

Abstract
Angelman syndrome (AS) and Prader–Willi syndrome (PWS) are distinct clinical phenotypes resulting from maternal and paternal deficiencies, respectively, in human chromosome 15q11–q13. Although several imprinted, paternally expressed transcripts have been identified within the PWS candidate region, no maternally expressed gene has yet been identified within the AS candidate region. We have developed an integrated physical map spanning the PWS and AS candidate regions and localized two breakpoints, including a cryptic t(14;15) translocation associated with AS and a non-AS 15q deletion, which substantially narrow the AS candidate region to ∼250 kb. Mapping data indicate that the entire transcriptional unit of the E6–AP ubiquitin–protein ligase (UBE3A) gene lies within the AS region. The UBE3A locus expresses a transcript of ∼5 kb at low to moderate levels in all tissues tested. The mouse homolog of UBE3A was cloned and sequenced revealing a high degree of conservation at nucleotide and protein levels. Northern and RT–PCR analysis of Ube3a expression in mouse tissues from animals with segmental, paternal uniparental disomy failed to detect substantially reduced or absent expression compared to control animals, failing to provide any evidence for maternal-specific expression from this locus. Recent identification of de novo truncating mutations in UBE3A taken with these observations indicates that mutations in UBE3A can lead to AS and suggests that this locus may encode both imprinted and biallelically expressed products.
[The sequence data described in this paper have been submitted to the GenBank data library under accession no. U82122.]
Angelman syndrome (AS) and Prader–Willi syndrome (PWS) are distinct neurobehavioral disorders associated with maternal and paternal deficiencies, respectively, of gene expression from human chromosome 15q11–q13, indicating that oppositely imprinted genes are involved in these disorders (Ledbetter and Ballabio 1995; Nicholls 1993). Patients with AS are characterized by mental retardation with absent speech, microcephaly, seizures, abnormal electroencephalograph (EEG), a puppet-like movement disorder, inappropriate laughter, and characteristic facies (Williams et al. 1995). PWS is characterized by hypotonia, hypogonadism, failure to thrive, and feeding difficulties in infancy. Older PWS patients develop obesity secondary to hyperphagia and exhibit moderate mental retardation, small hands and feet, and characteristic facies (Holm et al. 1993).
A number of paternally expressed transcripts have been identified in the PWS candidate region, and these include small nuclear ribonucleoprotein-associated polypeptide N (SNRPN) (Ozcelik et al. 1992; Glenn et al. 1993b; Reed and Leff 1994), a gene with intron–exon structure, but without an obvious open reading frame designated as imprinted in Prader–Willi (IPW) (Wvrick et al. 1994), and two large transcripts that lack intron/exon structure or substantial open reading frames and are designated PAR-5 and PAR-1 for Prader–Willi/Angelman region (Sutcliffe et al. 1994). Similar to observations at other imprinted loci, differential methylation is observed over a wide region in 15q11–q13, with the majority of sites being methylated on the maternally derived homolog. This includes substantial maternal methylation at a CpG island located at the 5′ end of SNRPN (Sutcliffe et al. 1994; Glenn et al. 1996), and discrete sites of maternal methylation at a marker designated PW71 (Dittrich et al. 1993). Southern blot probes detecting differential methylation have been particularly useful in facilitating molecular diagnosis of these two disorders (Cassidy et al. 1996).
