

Abstract
We report a simple method, the PinPoint assay, for detecting and identifying single-base variations (polymorphisms) at specific locations within DNA sequences. An oligonucleotide primer is annealed to the target DNA immediately upstream of the polymorphic site and is extended by a single base in the presence of all four dideoxynucleotide triphosphates and a thermostable DNA polymerase. The extension products are desalted, concentrated, and subjected to delayed-extraction MALDI-TOF mass spectrometry. The base at the polymorphic site is identified by the mass added onto the primer. Heterozygous targets produce two mass-resolved species that represent the addition of both bases complementary to those at the polymorphic site. The assay is suitable for double-stranded PCR products without purification or strand separation. More than one primer can be simultaneously extended and then mass-analyzed. The mass spectrometric method thus shows promise for high-volume diagnostic or genotyping applications.
The ability to detect genetic variations, or polymorphisms, is a key tool of the biotechnologist. Many inherited diseases, such as sickle cell anemia, are the result of single point mutations, whereas other diseases, such as breast cancer, are influenced by many widely dispersed mutations (Miki et al. 1994). Mutations not associated with disease (polymorphisms) are also widely studied, because this genetic diversity is the basis for DNA-based identity testing, typing of animals and plants, and evolutionary studies (Erlich et al. 1991). Only ∼20% of human polymorphisms are length polymorphisms, whereas ∼80% of human polymorphisms are single-nucleotide polymorphisms, or SNPs.
Perhaps the most widely employed techniques to identify polymorphisms are allele-specific hybridization probes or allele-specific reverse dot–blot hybridizations (Saiki et al. 1986; Keller et al. 1991). Discrimination of alleles can be difficult, requiring lengthy incubations, tedious washings, and stringent control of time, temperature, and salt concentration. Other common approaches rely on amplification, such as PCR, which can often be made allele-specific (Ballabio et al. 1990) and whose products can then be analyzed by gel electrophoresis. Similarly, ligase chain reaction (LCR) (for review, see Wiedmann et al. 1994) or ligase amplification reaction (Wu and Wallace 1989) can be employed to discriminate allelic differences. Automated fluorescent DNA sequencers can be applied to detection of these polymorphisms (Nickerson et al. 1990), but the speed of analysis is still limited by the relatively slow electrophoresis process. The products of these PCR or LCR amplifications can be detected by ELISA assays (Nickerson et al. 1990) or by mass spectrometry (Jurinke et al. 1996). However, not all PCR reactions can be made reliably allele-specific and LCR assays tend to have a high false-positive failure rates, traits that have prevented their more widespread dissemination. Detection of a base substitution by PCR is simple if a restriction sequence is created or destroyed, because the resultant length change can be detected by electrophoresis or mass spectrometry (Embury et al. 1987; Liu et al. 1995). However, only a small fraction of polymorphisms alter known restriction sites.
Perhaps the most suitable method to identify SNPs is to hybridize a primer adjacent to the polymorphic locus, extend it by a single 2′,3′ dideoxynucleotide, and identify the base added, which is the Watson–Crick complement of the base at the polymorphic site. Terms for this type of assay include primer-guided nucleotide incorporation assay (Syvanen et al. 1993), minisequencing (Nyren et al. 1993), genetic bit analysis (Nikiforov et al. 1994), and single nucleotide primer extension assay (Kuppuswamy et al. 1991). Common to all these procedures, a segment of DNA containing the polymorphism is first amplified by PCR or another amplification technique. Some of these assays require purification of the PCR product and processing to remove one of the complementary strands (Kuppuswamy et al. 1995). An oligonucleotide primer is annealed immediately upstream of the polymorphism, then, in the presence of a ddNTP complementary to the polymorphic site and a DNA polymerase, a single dideoxy residue is incorporated onto the 3′ end of the primer, or two different residues if the target DNA is heterozygous.
SNP assays have been performed historically with four separate extension reactions, each with a single ddNTP carrying a label that is radioactive, fluorescent, or a hapten. In a recent improvement (Nikiforov et al. 1994) the primer extension reaction was carried out simultaneously with two ddNTPs, each carrying a different hapten. Signal was developed with two different colorimetric detection reagents, and the entire process duplicated separately with the remaining two ddNTPs.
Recent technical improvements in MALDI-TOF (matrix-assisted laser desorption ionization mass spectrometry with time-of-flight) detection mass spectrometry have raised the promise of obviating the need for any labels and determining sequence based entirely upon the inherent mass differences of the four naturally occurring bases. For example, mass spectrometry analysis of Sanger dideoxynucleotide ladders was demonstrated recently following the introduction of delayed extraction (DE)-MALDI-TOF (Vestal et al. 1995) and improved methods for Sanger sample generation (Roskey et al. 1996). We demonstrate that the primer extension reactions can be conducted in a single vessel, with four unlabeled ddNPTs, followed by an analysis by DE-MALDI-TOF. The resulting assay, which we term the PinPoint assay, identifies the polymorphism at a given locus, and is faster and less laborious than non-mass spectrophotometric methods. The improvements to existing primer extension assays include use of a recently commercialized thermophilic DNA polymerase, MALDI-compatible enzyme reaction solutions, a simple method for sample concentration and desalting, and analysis by DE-MALDI-TOF.
RESULTS
Comparison of Thermal Stable DNA Polymerases
In the model system (Fig. 1), the sequence for the 20-mer SNP primer was chosen so that upon annealing, its 3′-terminal base was adjacent to the polymorphic site. The primer was typically annealed at 37°C and extended at 72°C in the presence of one or more ddNTPs to form an extension of 21 bases. After denaturing the complex at 95°C, the entire process can be repeated for multiple thermal cycles. As long as unextended primer remains in large excess, the concentration of the extension product should increase linearly in each thermal cycle by a value equal to the concentration of the target sequence. If primer is not in large excess, the primer annealing process will become less efficient in each cycle because of competition with extension product.
Figure 1.
