experiments (10 mM) was sufficient to reduce PDI and potentially could reduce the C-propeptide. However, control experiments with increasing concentrations of DTT demonstrated that the intra-chain disulfide bonds within the C-propeptide were not reduced under these conditions (results not shown). A complex between PDI and the C-propeptide remained after treatment with either DTT or DPS (Figure 4, lanes 3 and 7). These results clearly demonstrate that an interaction between PDI and the C-propeptide persists even under conditions where PDI is either reduced or oxidized. Hence the interaction of PDI with the C-propeptide within an intact ER is not regulated by the redox state of PDI.
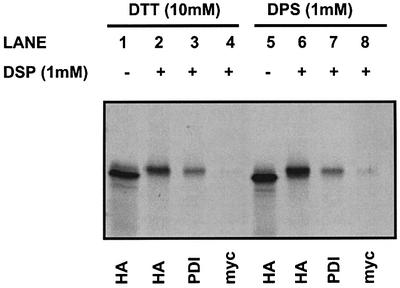
Fig. 4. Reduced and oxidized PDI associates with the S3 C-propeptide. The S3 C-propeptide was translated in a rabbit reticulocyte lysate in the presence of SP cells. SP cells were isolated by centrifugation, and then treated with DTT (lanes 1–4) or DPS (lanes 5–8). Excess DTT and DPS was removed by centrifugation before samples were chemi cally cross-linked with DSP or left untreated. Cross-linked and non-cross-linked samples were immunoprecipitated using the indicated antibodies before separation by SDS–PAGE on a 10% polyacrylamide gel, and translation products were visualized by autoradiography.
Regulation of binding of PDI to the α-subunit of P4H
If PDI acts as a redox-dependent chaperone as previously suggested, then when it is bound to its substrate it should be reduced and become oxidized upon dissociation. To determine the redox state of PDI when present in a P4H complex, we took advantage of the fact that P4H can be affinity purified specifically by binding to poly-l-proline–Sepharose beads. HT1080 cells were treated with NEM to freeze the disulfide status and then lysed, and P4H was purified to determine the redox state of P4H-associated PDI. PDI was dissociated from the α-subunit by treating with 2 M urea, and the resulting dissociated PDI was treated with DPS (lane 5), DTT (lane 6) or left untreated, and separated by native gel electrophoresis (Figure 5). As a comparison, total PDI from the cell lysate was also analysed. Both total PDI and the PDI present as a complex with the α-subunit of P4H co-migrated with oxidized PDI (lanes 1 and 2, and lanes 4 and 5). These results indicate that PDI is oxidized when present as a subunit of P4H. This also indicates that PDI does not need to be reduced to bind to a polypeptide substrate.

Fig. 5. PDI is oxidized when present as a subunit of the P4H complex. P4H was affinity purified by binding to poly-l-proline–Sepharose beads. Purified P4H was treated with 2 M urea, and the resulting dissociated PDI was treated with DPS (lane 5), DTT (lane 6) or left untreated. Similarly, intact HT1080 cells were treated with DPS (lane 2), DTT (lane 3) or left untreated. All samples were treated with NEM to prevent further disulfide exchange, and intact HT1080 cells were lysed. Proteins were separated by native gel electrophoresis on a 7.5% poly acrylamide gel, transferred to nitrocellulose, and PDI was detected using an anti-PDI antibody.
Purified P4H may also be eluted from poly-l-proline– Sepharose beads by competition with poly-l-proline peptide and analysed directly by native gel electrophoresis. This provides information regarding the oligomeric state of P4H and therefore the interaction of the α-subunit with PDI, which has been shown previously to be non-covalent (Nietfeld et al., 1981). The concentration of peptide used for the elution step is 4 mM, which is sufficient to release intact P4H from the affinity matrix. When P4H is eluted and separated directly, the protein migrates as a tetramer (Figure 6A, lane 1). When the purified P4H complex is incubated with DTT, PDI dissociates and migrates with the mobility of a monomer (lanes 2 and 3). However, when the purified complex is incubated with DPS, PDI does not dissociate, as judged by the absence of a monomer (lane 4). The mobility of the complex does become more diffuse, possibly due to charge modification of the polypeptides with the oxidizing agent. These results demonstrate that P4H cannot be dissociated in the presence of an oxidizing agent but can be dissociated in the presence of a reducing agent.