The majority (∼70%) of both AS and PWS patients exhibit large (3–4 Mb) interstitial deletions of a common interval in 15q11–q13. PWS deletions occur exclusively on the paternally derived homolog, whereas AS deletions are specific to the maternal chromosome. Most of the remainder of PWS patients demonstrate maternal uniparental disomy (UPD); however, the reciprocal paternal UPD is observed in only ∼4% of AS patients (Nicholls 1993; Ledbetter and Ballabio 1995). Imprinting defects, termed imprinting mutations, are seen in ∼2% of PWS and 7% of AS patients and are characterized by biparental inheritance but abnormal methylation and a deficiency of imprinted gene expression in the region (Glenn et al. 1993a; Reis et al. 1994; Sutcliffe et al. 1994; Cassidy et al. 1996). The observation of small deletions upstream of SNRPN in several imprinting mutation cases implies the existence of an imprinting center (IC), which may act to regulate the switching of the imprinting pattern during gametogenesis (Sutcliffe et al. 1994; Buiting et al. 1995; Dittrich et al. 1996). In addition to deletions, UPD, and imprinting mutations, a fourth class of patients exists for AS and is characterized by biparental inheritance and normal methylation at SNRPN and PW71. It has been hypothesized that this group of patients represents likely intragenic mutant cases of AS and includes rare families with multiple affected cases over several generations demonstrating an imprinted pattern of inheritance and linkage to 15q11–q13 (Meijers-Heijboer et al. 1992; Wagstaff et al. 1993).
Observation of rare deletion and translocation patients who are deficient for only a portion of the 15q11–q13 large deletion interval has allowed further definition of candidate regions for AS and PWS. One particularly important case involved familial segregation of an interstitial deletion that caused AS when maternally inherited but a normal phenotype when paternally inherited (family Se) (Hamabe et al. 1991; Saitoh et al. 1992). This most likely indicates that the deletion interval contains part or all of the AS gene but not those genes whose deficiency produces a PWS phenotype; this effectively separates the PWS and AS candidate regions. One breakpoint in this case was found to map just telomeric to the group of paternally expressed transcripts, with the deletion extending telomeric ∼1.5 Mb (Greger et al. 1993). In the course of positional cloning efforts to identify genes in the AS/PWS region, we found that several transcripts, including the gene encoding the E6-associated protein (now designated E6–AP ubiquitin–protein ligase; gene symbol UBE3A) mapped to the AS candidate region (Nakao et al. 1994). RT–PCR analysis of these transcripts for imprinted expression using cultured fibroblasts and lymphoblasts from AS and PWS patients with large deletions (Nakao et al. 1994) thus far has failed to demonstrate maternal-specific expression for any transcript in this region.
Two recently reported cases involving chromosome 15 abnormalities may further define the AS candidate region. The first case involves detection of a cytogenetically visible deletion in proximal 15q in a mother and her son, who was ascertained for developmental delay, but with no obvious features of AS (Michaelis et al. 1995). Fluorescent in situ hybridization (FISH) and microsatellite analysis had shown that the centromeric breakpoint maps within an ∼800-kb interval between markers D15S10 and D15S113. A second case involves a familial cryptic t(14;15) translocation associated with AS. In this family, the mother and unaffected sibling are chromosomally balanced with the affected sibling unbalanced and deficient for genetic material in proximal chromosome 15 (Burke et al. 1996). The simplest interpretation of the data for this family is that the der(15) inherited by the unaffected sibling retains all of the genetic information necessary to protect against AS. FISH analysis of this family suggested that the der(15) translocation breakpoint maps near D15S10. A third case in which two affected siblings with familial AS demonstrated different alleles at a marker locus (D15S122), which maps telomeric to the Se family breakpoint, may narrow the AS candidate region even further (Greger et al. 1994).
Positional cloning efforts in the PWS/AS region have led to the generation of physical maps comprised of a yeast artificial chromosome (YAC) contig of the 3 to 4-Mb common deletion interval (Mutirangura et al. 1993), as well as cosmid and pulsed-field gel electrophoresis (PFGE) maps of the region surrounding the Se breakpoint, extending centromeric from the IC region, through the PWS paternal expression domain and part of the AS candidate region telomeric to the breakpoint (Sutcliffe et al. 1994). Coverage of this interval was incomplete, however, with several gaps present in the YAC-derived cosmid contig. We now report the development of a complete and integrated physical map of the IC, PWS, and AS candidate regions comprised of YACs, P1 artificial chromosomes (PACs) and cosmids, annotated with genes and other marker loci. The t(14;15) and non-AS 15q deletion breakpoints have been localized, defining a narrowed AS critical region containing the UBE3A gene. Although evidence for any maternally expressed product from this locus is lacking, recent identification of de novo truncating mutations in a few AS patients indicates that UBE3A is the AS gene and argues that a maternal-specific product from this locus should exist (Kishino et al. 1997; Matsuura et al. 1997).