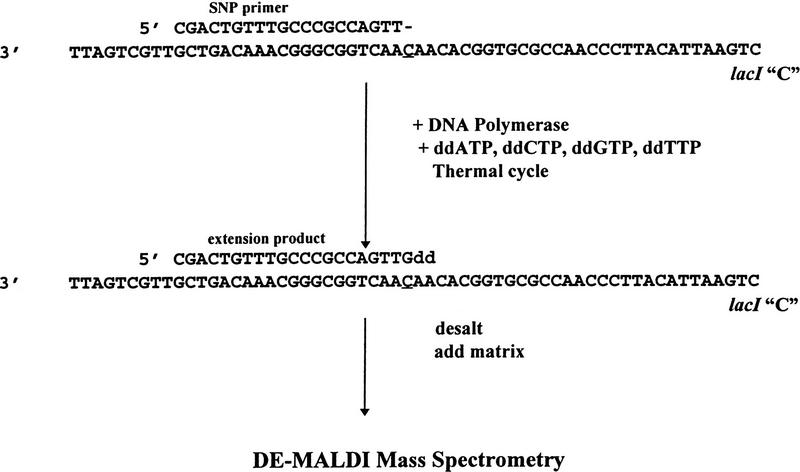
Model system for single nucleotide polymorphism detection. SNP primer was annealed as indicated to the test sequence containing a polymorphic site at the underlined position. In the presence of a DNA polymerase and four ddNTPs, the primer is extended by a single base complementary to the polymorphic site. When subjected to DE-MALDI-TOF, the difference in mass between unextended and extended primer indicates which bases were added.
After the primer extension reaction is completed, samples were concentrated and desalted by reversed phase absorption/elution, mixed with matrix, and subjected to DE-MALDI-TOF mass spectrometry. Because duplex structures fully dissociate under MALDI conditions, the target DNA, which is generally much longer than the primer, does not interfere with detection of primer or its extension product. In these experiments, the signal from the target sequence was generally not observed because it was lower in concentration than the primer and because MALDI-TOF response is lower for higher mass DNAs. The base added onto the extended primer is determined by the incremental mass of ddNMP residues added to the primer (Table 1). The smallest mass difference between the naturally occurring bases is between A and T residues, 9 daltons. In all cases, the difference between measured and calculated mass differences in primer extension products was <1 dalton.
Table 1.
Mass of Dideoxy NMP Additions to SNP Primers
| Base | Formula | Mass (daltons) |
|---|---|---|
| ddC | C9H12N3O5P | 273.19 |
| ddT | C10H13N2O6P | 288.20 |
| ddA | C10H12N5O4P | 297.21 |
| ddG | C10H12N5O5P | 313.21 |
Volatile buffers were used in the PCR and primer extension reactions because it was more difficult to obtain good MALDI-TOF spectra if the PCR or extension reactions were conducted with manufacturer’s reaction mixtures. There are a number of components in these reaction mixtures known to interfere with MALDI, such as tris(hydroxymethylaminomethane) buffer, alkali metal salts, detergents, bovine serum albumin, and glycerol. The POROS concentration/desalting procedure would be expected to remove many of these components, although a systematic study to determine which were removed was not done. We found that high-quality spectra were more easily obtained by substituting reaction mixtures composed of ammonium acetate (at the pH recommended by the manufacturer) and magnesium chloride, leaving out all the other components. The optimal ionic strength of the ammonium acetate buffer was determined individually for each enzyme (see Methods) by employing each enzyme in a model PCR. Under the optimal conditions of pH and ionic strength, the yields from PCRs and primer extension reactions were indistinguishable from those obtained with the manufacturer’s reaction mixtures (data not shown). With the ammonium acetate reaction buffers, either desalting with a Sephadex G-25 spin column or absorption/elution from POROS were effective in desalting the sample, although only the POROS procedure provided additional sample concentration.
To determine the most suitable enzyme for the PinPoint assay, experiments were conducted with the C (wild-type allele ) target. The assay utilized a 2.5-fold excess of primer over target, so if each thermal cycle were 100% efficient, three thermal cycles should fully convert primer into extension product. The initial enzymes tested were AmpliTaq DNA polymerase, and the Exo+ and Exo− Pfu DNA polymerases. With as many as 25 thermal cycles and 1 μm ddGTP, only a small proportion of primer was extended with AmpliTaq (Fig. 2C). The Exo+ Pfu DNA polymerase, possessing 3′ → 5′ proofreading activity, produced extensive hydrolysis products of the primer (Fig. 2E). Both AmpliTaq and Exo− DNA polymerases produced minor amounts of hydrolysis products, the masses of which were consistent with 5′ → 3′ exonuclease activity. A high percentage of the primer was extended by Exo− Pfu DNA polymerase (Fig. 2D) and quantitative extension was obtained with ThermoSequenase (Fig. 2B). The enzymes that failed to extend efficiently in 10 mm ddGTP also failed to extend efficiently in concentrations of ddGTP up to 1 mm.
Figure 2.
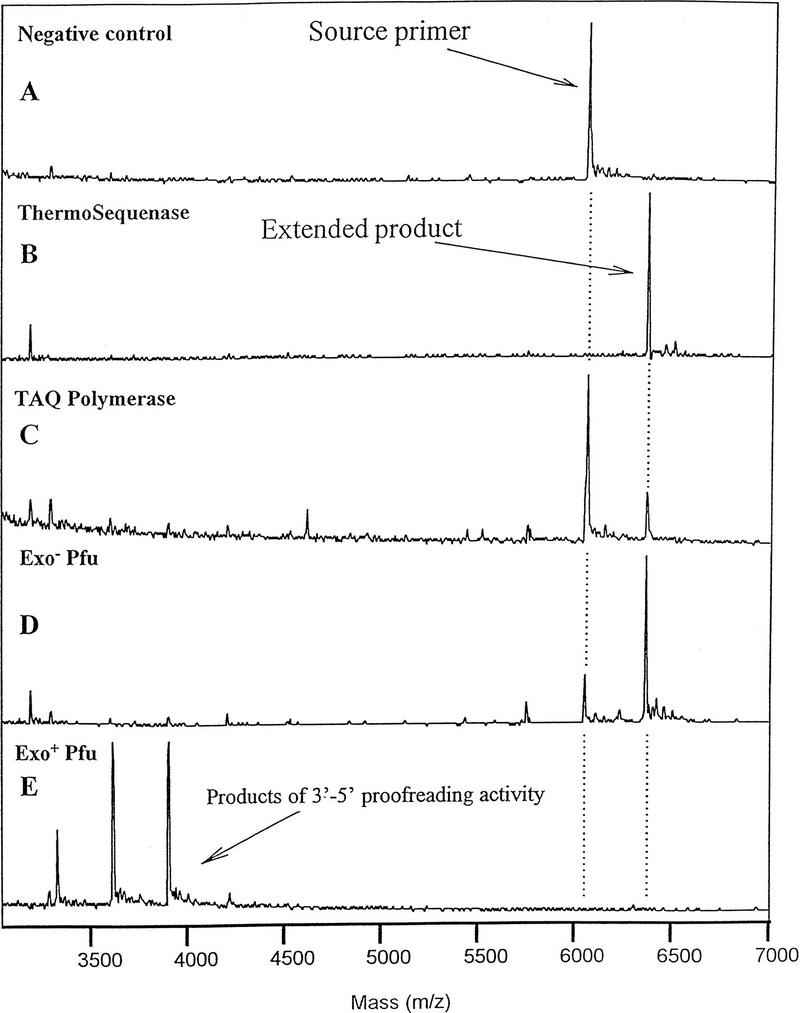
Comparison of enzymes in primer extension. Primer extension product was produced in 25 thermal cycles. (A) negative enzyme control; (B) ThermoSequenase; (C) AmpiTaq DNA polymerase; (D) Exo− Pfu DNA polymerase; (E) exo+ Pfu DNA polymerase. Conditions were as described in Methods and in Fig. 1.