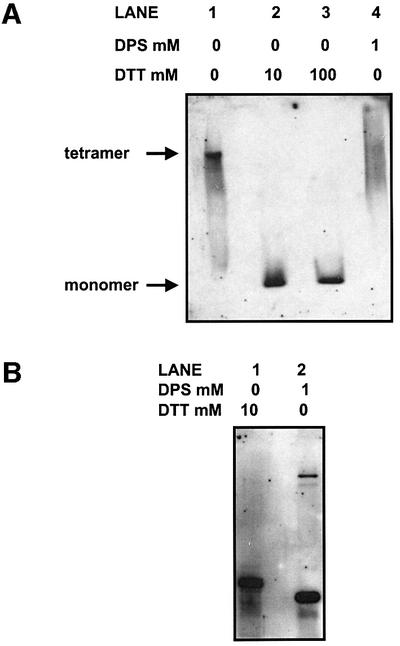
Fig. 6. P4H is not dissociated in the presence of an oxidizing agent but dissociates in the presence of a reducing agent. P4H was affinity purified by binding to poly-l-proline–Sepharose beads, and eluted with excess poly-l-proline. (A) Released P4H was left untreated (lane 1), or treated with 10 or 100 mM DTT (lanes 2 and 3) or DPS (lane 4). Proteins were separated by native gel electrophoresis on a 7.5% poly acrylamide gel, transferred to nitrocellulose, and PDI was detected using an anti-PDI antibody. (B) Released P4H was treated with DTT (lane 1) or DPS (lane 2), and proteins were separated by SDS–PAGE on a 10% polyacrylamide gel, transferred to nitrocellulose, and PDI was detected using an anti-PDI antibody.
The effect of DTT on the stability of P4H was investigated further by assessing the redox state of the α-subunit following reduction. The purified complex was treated with either DTT or DPS and separated by SDS–PAGE under non-reducing conditions, and the subsequent western blot was probed with an α-subunit antibody (Figure 6B). The mobility of the α-subunit was slower after DTT treatment than after DPS treatment (lanes 1 and 2), indicating that intra-chain disulfide bonds within the α-subunit were broken after DTT treatment. These results are consistent with those published previously (John and Bulleid, 1994) and suggest that the effect of DTT on the P4H complex is probably due to a reduction of essential disulfide bonds within the α-subunit, rather than an effect on the redox state of PDI.
It has been demonstrated previously that there is a glutathione-dependent dissociation of PDI from cholera toxin (Tsai et al., 2001). Here we have shown that the alternative oxidizing agent DPS does not lead to a dissociation of PDI from the α-subunit of P4H. To address this apparent discrepancy, we evaluated the effect of glutathione disulfide (GSSG) on the stability of the complex between PDI and the α-subunit of P4H. The purified P4H was incubated with increasing concentrations of GSSG and then separated by native gel electrophoresis. At concentrations of 30–50 mM GSSG, we observed a dissociation of the P4H complex as judged by the appearance of the PDI monomer after native gel electrophoresis (Figure 7A, lanes 3 and 4). This is the same concentration range reported previously to cause a dissociation of PDI from cholera toxin. The dissociated PDI migrated more slowly than the reduced form, indicating that it remained oxidized under these conditions.

Fig. 7. Dissociation of P4H in the presence of GSSG or mastoparan. P4H was affinity purified by binding to poly-l-proline–Sepharose beads, and eluted with excess poly-l-proline. Released P4H was left untreated (lane 1), treated with increasing concentrations of (A) glutathione disulfide or (B) mastoparan (lanes 2–4), or treated with DTT (lane 5). Samples were separated by native gel electrophoresis on a 7.5% polyacrylamide gel, transferred to nitrocellulose and PDI was detected using an anti-PDI antibody.