RESULTS
PAC clones were isolated following PCR and hybridization-based screening for the following loci: PW71 (D15S63), SNRPN 3′-untranslated region (UTR), PAR-5, IPW, PAR-1, UBE3A, and D15S10. Multiple clones were identified for each locus and were assessed by PCR for STS (sequence-tagged site) content using the above loci and confirmed by hybridization using these and other probes in the region. STS mapping of other markers included D15S174, corresponding to the Se breakpoint (Greger et al. 1994), a new microsatellite marker (D15S1506) derived from cosmid 17, and D15S122, which was found to map just centromeric to D15S10 (Fig. 1B). Additionally, relationships between PAC clones and previously isolated cosmids were determined and are shown in Figure 1B.
Figure 1.
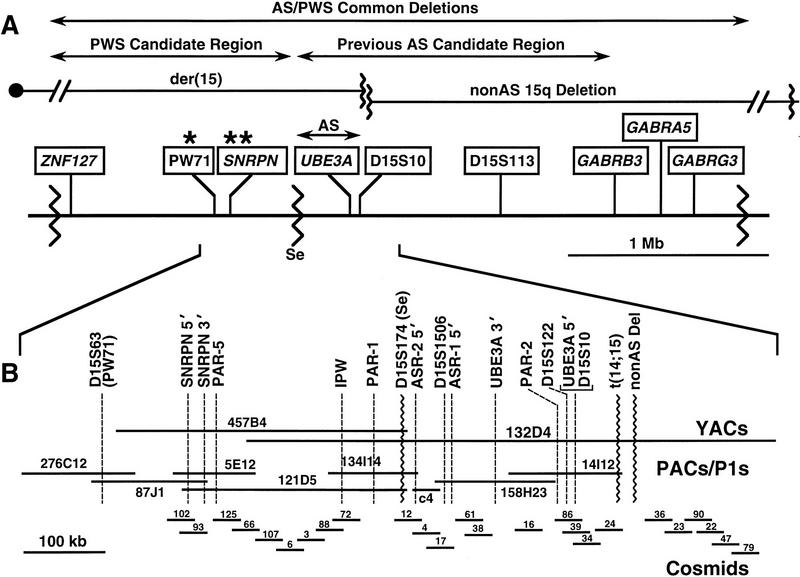
(A) A schematic physical map of the 15q11–q13 PWS/AS common deletion interval is shown with the centromere toward the left and the telomere toward the right. Genes and genomic markers are shown in boxes. Sites of differential methylation are indicated by asterisks (*) over PW71 and SNRPN. Wavy vertical lines represent chromosomal breakpoints. The common PWS and AS deletion breakpoints are near either end of the map, and the Se family centromeric deletion breakpoint maps between SRRPN and UBE3A, defining the PWS and AS candidate regions, indicated over the map. Breakpoints in the 15q non-AS deletion case and the t(14;15) translocation case, together with the Se breakpoint, define the narrowed AS critical region indicated above UBE3A. (B) YAC, PAC, cosmid, STS, and gene map of the ∼1-Mb region surrounding the Se breakpoint. Genomic clones are indicated by horizontal lines; gene and STS markers are indicated by broken vertical lines.