The failure of AmpliTaq DNA polymerase to produce substantial quantities of extension product with ddGTP is probably attributable to the observation that it incorporates all deoxynucleotides relatively slowly (Tabor and Richardson 1995). If 1 μm dGTP was substituted for ddGTP, AmpliTaq DNA polymerase quantitatively extended the primer by a single dG residue (this is the expected extension product in the presence of dGTP alone; see Fig. 1). This was also true of the other enzymes tested, except for Exo+ Pfu DNA polymerase, which degraded primer unless supplied with all four dNTPs (data not shown). It is expected that with proofreading DNA polymerases, the exonuclease activity will predominate if the enzyme is not actively supporting primer extension. The observation of inefficient primer extension with AmpliTaq DNA polymerase does not conflict with the previously described non-mass spectrometric approaches to SNP assays employing this enzyme, because they all utilized enzyme amplification detection techniques or high specific activity radioisotopes and therefore were probably more sensitive detection techniques than MALDI-TOF.
The advantage observed with ThermoSequenase, presumably a mutated Taq DNA polymerase, was because it has virtually the same kinetics of addition of dideoxynucleotides as deoxynucleotides (Tabor and Richardson 1995). In our hands, the kinetics of primer extension from 1 μm to 1 mm ddGTP versus dGTP were indistinguishable. Under the stated conditions, with 0.4 μm target sequence and 1 μm primer, ∼50% of the primer was converted to the extended form in a single cycle (data not shown) This indicates that the efficiency of the primer extension was very high, at least in the initial thermal cycles in which there is a large excess of unextended primer.
Detection Limit
To estimate the detection limit of the assay, primer extension was repeated at lower target concentrations, employing 25 thermal cycles, 1 μm ddGTP, and other conditions as described in Methods. As shown in Figure 3, primer extension was clearly detectable at target concentrations as low as 400 pm. Additional experiments were conducted with 400 pm target to determine whether the yield of the assay could be improved. The reaction yield, estimated from the relative heights of the primer and primer extension products in the DE-MALDI-TOF spectra, could be increased by running 50 thermal cycles instead of 25. However, other changes such as increasing enzyme concentration twofold, increasing extension times twofold, or increasing the times for primer annealing or denaturation all had no discernible effect. Also, primer annealing temperatures between 30 and 45°C and extension temperatures between 60 and 72°C produced equivalent results. These results indicated that the initial assay conditions were nearly optimal for yield and, further, that yield was not very sensitive to the parameters of temperature and incubation times.
Figure 3.
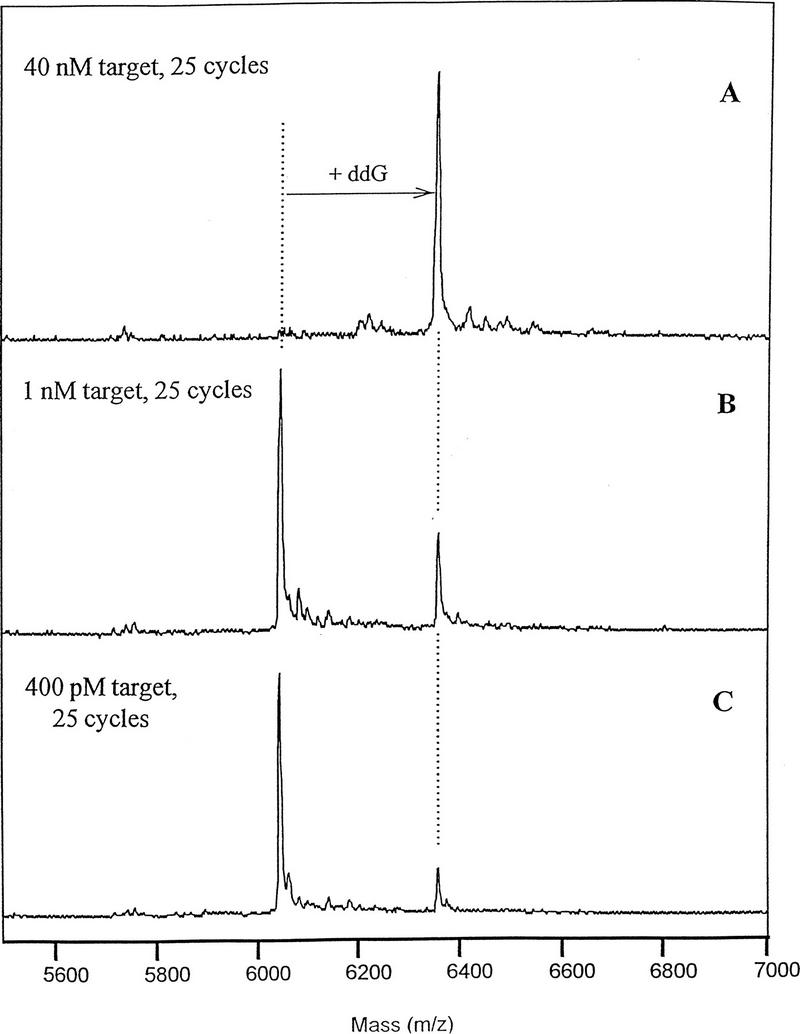
Identification of polymorphism at low level of target DNA. The primer extension assay was conducted with ThermoSequenase as described in Methods in the presence of 10 μm ddNTPs. (A) Forty nanomolar target sequence; (B) 1 nm target sequence; (C) 400 pm target sequence.