Glutathione is a tripeptide as well as an oxidizing agent, so a possible explanation for the different affect of GSSG and DPS could be that GSSG is competing directly for the peptide-binding site on PDI and at high concentrations causes PDI to dissociate from its substrate. To test this hypothesis, we incubated purified P4H with the peptide mastoparan. Mastoparan is a non-thiol-containing peptide that has been shown previously to bind to PDI (Klappa et al., 1997). High concentrations (30–50 mM) of mastoparan lead to the dissociation of the P4H complex (Figure 7B). Again the dissociated PDI remained in an oxidized state after treatment with mastoparan (compare lanes 4 and 5). Hence it would appear that peptides that do not contain thiol groups could compete for binding to PDI, resulting in the dissociation of PDI from its substrate. This clearly demonstrates that the effect of GSSG on the binding of PDI to its substrate is due to it competing for the peptide-binding site rather than an effect on the redox state of PDI.
Discussion
The folding and assembly of polypeptide chains into their correct three-dimensional structure is facilitated by a number of folding enzymes and molecular chaperones. PDI is unique amongst these proteins in that it acts both as an enzyme and as a molecular chaperone or polypeptide-binding protein. What is more, the enzymatic activity of PDI can change depending on the redox state, such that it can behave as an oxidase, isomerase or reductase (Freedman et al., 1994). Given the complexity of the various functions of PDI, it has proven difficult to determine how the polypeptide-binding activity is regulated. A major breakthrough in the quest to understand this activity came when work on the unfolding of cholera toxin highlighted the role of PDI in this process and suggested that the molecular chaperone activity of PDI could be regulated by its redox state (Tsai et al., 2001). This is a particularly attractive hypothesis as it could explain how proteins destined for retrograde transport through the translocon are unfolded. Unfortunately, the work presented here does not support this mechanism of regulation for the binding of PDI to two different substrates, but does, however, provide an explanation for the previous observations.
To determine whether PDI acts as a redox-dependent molecular chaperone, it is important to be able to assay and manipulate the redox state of PDI. Mammalian PDI contains six cysteine residues, four of which are within the two active sites, whereas two are present within the b′ domain. All these residues have the potential to form intra-chain disulfide bonds, though it is clear from our cross-linking data with BMH that there must be at least one free thiol residue within PDI when it is present in the ER. Indeed, previous measurements of the thiol content of purified PDI would indicate that there are two free thiol groups per PDI molecule (Quan et al., 1995). Low concentrations of reducing agents are required to activate the isomerase activity of PDI, indicating that when isolated, the active sites of PDI are oxidized (Lyles and Gilbert, 1991). This conclusion is supported by both our results and those published previously (Mezghrani et al., 2001), which demonstrate that at steady state the active sites of PDI are oxidized in mammalian cells. Therefore, the free thiols are most likely to be the non-active site cysteine residues within the b′ domain. The low concentrations of reduced glutathione (GSH) required to ensure PDI acts as a reductase may explain the requirement for GSH during the unfolding of cholera toxin, particularly as this unfolding requires the reduction of a disulfide bond. Thus, under normal steady-state conditions within the cell, PDI is most probably predominantly oxidized but would be reduced transiently to allow both isomerase and reductase activities.
In the work presented here, we were careful to determine the redox state of PDI to ensure that any effect that we observed was due to an alteration of its redox state rather than a direct effect on the substrate or a non-redox specific effect of the reagent used. The change in the redox state of PDI can be monitored most easily by native gel electrophoresis that demonstrates that a conformational change to PDI takes place upon oxidation. This supports previous work that indicated that PDI becomes more sensitive to protease digestion upon reduction (Tsai et al., 2001). Our results, however, clearly demonstrate that under conditions where PDI is either reduced or oxidized, it is still able to bind to the collagen C-propeptide and when it is oxidized it still binds to the α-subunit of P4H. Hence the change in conformation observed by native gel electrophoresis or protease sensitivity does not result in the release of these substrates from PDI. Indeed, PDI is oxidized when associated with the α-subunit of P4H, clearly showing that PDI does not need to be reduced in order to bind to its substrate.