To localize breakpoints in the t(14;15) and non-AS 15q deletion cases, PACs and cosmids from the region including and telomeric to the Se breakpoint were utilized as hybridization probes on Southern blots containing genomic DNA from the respective families and normal controls. Any clone crossing a breakpoint would be expected to detect a variant restriction fragment because of a rearrangement of restriction sites at the translocation or deletion junction. PAC 14I12 and overlapping cosmid 24 detected a possible EcoRI junction fragment when used as probes on blots containing DNA from the t(14;15) family (data not shown). To evaluate this possibility, each of four individual EcoRI fragments constituting the c24 insert was tested individually on Southern blots containing normal and t(14;15) samples. One 8-kb EcoRI fragment, designated 24E8, detected variant fragments with multiple enzymes in the mother and affected child (Fig. 2). With some enzyme–probe combinations, such as with HindIII, two abnormal fragments were seen in the mother, one of which was common to the affected daughter. This result is consistent with the mother having two derivative chromosomes and the child having only the der(14). The fact that one probe can detect both breakpoints suggests that there was not a substantial loss of genetic material from the site of breakage on chromosome 15.
Figure 2.
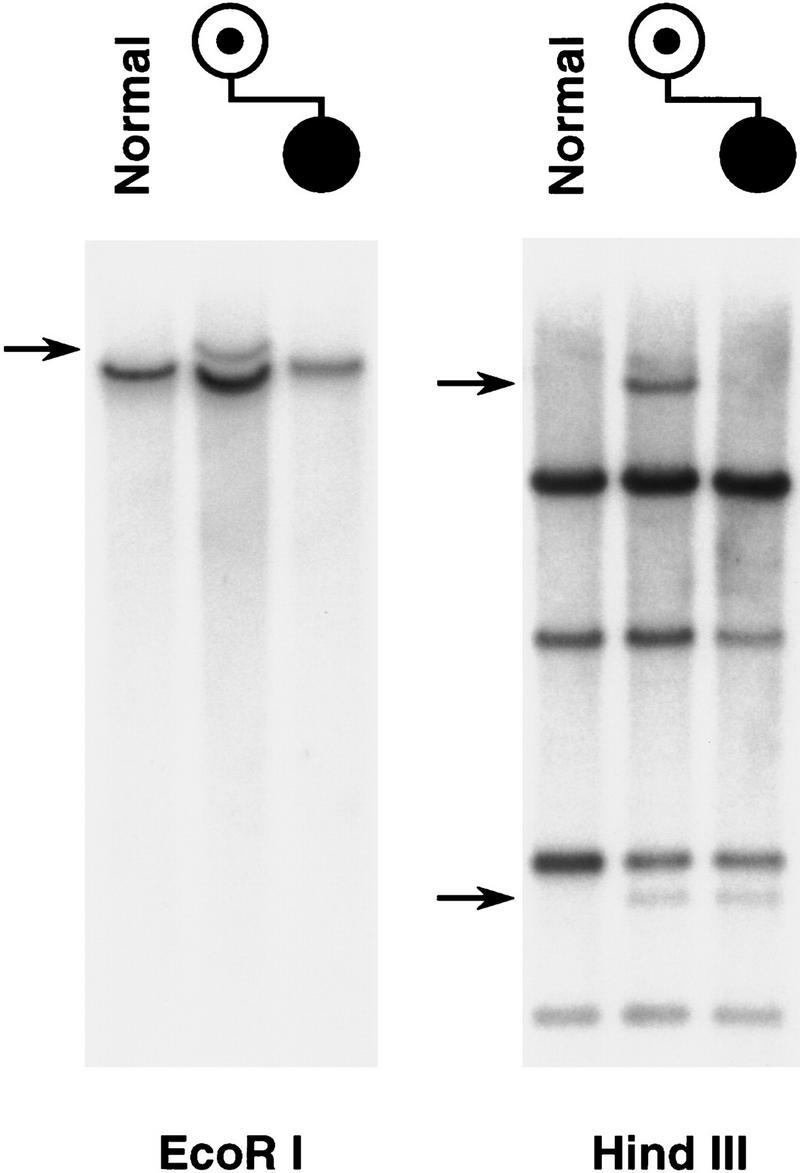
Detection of the t(14;15) cryptic translocation breakpoints. An 8-kb EcoRI fragment from cosmid 24 detects a single translocation junction fragment in EcoRI-digested DNA from the mother but not from the child, who is unbalanced. Both der(14) and der(15) breakpoints are seen in the HindIII panel, but the unbalanced, affected child has only the der(14) and not the der(15) chromosome, therefore displaying only one of the two breakpoint fragments seen in the mother.