Specificity of Incorporation of Dideoxynucleotides
For the PinPoint assay to be specific, the primer must anneal specifically and be extended only by the bases complementary to the target sequences. The first criterion is controlled by well-known parameters governing DNA hybridization and the second by the fidelity of the DNA polymerase. The specificity of extension was tested by thermal cycling ThermoSequenase reaction mixtures with mixtures of ddNTPs. The target sequence was also varied to include all four possible bases. As expected, in the presence of all four ddNTPs, only the base complementary to the polymorphic site was added (Fig. 4). The results were only slightly different when the assay was carried out with each ddNTP separately. In the presence of ddATP, ddCTP, or ddGTP primer extension was observed only with the target carrying the complementary base at the polymorphic site (data not shown). However, in the presence of ddTTP (Fig. 5), a small amount of nonspecific extension was observed with templates containing either a C or G at the site of the polymorphism (Fig. 5). This phenomenon might be a result of either enzyme nonspecificity or nonspecificity of primer annealing. In the model system, some mispriming attriubtable to incorrectly annealed primer is possible because the target sequence contains another three-base sequence coincidentally identical to the sequence adjacent to the polymorphic site (a 5′-CAA sequence starting at position 15 from the 3′ end). If mispriming occurred at this sequence, it would result in incorporation of a T, rather than C residue, as was observed. Some misspriming may occur because the annealing temperature employed, 37°C, is relatively nonstringent for a primer of this length. However, nonspecific incorporation of T residues in the model system was observed only when the reaction mixture was starved for incorporation of the correct base: It was never observed when the reaction mixture contained the ddNTP base complementary to the polymorphic site.
Figure 4.
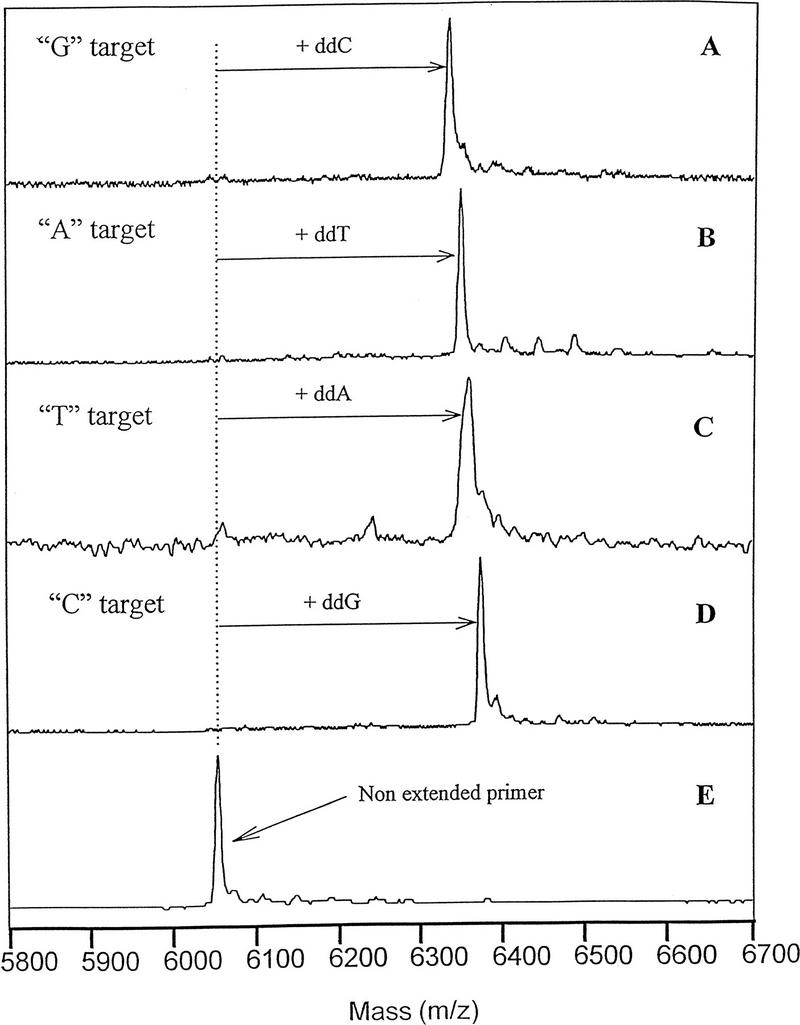
Specificity of primer extension reaction in the presence of 4 ddNTPs. The lacI model system was tested in the presence of ThermoSequenase, all four ddNTPs, and targets containing a different base at the polymorphic site. (A) G target; (B) A target; (C) T Target; (D) C target; (E) negative enzyme control.
Figure 5.
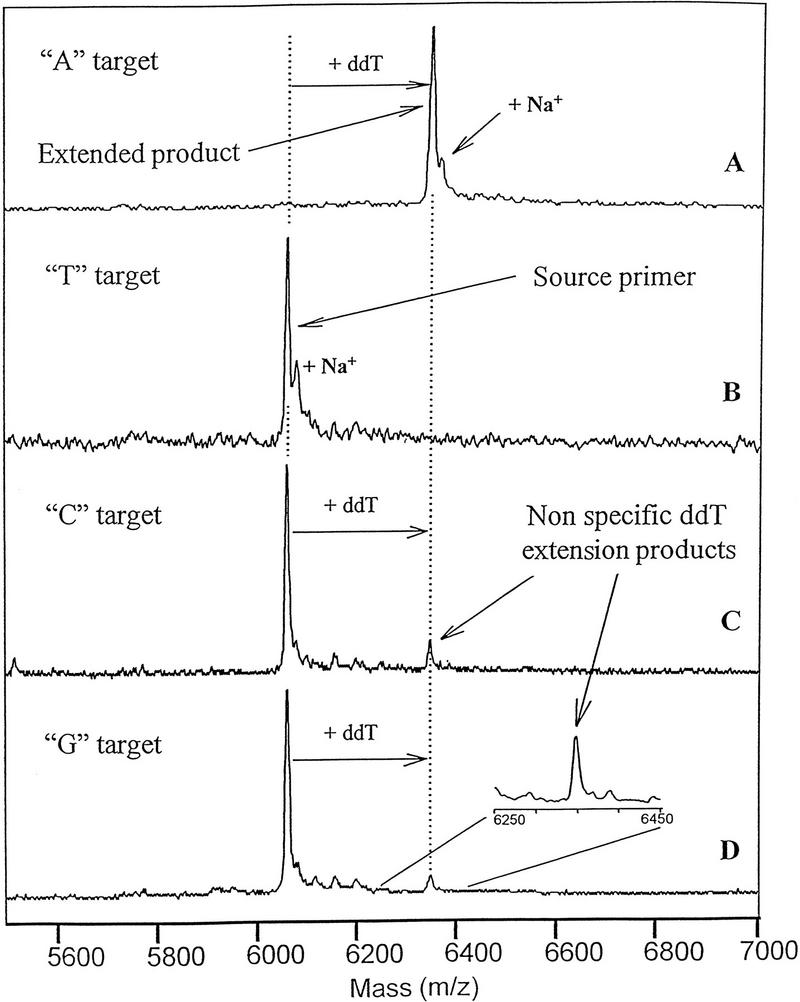
Specificity of extension in the presence of ddTTP. The lacI model system was tested in the presence of ThermoSequenase, ddTTP, and targets containing a different base at the polymorphic site. (A) A target; (B) T target; (C) C target; (D) G target.