It has been known for some time that PDI can bind to a wide and diverse range of peptides in vitro, an interaction that is independent of the thiol content of the binding peptide. We show here that the interaction between PDI and the α-subunit of P4H can be disrupted by the addition of GSSG or mastoparan. This disruption is most likely to be a consequence of the competition of binding to the peptide-binding site of PDI and not a result of a change in the redox state of PDI. The high concentration of peptide required indicates that these are not preferential substrates for PDI and are unlikely to bind under normal physiological conditions. The dissociation results in the release of PDI that is in the same oxidized redox state as when bound to the α-subunit. We also see a similar dissociation of P4H if the concentration of poly-l-proline used for elution from the affinity matrix exceeds 10 mM (results not shown). These results suggest that the GSSG-dependent dissociation of PDI from cholera toxin can be explained by peptide competition rather than an effect on the redox state of PDI. Hence there is a clear alternative explanation for the results reported previously that bring into question the generalized concept that the polypeptide-binding activity of PDI is redox dependent.
If the polypeptide-binding function of PDI is not regulated by redox, then how is it regulated? The association/dissociation of PDI and its substrate could be in equilibrium, or significant conformational changes occurring in either the substrate or PDI could cause release. Our experiments do not rule out the possibility that a conformational change in PDI regulates binding, but only that this conformational change is not necessarily a consequence of a change in the redox state of PDI. Clearly a conformational change in the substrate could also occur during either folding or assembly to cause release. Further work needs to be carried out to determine the actual mechanism for regulation of polypeptide binding to PDI.
Materials and methods
Construction of recombinant plasmids
Recombinant α1(III) and α2(I) C-propeptide constructs were generated from the proα1(III) and proα2(I) procollagen chains by PCR and subcloned into the expression vector pCEP4. Both constructs contained an influenza HA epitope tag between the signal sequence and the start of the C-propeptide sequence to allow for immunoprecipitation. Introduction of the mutation S3 into the α2(I) C-propeptide was achieved by site-directed mutagenesis using a Quickchange mutagenesis kit (Stratagene) with the following primers: 5′-CCCAACCAAGGAA GCACTATGGAAGCC-3′ and 5′-GGCTTCCATAGTGCTTCCTTG GTTGGG-3′, creating the plasmid α2(I) S3 C-propeptide.
Transcription in vitro
Transcription reactions were carried out as previously described (Gurevich et al., 1991). Recombinant plasmids were linearized with BamHI and transcribed using T7 RNA polymerase (Promega, Southampton, UK). Reactions (100 µl) were incubated at 37°C for 2 h, followed by phenol/chloroform extraction and ethanol precipitation. RNA was resuspended in 50 µl of RNase-free water containing 1 mM DTT and 40 U of RNasin (Promega).
Preparation of semi-permeabilized cells
The human fibrosarcoma cell line HT1080 was obtained from the European Collection of Animal Cell Cultures (ECACC). Cell lines were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen Ltd, UK) supplemented with 10% fetal calf serum. SP cells were prepared as described previously (Wilson et al., 1995).
Translation in vitro
RNA was translated using a rabbit reticulocyte lysate (Flexilysate, Promega, Southampton, UK) for 90 min at 30°C. The translation reaction (25 µl) contained 16.5 µl of reticulocyte lysate, 0.4 µl of 1 mM amino acids (minus methionine), 0.5 µl of 100 mM KCl, 15 µCi of [35S]l-methionine (NEN, Dreiech, Germany), 1 µl of transcribed RNA and ∼105 SP cells. Following translation, SP cells were isolated by centrifugation at 16 000 g for 4 min, and resuspended in KHM buffer (100 mM KOAc, 2 mM MgOAc, 20 mM HEPES buffer pH 7.2).