Use of all cosmids and PACs from the region failed to detect any variant junction fragments on blots with the non-AS 15q deletion family. However, dosage analysis following multiple hybridizations provided a consistent pattern of reduced intensity with cosmids telomeric to c24 compared to normal controls, whereas cosmid 24 and all clones centromeric consistently demonstrated equal intensities on Southern blot analysis (data not shown). We conclude that the breakpoint in this case must lie in the gap between c24 and its telomeric neighbor c36. This would indicate that the non-AS 15q deletion breakpoint maps more telomeric than the t(14;15) breakpoint and that the latter provides a more narrowed definition of the AS candidate region. Based on chromosomal breakpoints, the AS candidate region is considered to extend from the Se breakpoint telomeric to the t(14;15) breakpoint covering a region of ∼250 kb.
Significant efforts have been made over the past few years to identify novel genes within the AS candidate region, which have included exon trapping, cDNA selection, and genomic sequencing. Exon trapping, in particular, has led to the identification of two novel transcripts, designated ASR-1 and ASR-2, for Angelman syndrome region (Fig. 1B; P. Fang, unpubl.). Despite intensive efforts, however, we have failed to identify any novel maternally expressed gene in this region, which has prompted reevaluation of UBE3A.
It became clear that the entire transcriptional unit of UBE3A was contained within the narrowed AS candidate region. Whereas previous data indicated a lack of maternal-specific expression in human fibroblasts and lymphoblasts, the possibility of tissue- or temporal-specific expression was not excluded. Given the continued status of UBE3A as a positional candidate for AS, we began further characterization of the gene to evaluate this possibility. Previously published Northern analysis demonstrated expression of a 5-kb transcript in primary foreskin keratinocytes (Huibregtse et al. 1993). Northern analysis in a more extended panel of tissues confirms the size of the transcript and shows low to moderate levels of expression in most tissues (Fig. 3). Although heart and skeletal muscle exhibit substantially stronger signals than other tissues, comparison to control gene [glyceraldehyde-3-phosphate dehydrogenase (GAP3D)] expression indicates that relative loading variation accounts for much of this difference. The transcriptional orientation of UBE3A became apparent, when it was found that the 3′ UTR mapped centromeric to more 5′ exons, indicating transcription from telomere toward centromere, opposite that for SNRPN (Buiting et al. 1993).
Figure 3.