Heterozygote Analysis
A desired capability of this technique would include the analysis of heterozygotes, carrying two chromosomes with different bases at the polymorphic site. As shown in Figure 6, when the assay was carried out on a sample containing an equimolar mixture of target DNAs containing a C and a T at the polymorphic site, the analysis produced two partially resolved peaks of primer extension products with a mass difference between them very close to the expected value (16.00 daltons). Experiments with other model systems have confirmed that there is adequate resolution to identify A/T heterozygotes that possess the smallest mass difference between the four bases, 9 daltons.
Figure 6.
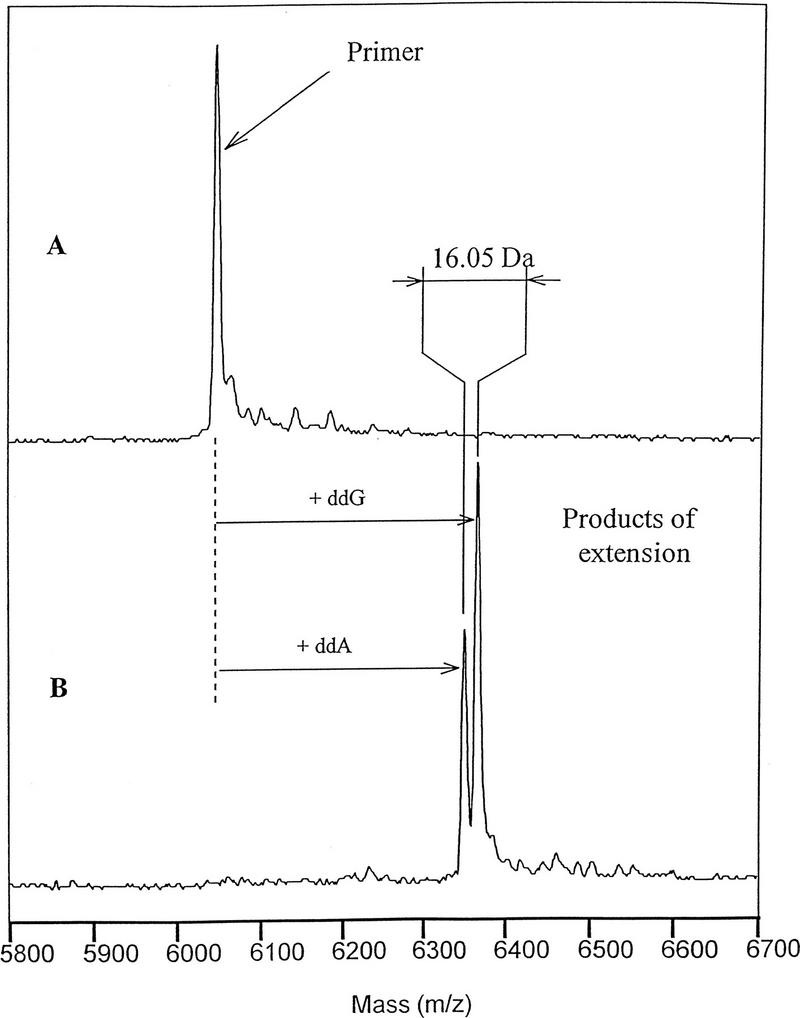
Primer extension assay with heterozygous sample. The SNP assay was conducted with ThermoSequenase as described in Methods in the presence of 10 μm ddNTPs and 0.4 μm each of target sequences with C (wild-type) and T (mutant) at polymorphic site. (A) Negative enzyme control; (B) Plus ThermoSequenase.
Pinpoint Assay of Double-Stranded DNA
The model system described previously consisted of a single-stranded DNA target sequence. In the practical application of the technique, the target sequence would most often be a double-stranded DNA, the product of PCR or another amplification technique. A possible complication to the assay is that the reannealing of the two complementary strands of target DNA could compete with the primer annealing to the target sequence and lower the amount of extension product.
To determine whether this technique would be applicable to double-stranded DNA, reaction mixtures were prepared containing 0.4 μm of the target sequence and a 10% molar excess of its complement. The results with double-stranded target demonstrated no significant diminution of the extension product (Fig. 7). It was anticipated that self-annealing of the target would not be a problem, because the primer concentration was significantly higher than the target concentration. Therefore, it should anneal to the template faster than the denatured template strands should reanneal to themselves. At lower target concentrations, the competition attributable to template reannealing should be even less effective. These results indicate no need to convert double-stranded DNA targets to single strands.
Figure 7.
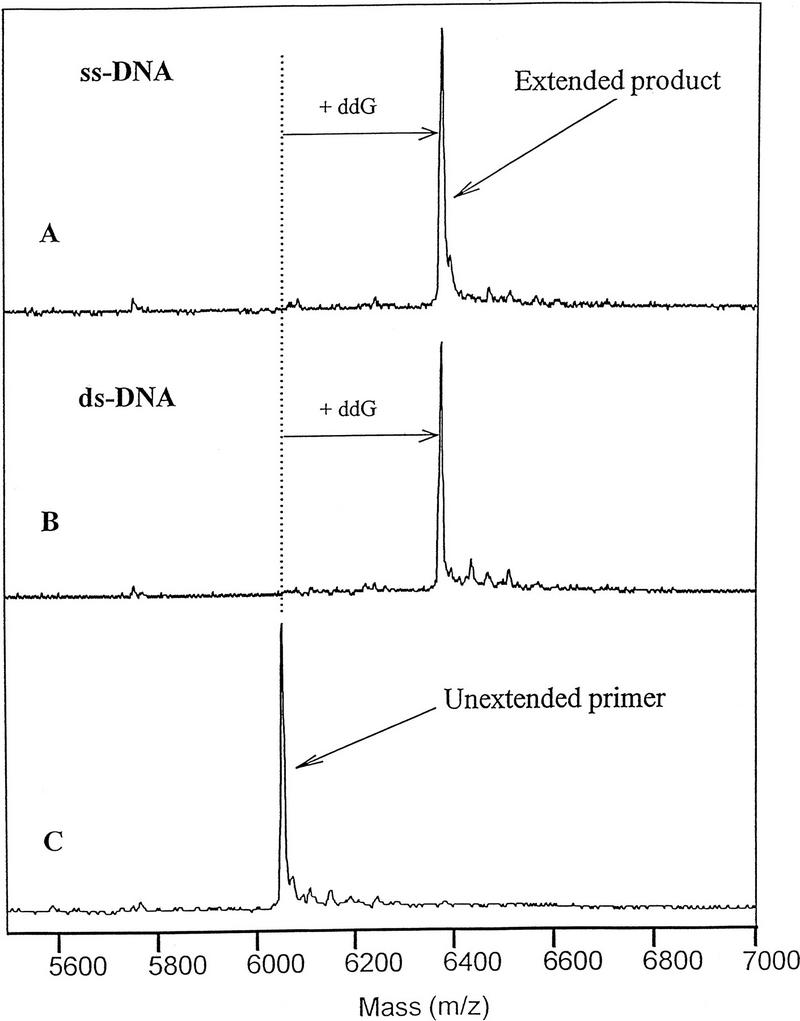
Single nucleotide extension with single and double-stranded target. The single nucleotide extension assay was conducted with ThermoSequenase as described in Methods in the presence of 10 μm ddNPTs. (A) Single-stranded target; (B) Target with 10% excess of complementary sequence; (C) Negative enzyme control.