Chemical cross-linking
After translation, SP cell pellets were resuspended in a final volume of 20 µl of KHM buffer. BMH (Pierce, IL) was added from a 2.5 mM stock [prepared fresh in dimethylsulfoxide (DMSO)] to a final concentration of 25 µM. DSP (Pierce) was added from a 20 mM stock (prepared fresh in DMSO) to a final concentration of 1 mM. Cross-linking of samples was performed for 20 min at room temperature, followed by a further 5 min incubation after addition of 50 mM glycine or 5 mM DTT to quench the DSP or BMH reactions, respectively.
Immunoprecipitation
Following translation, cells were isolated by centrifugation for 4 min, and resuspended in 0.5 ml of immunoprecipitation (IP) buffer [50 mM Tris–HCl buffer pH 7.4, containing 0.15 M NaCl, 10 mM EDTA, 1% (v/v) Triton X-100]. Immunoprecipitations were pre-incubated at 4°C for 30 min in IP buffer containing 50 µl of protein A–Sepharose [10% (w/v) in phosphate-buffered saline (PBS)] (Zymed Laboratories Inc., San Francisco, CA), and the samples were centrifuged for 5 min at 16 000 g to remove protein A-binding components. Immunoprecipitation of products was carried out at 4°C in the presence of an anti-HA antibody (Santa Cruz Biotechnology, CA) and 50 µl of protein A–Sepharose [10% (w/v) in PBS]. Precipitates were washed in immunoprecipitation buffer, resuspended in SDS–PAGE loading buffer [0.0625 M Tris–HCl pH 6.8, 2% (w/v) SDS, 10% (w/v) glycerol, 0.001% (w/v) bromophenol blue] in the absence or presence of 50 mM DTT, and boiled for 4 min. Samples were resolved by SDS–PAGE. After electrophoresis, gels were dried, processed for autoradiography and exposed to Kodak BioMax MR film (Eastman Kodak Company, NY).
Determination of the oxidation state of PDI using AMS
Cells were incubated at room temperature for 20 min with or without DTT (10 or 50 mM) (BDH Laboratory Supplies, Poole, UK) or DPS (1 or 5 mM) (Sigma). To prevent post-treatment disulfide exchange, cells were precipitated in 10% (w/v) trichloroacetic acid (TCA). Precipitates were washed twice in 70% acetone and resuspended in 10 µl of SDS–PAGE loading buffer supplemented or not with 25 mM freshly prepared AMS (Molecular Probes, Leiden, The Netherlands). Samples were incubated for 1 h at room temperature and resolved by SDS–PAGE under reducing conditions. Following separation, samples were transferred onto nitrocellulose membrane, and subsequently immunodecorated with a polyclonal antibody raised against PDI (John et al., 1993), and a secondary horseradish peroxidase (HRP)-conjugated anti-rabbit antibody (Dako, Denmark). Chemiluminescent detection was performed using supersignal reagent (Pierce, IL).
Preparation of poly-l-proline Sepharose
A 0.3 g aliquot of cyanogen bromide-activated Sepharose (Amersham) was added gradually to 10 vols of 1 mM HCl in a scintered glass funnel, filtered and allowed to swell for 15 min at room temperature. The Sepharose was then flushed with 10 ml of 1 mM HCl under suction, and washed with 10 ml of chilled coupling solution (0.1 M sodium hydrogen carbonate pH 8.3, 0.5 M NaCl). The Sepharose was then added to 5 ml of coupling buffer containing 10 mg of poly-l-proline (mol. wt 30 000 Da) (Sigma), which was incubated on a continual mixing roller for 16–20 h at 4°C. The poly-l-proline matrix was then filtered, and washed with a further 5 vols of coupling buffer. The matrix was incubated with 6 ml of blocking solution (0.1 M Tris–HCl pH 8.0) for 2 h at room temperature with continual rolling. The matrix was then washed with three alternating cycles of 10 gel volumes of high (pH 8.3) and low (pH 4) pH buffers, and allowed to stand for 5 min between each cycle. Finally, the matrix was then washed and stored in PBS containing 0.3 mM phenylmethylsulfonyl fluoride (PMSF) and 0.1% (w/v) sodium azide at 4°C.