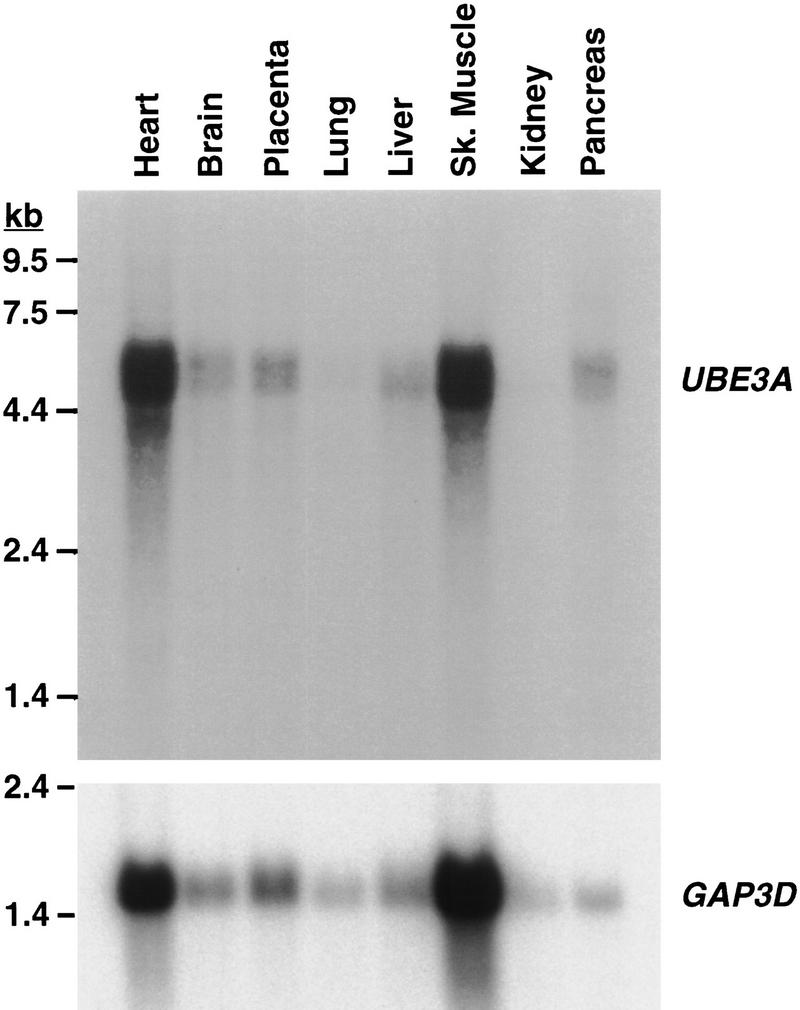
Northern analysis of UBE3A gene expression. Hybridization of UBE3A and control GAP3D cDNA probes to a Northern filter (Clontech) containing 2 μg poly(A)+ RNA per lane is shown.
To evaluate UBE3A further, the mouse homolog was isolated by screening a mouse brain cDNA library. One cDNA clone of ∼2.9 kb included the entire coding region, and this clone was completely sequenced (GenBank accession no. U82122). Comparison to the human gene revealed 93% identity at the nucleotide level and 94% at the peptide level (Fig. 4). A stretch of 9 amino acids in the human protein was not observed in the translation of this mouse sequence. This sequence in the human corresponds to the middle of exon 3 and is not adjacent to a splice boundary. Generally, a higher degree of conservation is observed toward the 3′ end than at the 5′ end.
Figure 4.

Comparison of peptide sequences for human and mouse UBE3A. The deduced peptide sequence of the mouse cDNA was compared to the published human peptide sequence, using the Wisconsin package; this analysis revealed 94% amino acid identity between human and mouse for UBE3A.
Progress toward characterization of the 5′ end of the human and mouse genes led to the detection of CpG islands in both species. This CpG island had been identified previously by PFGE mapping in the region (Sutcliffe et al. 1994). Sequencing of mouse and human cDNAs containing 5′ exons and genomic fragments containing the CpG island telomeric to the Se breakpoint identified a 5′ exon and detected a sequence in GenBank (L23501) reported previously (Woodage et al. 1994) following BLAST search. In the report by Woodage and colleagues, a long-range PFGE map of the PWS/AS region was presented along with a higher resolution map of the DNA surrounding this CpG island, including ∼3 kb of DNA sequence. This mapping information is quite useful, because it positions D15S10 within ∼10 kb of the CpG island. Using a probe within the CpG island, these investigators detected a transcript of ∼4.5 kb that was expressed with greatest abundance in skeletal muscle. Hybridization of a 1.5-kb PstI fragment containing the CpG island detects the identical transcript identified by a UBE3A cDNA clone (data not shown). We conclude that the CpG island in question contains a 5′ untranslated exon of UBE3A and that the transcript detected by Woodage et al. (1994) was UBE3A.