Primer Extension from BRCA1 PCR Product
The primer extension assay was carried out on PCR product from the BRCA1 exon 13 locus (Fig. 8) treated with shrimp alkaline phosphatase to hydrolyze residual dNTPs in the PCR mixture. Single-base extensions were observed from primer R377 and from the carried-over PCR primers F320 and R480 as well (Fig. 8). In each case, the predominant extension product was the complement of the base immediately downstream from the primer. dNTP carryover from the PCR was controlled by using a relatively low concentration of dNTPs in the PCR, 50 μm, and by the shrimp alkaline phosphatase treatment. Single-base extension (without ladder formation, which would be expected with dNTP carryover) has been obtained with up to 50% (vol/vol) PCR product in the reaction mixture, provided the PCR product was treated by alkaline phosphatase. Sephadex G-25 spin column gel filtration, ethanol precipitation, and a variety of other techniques are also satisfactory to remove the residual dNTPs (data not shown). To conduct the assay with unpurified PCR product, it is only necessary that the primers and their single-base extension products differ in mass sufficiently to be resolved in the mass spectrometer. In the example of Figure 8, all the species were well resolved because primer R377 was 18 bases long wheras PCR primers F320 and R480 were 20 and 25 bases long, respectively. PCR primers can also be removed, if desired, by Sephadex G-50 spin column purification.
Figure 8.
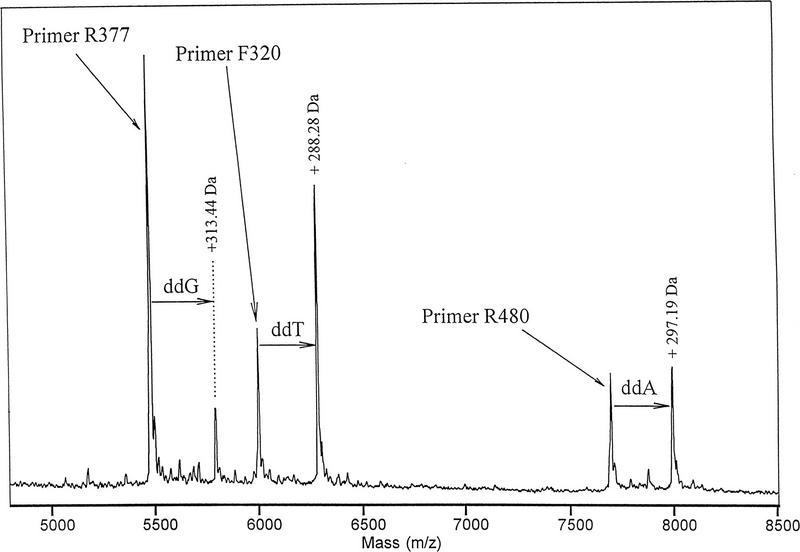
Single nucleotide extension of unpurified BRCA1 PCR product. BRCA1 PCR product was amplified with primers F320 and R480 as described in Methods. Following the primer extension assay, an aliquot of unextended primers F320 and R480 was added to the mixture at a concentration of 0.2 μm to provide a mass reference for their single-base extension products. Measured mass differences between primers and their expected extension products differed from calculated values by a maximum of 0.14 dalton.
DISCUSSION AND CONCLUSIONS
Identification of single nucleotide polymorphisms by mass spectrophotometric analysis of primer extension products provides a facile method for identifying single base polymorphisms. The method offers several advantages over non-mass spectrometric approaches: The samples are prepared in a single tube and analyzed on the MALDI sample plate. Although the current approach has been based upon MALDI-TOF, with slight variations in sample preparation to more thoroughly remove salts, detection could probably be accomplished with electrospray mass spectrometry as well. Unlike radioactive or ELISA approaches (Kuppuswamy et al. 1991; Nikiforov et al. 1994), the mass spectrophotometric process does not require labeling the primer. The entire assay procedure is relatively fast: The primer extension assay currently requires ∼3 min per thermal cycle, but with relatively high concentration of PCR product even a single thermal cycle may often suffice. The thermal cycling time can likewise probably be much reduced. Sample desalting with the reversed phase POROS requires only a few minutes and is amenable to further automation. The samples can be analyzed in the DE-MALDI-TOF instrument without user intervention, in automated mode, at the rate of about two per minute, with further increases in speed likely. If there are only a few samples to process, the turnaround time of the PinPoint assay (following PCR) can be reduced to ∼90 min, much shorter than non-mass spectrometer assays described previously (Nikiforov et al. 1994; Nyren et al. 1993; Syvanen et al. 1993).
The approach described here produces excellent signal to noise ratios for several reasons. In comparison to other DNA analysis techniques, such as forming Sanger ladders (Roskey et al. 1996), all of the primer extension products are of a single length. A second factor in the improved signal is that ThermoSequenase produces a more efficient incorporation of ddNTPs than other thermal stable polymerases. The third factor is that the SNP extension products are only one base longer than the primer itself, so these products are always relatively short and therefore produce better signal response in MALDI-TOF than longer oligonucleotides.