Cell lysate preparation and prolyl 4-hydroxylase isolation
HT1080 cells at 90–100% confluence were washed twice in PBS, suspended by trypsinization and washed in KHM buffer. Cells were isolated by centrifugation and resuspended in 0.5 ml of lysis buffer [50 mM Tris–HCl pH 7.4, containing 0.15 M NaCl, 10 mM EDTA, 1% (v/v) Triton X-100]. Cells were incubated on ice for 30 min, and solubilized material was separated by centrifugation at 16 000 g for 30 min at 4°C. Supernatants were recovered and stored, or incubated with gentle agitation for 16 h with 10% (w/v) poly-l-proline–Sepharose. The poly-l-proline–Sepharose was isolated by centrifugation at 16 000 g for 5 min and washed three times with lysis buffer. Proteins were eluted from the poly-l-proline–Sepharose by incubating the beads with gentle agitation for 30 min in KHM buffer containing 4 mM poly-l-proline (5000 kDa). Eluted material was separated from poly-l-proline–Sepharose by centrifugation at 13 000 g for 5 min.
Redox manipulation of prolyl 4-hydroxylase
Eluted P4H or HT1080 cell lysate was incubated at room temperature for 20 min with DTT (10 or 50 mM) or DPS (1 or 5 mM). Samples were treated with 25 mM NEM at room temperature for 5 min, and 10 µl of PAGE loading buffer [0.0625 M Tris–HCl pH 6.8, 10% (v/v) glycerol, 0.001% (w/v) bromophenol blue] was added. Samples were resolved by native gel electrophoresis (PAGE performed in the absence of SDS). Following separation, samples were transferred onto a nitrocellulose membrane, and subsequently immunodecorated with a polyclonal antibody raised against PDI or a monoclonal antibody raised against the α-subunit of P4H (ICN Biomedicals, CA), and a secondary HRP-conjugated anti-rabbit or anti-mouse antibody (Dako, Denmark), respectively. Chemiluminescent detection was performed using supersignal reagent.
Peptide binding to prolyl 4-hydroxylase
P4H eluted from the poly-l-proline–Sepharose was incubated at room temperature for 20 min with 5, 30 or 50 mM GSSG (Sigma) or mastoparan (Sigma). Following treatment, proteins were resolved by native gel electrophoresis, transferred to nitrocellulose and PDI detected with a polyclonal antibody to PDI as described above.
Acknowledgments
Acknowledgements
We thank Stephen High for critical reading of the manuscript. This work was supported by a grant from The Wellcome Trust (ref: 61944). R.A.L. is the recipient of a BBSRC studentship.