Given observations of differential methylation at the SNRPN 5′ CpG island and correlation of methylation with transcriptional silencing of the maternal allele, it was of interest to examine the methylation status of the UBE3A CpG island. Hybridization of the 1.5-kb genomic PstI fragment spanning the CpG island to Southern blots containing DNA isolated from peripheral leukocytes of deletion PWS or deletion AS patients and controls digested with XbaI in combination with methylation-sensitive enzymes such as NotI or SacII revealed no difference (data not shown), indicating the absence of preferential methylation of either homolog at this locus.
To address the possibility that UBE3A might be expressed in a maternal-specific manner in tissues other than fibroblasts or lymphoblasts, expression of the mouse gene was assessed in controls and animals with segmental, paternal UPD for the region known to contain the Ube3a gene (Cattanach et al. 1997). If Ube3a exhibited maternal-specific expression, one would expect absence of expression in the UPD mice compared with controls. Hybridization of cDNA probes corresponding to coding regions (Fig. 5A) and the 3′ UTR (Fig. 5B) to Northern blots containing total RNA from brain, liver, or testis failed to detect substantially reduced or absent expression in the UPD animals. Similar results were obtained by RT–PCR analysis (data not shown). The coding region probe detected three major transcripts, whereas hybridization with a 3′ UTR probe detected only the two larger transcripts of ∼4.5 and ∼10 kb. Given what is known about the size of the UBE3A/Ube3a coding region, the significance of the smaller ∼1-kb transcript detected by the coding region probe is unknown but could reflect cross-hybridization to a related gene; the difference between the two larger transcripts is also unknown but may reflect length variation in the 3′ UTR, presumably from alternate polyadenylation signals.
Figure 5.
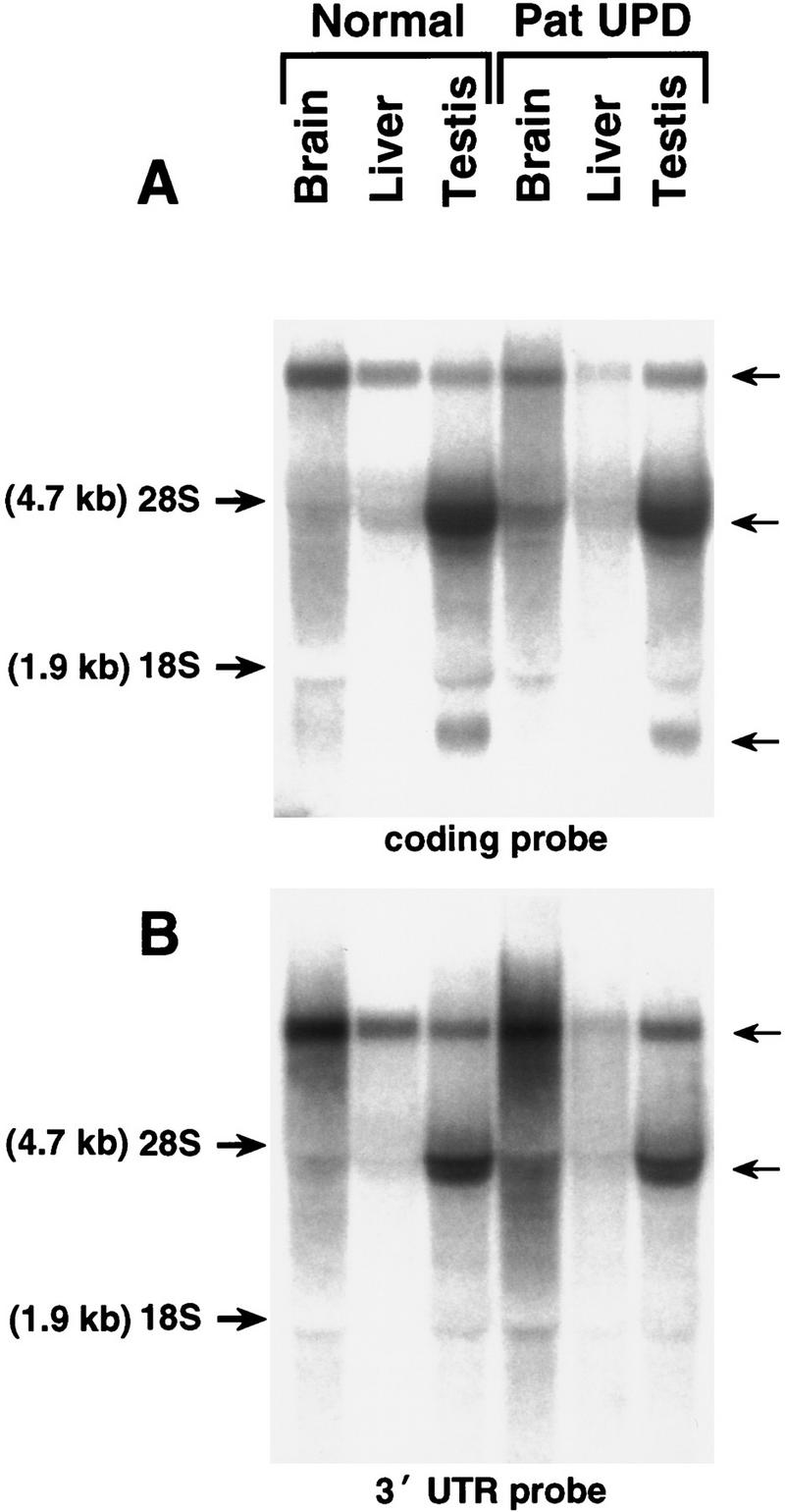
Northern analysis of Ube3a gene expression in mouse tissues from control and paternal UPD mice using probes corresponding to the coding region (A) and the 3′ UTR region (B). Comparison to the ethidium bromide-stained gel (not shown) reveals no significant difference between the control and UPD animals.
DISCUSSION