In many of the examples illustrated here, nearly 100% of primer, normally at 1 μm, was fully converted to extension product in 25 thermal cycles. In our hands, the assay was robust and reliable down to at least 400 pm target sequence (4 fmoles in a 10-μl PCR). Because a typical PCR produces product at a significantly higher concentration by plateau phase, from 10−8 to 10−7 m, (Mathieu-Daude et al. 1996), it was not surprising that 25 cycles of primer extension on PCR product normally extended a high proportion of the SNP primer.
Ongoing work with polymorphism typing of PCR products in our laboratory indicates that it is feasible to extend many primers simultaneously and analyze them together, as long as the primers are resolved in mass. We are developing improved methods to destroy both residual dNTPs and PCR primers so that the amount of PCR product accommodated by the primer extension assay can be increased without requiring any purification steps. Increasing the amount of PCR product in the assay will further increases its sensitivity and applicability to polymorphism analysis in PCR reactions that have amplified only weakly. If multiple polymorphic sites can be assayed simultaneously, this technique may prove a useful high-volume assay for multiple polymorphic loci for diagnostic or DNA typing applications.
METHODS
SNP assays were characterized initially with a model system in which the targets were synthetic DNAs containing a partial sequence of the Escherichia coli lacI gene (Fig. 1), also described previously in a ligase chain reaction kit (Stratagene, LaJolla, CA). Oligonucleotides were synthesized by the 0.2 μm β-cyanoethylphosphoramidite protocol on the Expedite Nucleic Acid Synthesis System (PerSeptive BioSystems, Framingham, MA) or purchased from GIBCO-BRL (Gaithersburg, MD). 3-Hydroxypicolinic acid and ion exchange resin in ammonium form were obtained from a Sequazyme DNA Sequencing Kit (PerSeptive Biosystems); other sources for these reagents were described previously (Smirnov et al. 1996). AmpliTaq DNA polymerase was obtained from Perkin Elmer (Norwalk, CT), Exo+ and Exo− Pfu DNA polymerases from Strategene (LaJolla, CA), and shrimp alkaline phosphatase and ThermoSequenase from Amersham (Arlington Heights, IL). Deoxy- and dideoxynucleotides were obtained from GIBCO-BRL.
lacZ Primer and Target Sequences
The sequence of the synthetic wild-type target lacI C was: 5′-CTGAATTACATTCCCAACCGCGTGGCACAACAACTGGCGGGCAAACAGTCGTTGCTGATT-3′. Other targets, lacI A, lacI G, and lacI T, were identical to lacI C except for an A, G, or T, respectively, at the polymorphic site at position 31 (underlined). The complementary sequence to lacI C was 5′-AATCAGCAACGACTGTTTGCCCGC-CAGTTGTTGTGCCACGCGGTTGGGAATGTAATTCAG-3′. The sequence of the 20-mer primer, SNP B, is underlined.
PCR of BRCA1 Exon 13
A 648-bp PCR product from BRCA1 exon 13 was obtained from Myriad Genetics, Inc. (Salt Lake City, UT). This material was amplified further with primers F320 (5′-AGCCTTCTAACAGCTACCCT-3′) and R480 (5′-CATAAAATGTTGGAGCTAGGTCCTT-3′). The reaction mixture contained, in a volume of 100 μl, 50 mm ammonium acetate at pH 8.8, 2 mm magnesium chloride, 0.4 μm each PCR primer, 2.5 units of Exo− Pfu DNA polymerase, 50 μm each of dATP, dCTP, dGTP, and dTTP, and 1 μl of a 100-fold dilution of 648-bp PCR product amplified previously. The mixture was thermal-cycled in a Perkin Elmer DNA Thermal Cycler Model 9600 according to the following program: (95°C for 20 sec, 64°C for 30 sec, 72°C for 60 sec) for 25 cycles. Excess dNTPs in the sample were hydrolyzed by the addition of 0.1 units of shrimp alkaline phosphatase per microliter of PCR reaction mixture, incubation at 37°C for 20 min, followed by incubation at 80°C for 20 min. The sequence of primer R377 employed in the BRCA1 primer extension assays was 5′-TGCTTTGTTCTGGATTTC-3′.
Primer Extension Reactions
Reaction mixtures contained, except as noted, 2 mm magnesium chloride, 1 μm SNP primer, 10 μm of dideoxynucleotides, and synthetic oligonucleotide or PCR product as the target sequence. The buffer in the reaction mixtures containing ThermoSequenase was 25 mm ammonium acetate at pH 9.3. The buffer with either of the Pfu DNA polymerases was 50 mm ammonium acetate at pH 8.8, and the buffer with AmpliTaq DNA polymerase was 100 mm ammonium acetate at pH 8.3. The concentration of each enzyme was 20 U/ml of ThermoSequenase or 25 U/ml of AmpliTaq DNA polymerase, Exo+ or Exo− Pfu DNA polymerases. The reaction mixtures (10–100 μl) were thermal-cycled in a Perkin Elmer DNA Thermal Cycler 9600 (95°C for 10 sec, 37°C for 60 sec, 72°C for 60 sec). In some cases, an aliquot of unextended primers was added following primer extension; this assured the presence of unextended primer of known mass in the mixture as a calibration standard.
Reversed Phase Sample Desalting and Concentration
The sample purification method was modified from one described previously (Roepstorff et al. 1996). Gel-loading pipette tips (Type GT-1514, Rainin Instruments, Woburn, MA) were packed with POROS 50 R1 (PerSeptive Biosystems). They were packed by briefly mixing a 40 mg/ml suspension of POROS R1 in 20% acetonitrile with a Vortex Genie (VWR Scientific, Plainfield, NJ) and immediately dispensing 5 μl of the suspension to each tip. Solvent was removed by applying compressed air to the tip. To use these packed pipette tips, solutions were introduced as a slug into the barrel end of the tip and forced out with compressed air.
Each tip was washed first with a 10-μl slug of 100 mm triethylammonium acetate (pH 6.5), then sample was applied in 20-μl aliquots of a 1:1 mixture of sample and 100 mm triethylammonium acetate. It was then washed with 10 μl of 100 mm triethylammonium acetate, followed by 10 μl of 10 mm triethylammonium acetate (pH 6.5), and then compressed air to remove the remaining solution. The sample was eluted with 2-μl slugs of 20% acetonitrile in water.