References
- Bottomley M.J., Batten,M.R., Lumb,R.A. and Bulleid,N.J. (2001) Quality control in the endoplasmic reticulum of unassembled procollagen C-propeptides. Curr. Biol., 11, 1–5. [DOI] [PubMed] [Google Scholar]
- Bulleid N.J., Dalley,J.A. and Lees,J.F. (1997) The C-propeptide of procollagen can be replaced with a transmembrane domain without affecting trimer formation or collagen triple helix folding during biosynthesis. EMBO J., 16, 6694–6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H., Wang,C.-C. and Tsou,C.-L. (1994) Chaperone-like activity of protein disulfide isomerase in the refolding of a protein with no disulfide bonds. J. Biol. Chem., 269, 24550–24552. [PubMed] [Google Scholar]
- Chessler S.D. and Byers,P.H. (1992) Defective folding and stable association with protein disulfide isomerase/prolyl hydroxylase of type I procollagen with a deletion in the proα2(I) chain that preserves the gly-X-Y repeat pattern. J. Biol. Chem., 267, 7751–7757. [PubMed] [Google Scholar]
- Chessler S.D. and Byers,P.H. (1993) BiP binds type I procollagen proαchains with mutations in the carboxy-terminal propeptide synthesized by cells from patients with osteogenesis imperfecta. J. Biol. Chem., 268, 18226–18233. [PubMed] [Google Scholar]
- Freedman R.B., Hirst,T.R. and Tuite,M.F. (1994) Protein disulphide isomerase; building bridges in protein folding. Trends Biochem. Sci., 19, 331–336. [DOI] [PubMed] [Google Scholar]
- Gurevich V.V., Pokrovskaya,I.D., Obukhova,T.A. and Zozulya,S.A. (1991) Preparative in vitro mRNA synthesis using SP6 and T7 polymerases. Anal. Biochem., 195, 207–213. [DOI] [PubMed] [Google Scholar]
- John D.C.A. and Bulleid.N.J. (1994) Prolyl 4-hydroxylase: defective assembly of α-subunit mutants indicates that α-subunit disulphide bonds are required for complex assembly. Biochemistry, 33, 14018–14025. [DOI] [PubMed] [Google Scholar]
- John D.C.A. and Bulleid,N.J. (1996) Intra-cellular dissociation and reassembly of prolyl 4-hydroxylase: the α-subunits associates with BiP allowing reassembly with the β-subunit. Biochem. J., 317, 659–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John D.C.A., Grant,M.E. and Bulleid,N.J. (1993) Cell-free synthesis and assembly of prolyl 4-hydroxylase: the role of the β-subunit (PDI) in preventing misfolding and aggregation of the α-subunit. EMBO J., 12, 1587–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivirikko K.I., Myllyla,R. and Pihlajaniemi,T. (1992) Hydroxylation of proline and lysine residues in collagens and other animal and plant proteins. In Harding,J.J. and Crabbe,M.J.C. (eds), Post Translational Modification of Proteins. CRC Press, London, UK, pp. 1–51.
- Klappa P., Hawkins,H.C. and Freedman,R.B. (1997) Interactions between protein disulphide isomerase and peptides. Eur. J. Biochem., 248, 37–42. [DOI] [PubMed] [Google Scholar]
- Klappa P., Ruddock,L.W., Darby,N.J. and Freedman,R.B. (1998) The b′ domain provides the principle peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J., 17, 927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide T., Takahara,Y., Asada,S. and Nagata,K. (2002) Xaa-Arg-Gly triplets in the collagen triple helix are dominant binding sites for the molecular chaperone HSP47. J. Biol. Chem., 277, 6178–6182. [DOI] [PubMed] [Google Scholar]
- Lees J.F. and Bulleid,N.J. (1994) The role of cysteine residues in the folding and association of the COOH-terminal propeptide of types I and II procollagen. J. Biol. Chem., 269, 24354–24360. [PubMed] [Google Scholar]
- Lyles M.M. and Gilbert,H.F. (1991) Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: pre-steady state kinetics and the utilisation of the oxidising equivalents of the isomerase. Biochemistry, 30, 619–625. [DOI] [PubMed] [Google Scholar]
- Mezghrani A., Fassio,A., Benham, A, Simmen,T., Braakman,I. and Sitia,R. (2001) Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J., 20, 6288–6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nietfeld J.J., van der Kraan,J. and Kemp,A. (1981) Dissociation and reassociation of prolyl4-hydroxylase subunits after cross-linking of monomers. Biochim. Biophys. Acta, 661, 21–27. [DOI] [PubMed] [Google Scholar]
- Pihlajaniemi T., Helaakoski,T., Tasanen,K., Myllylä,R., Huhtala,M.-L., Koivu,J. and Kivirikko,K.I. (1987) Molecular cloning of the β-subunit of human prolyl 4-hydroxylase. This subunit and protein disulphide isomerase are products of the same gene. EMBO J., 6, 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirneskoski A., Ruddock,L.W., Klappa,P., Freedman,R.B., Kivirikko,K.I. and Koivunen,P. (2001) Domains b′ and a′ of protein disulfide isomerase fulfil the minimal requirement for function as a subunit of prolyl 4-hydroxylase. J. Biol. Chem., 276, 11287–11293. [DOI] [PubMed] [Google Scholar]
- Prockop D.J. and Kivirikko,K.I. (1995) Collagens: molecular biology, diseases and potential for therapy. Annu. Rev. Biochem., 64, 403–434. [DOI] [PubMed] [Google Scholar]
- Puig A. and Gilbert,H.F. (1994) Protein disulfide isomerase exhibits chaperone and anti-chaperone activity in the oxidative refolding of lysozyme. J. Biol. Chem., 269, 7764–7771. [PubMed] [Google Scholar]
- Quan H., Fan,G. and Wang,C.-C. (1995) Independence of the chaperone activity of protein disulphide isomerase from its thioredoxin-like active site. J. Biol. Chem., 270, 17078–17080. [DOI] [PubMed] [Google Scholar]
- Tasab M., Batten,M.R. and Bulleid,N.J. (2000) Hsp47: a molecular chaperone that interacts with and stabilises correctly folded procollagen. EMBO J., 19, 2204–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B., Rodighiero,C., Lencer,W.I. and Rapoport,T.A. (2001) Protein disulphide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell, 104, 937–948. [DOI] [PubMed] [Google Scholar]
- Vuori K., Pihlajaniemi,T., Myllyla,R. and Kivirriko,K.I., (1992) Site-directed mutagenesis of human protein disulphide isomerase: effect on the assembly, activity and endoplasmic reticulum retention of human prolyl 4-hydroxylase in Spodoptera frugiperda insect cells. EMBO J., 11, 4213–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmsley A.R., Batten,M.R., Lad,U. and Bulleid,N.J. (1999) Intra-cellular retention of procollagen within the endoplasmic reticulum is mediated by prolyl 4-hydroxylase. J. Biol. Chem., 274, 14884–14892. [DOI] [PubMed] [Google Scholar]
- Wetterau J.R., Combs,K.A., Spinner,S.N. and Joiner,B.J. (1990) Protein disulfide isomerase is a component of the microsomal triglyceride transfer protein complex. J. Biol. Chem., 265, 9800–9807. [PubMed] [Google Scholar]
- Wetterau J.R., Combs,K.A., McLean,L.R., Spinner,S.N. and Aggerbeck, L.P. (1991) Protein disulphide isomerase appears necessary to maintain the catalytically active structure of the microsomal triglyceride transfer protein. Biochemistry, 30, 9728–9735. [DOI] [PubMed] [Google Scholar]
- Wilson R., Allen,A.J., Oliver,J., Brookman,J.L., High,S. and Bulleid,N.J. (1995) The translocation, folding, assembly and redox-dependent degradation of secretory and membrane-proteins in semi-permeabilized mammalian-cells. Biochem. J., 307, 679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R., Lees,J.F. and Bulleid,N.J. (1998) Protein disulfide isomerase acts as a molecular chaperone during the assembly of procollagen. J. Biol. Chem., 273, 9637–9643. [DOI] [PubMed] [Google Scholar]
- Winter J., Klappa,P., Freedman,R.B., Lilie,H. and Rudolph,R. (2002) Catalytic activity and chaperone function of human protein disulphide isomerase are required for the efficient refolding of proinsulin. J. Biol. Chem., 277, 310–317. [DOI] [PubMed] [Google Scholar]
- Yao Y., Zhou,Y.-C. and Wang,C.-C. (1997) Both the isomerase and chaperone activities of protein disulfide isomerase are required for the reactivation of reduced and denatured acidic phospholipase A2. EMBO J., 16, 651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]