The acetonitrile fraction containing the concentrated and desalted sample was mixed with 0.5–1 μl of 3-hydroxypicolinic acid (50 grams/liter solution in water) and treated on a piece of Parafilm with an approximately equal volume of cation exchange beads in ammonium form from a Sequazyme DNA Sequencing Kit (PerSeptive Biosystems; see also Roskey et al. 1996). The samples and beads were mixed for ∼15 sec with a pipette tip, the beads were allowed to settle, and the supernatant solution was removed and applied to a polished stainless steel MALDI sample and dried under a heat gun or under ambient conditions. Experiments were carried out on a DE-Voyager MALDI-TOF mass spectrometer (PerSeptive Biosystems, Framingham, MA) operating in the positive ion and linear modes. This instrument has a drift length of 1.3 m and uses a nitrogen laser for sample desorption and ionization. Other aspects of instrument operation were described previously (Roskey et al. 1996).
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance witht 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lhaff@pbio.com; FAX (508) 383-7883.
REFERENCES
- Ballabio A, Gibbs RA, Casky CT. PCR test for cystic fibrosis deletion. Nature. 1990;343:220. doi: 10.1038/343220a0. [DOI] [PubMed] [Google Scholar]
- Embury SH, Scharf SJ, Saiki RK, Randall K, Gholson MA, Golbus M, Arnheim N, Erhlich HA. Rapid prenatal diagnosis of sickle cell anemia by a new method of DNA analysis. New Engl J Med. 1987;316:656–661. doi: 10.1056/NEJM198703123161103. [DOI] [PubMed] [Google Scholar]
- Erlich HA, Gelfand D, Sninsky JJ. Recent advances in the polymerase chain reaction. Science. 1991;252:1643–1651. doi: 10.1126/science.2047872. [DOI] [PubMed] [Google Scholar]
- Jurinke C, van den Boom DA, Jacob K, Tang K, Worl R, Koster H. Analysis of ligase chain reaction products via matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal Biochem. 1996;237:174–181. doi: 10.1006/abio.1996.0225. [DOI] [PubMed] [Google Scholar]
- Keller GH, Huang D-P, Manek MM. Detection of human immunodeficiency virus type I DNA by polymerase chain reaction amplification and capture hybridization in microtiter well. J Clin Microbiol. 1991;29:638–641. doi: 10.1128/jcm.29.3.638-641.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppuswamy MN, Hoffmann JW, Kasper CK, Spitzer SG, Groce SL, Bajaj SP. Single nucleotide primer extension to detect genetic diseases: Experimental application to hemophilia B (factor IX) and cystic fibrosis genes. Proc Natl Acad Sci. 1991;88:1143–1147. doi: 10.1073/pnas.88.4.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y-H, Bai J, Zhu Y, Liang X, Siemieniak D, Venta PJ, Lubman DM. Rapid screening of genetic polymorphisms using buccal cell DNA with detection by matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom. 1995;9:735–743. doi: 10.1002/rcm.1290090905. [DOI] [PubMed] [Google Scholar]
- Mathieu-Daude F, Welsh J, Vogt T, McClelland M. DNA rehybridization during PCR: The CoT effect and its consequences. Nucleic Acids Res. 1996;24:2080–2086. doi: 10.1093/nar/24.11.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eldens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, Bell R, Rosenthal J, Hussey C, Tran T, McClure M, Frye C, Hattier T, Phelps R, Haugen-Strano A, Katcher H, Yakumo K, Gholami Z, Shaffer D, Stone S, Bayer S, Wray C, Bogden R, Dayananth P, Ward J, Tonin P, Narod S, Bristow PK, Norris FH, Helvering L, Morrison P, Rosteck P, Lai M, Barrett JC, Lewis C, Neuhausen S, Cannon-Albright L, Goldgar D, Wiseman R, Kamb A, Skolnick MH. A strong candidate for the breast cancer and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- Nickerson DA, Kaiser R, Lappin S, Stewart J, Hood L. Automated DNA diagnostics using an ELISA-based oligonucleotide ligation assay. Proc Natl Acad Sci. 1990;87:8923–8927. doi: 10.1073/pnas.87.22.8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov TT, Rendle RB, Goelet P, Rogers H-H, Kotewicz ML, Anderson S, Trainor GL, Knapp MR. Genetic bit analysis: A solid phase method for typing single nucleotide polymorphisms. Nucleic Acids Res. 1994;22:4167–4175. doi: 10.1093/nar/22.20.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyren PB, Pettersson B, Uhlen M. Solid phase minisequencing by an enzymatic luminometric inorganic pyrophosphate detection assay. Anal Biochem. 1993;208:171–175. doi: 10.1006/abio.1993.1024. [DOI] [PubMed] [Google Scholar]
- Roskey MT, Juhasz P, Smirnov IP, Takach EJ, Martin SA, Haff LA. DNA sequencing by delayed extraction matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Proc Natl Acad Sci. 1996;93:4724–4729. doi: 10.1073/pnas.93.10.4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiki RK, Bugawan TL, Horn GT, Mullis KB, Erlich HA. Analysis of enzymatically amplified beta globin and HLA-DQalpha DNA with allele-specific oligonucleotide probes. Nature. 1986;324:163–166. doi: 10.1038/324163a0. [DOI] [PubMed] [Google Scholar]
- Smirnov IP, Roskey MT, Juhasz P, Takach EJ, Martin SA, Haff LA. Sequencing oligonucleotides by exonuclease digestion and delayed extraction matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal Biochem. 1996;238:19–25. doi: 10.1006/abio.1996.0243. [DOI] [PubMed] [Google Scholar]
- Syvanen A-C, Sajantila A, Lukka M. Identification of individuals by analysis of biallelic DNA markers using PCR and solid-phase mini-sequencing. Am J Hum Genet. 1993;52:46–59. [PMC free article] [PubMed] [Google Scholar]
- Tabor S, Richardson CC. A single residue in DNA polymerases of the Escherichia coli DNA polymerase I family is critical for distinguishing between deoxy- and dideoxyribonucleotides. Proc Natl Acad Sci. 1995;92:6339–6343. doi: 10.1073/pnas.92.14.6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestal ML, Juhasz P, Martin SA. Delayed extraction matrix-assisted laser desorption time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 1995;9:1044–1050. [Google Scholar]
- Wiedmann M, Wilson JJ, Czajka J, Jy L, Barany F, Batt CA. Ligase chain reaction (LCR) overview and applications. PCR Methods Applic. 1994;3:S51–S64. doi: 10.1101/gr.3.4.s51. [DOI] [PubMed] [Google Scholar]
- Wu DY, Wallace RB. The ligation amplification reaction (LAR)—Amplification of specific DNA sequences using sequential rounds of template-dependent ligation. Genomics. 1989;4:560–569. doi: 10.1016/0888-7543(89)90280-2. [DOI] [PubMed] [Google Scholar]