Detection of the t(14;15) breakpoint and localization of the 15q deletion breakpoint, which narrow the AS candidate region substantially, are significant for several reasons. A previous report had suggested that the deletion of a region surrounding STS marker D15S113 was involved in producing the AS phenotype in a probable intragenic mutant case (Buxtion et al. 1994). Data had indicated the loss of an informative maternally inherited allele at this locus. It was subsequently determined that this patient was not deleted, and two alleles were detected at D15S113 (S. Christian and D.H. Ledbetter, unpubl.). The t(14;15) translocation family provides the most compelling evidence for a more centromeric location for the putative AS gene. The presence of a maternally inherited der(15) in the unaffected sibling in this family indicates that this chromosome most likely contains the putative AS gene in the appropriate cis relationship with the IC, to protect against AS. This is supported further by the localization of the 15q non-AS deletion breakpoint just telomeric to the t(14;15) breakpoint. Together with the Se family AS deletion breakpoint, these two additional cases provide a well-defined AS candidate region of ∼250 kb that is represented completely by cosmid and PAC clones.
Physical mapping within this region indicates that the entire UBE3A transcriptional unit is positioned within the narrowed AS candidate region. Substantial efforts, including exon trapping, cDNA selection, and genomic sequencing techniques, were applied toward the identification of genes in this region, but no unique maternally expressed transcript has been defined yet. Failure to detect a novel maternally expressed gene and the continued status of UBE3A as a positional candidate for AS has prompted us to reevaluate this gene for involvement in AS.
Because of the concerns that UBE3A might be subjected to tissue-specific or developmental imprinting, which could not be assessed by expression analysis in fibroblasts or lymphoblasts, we chose to pursue studies in the mouse, which is a more amenable system for addressing these questions. By analyzing expression in various mouse tissues, particularly brain, the question of tissue-specific imprinting can be examined. The availability of mice with segmental, paternal UPD for the AS and PWS candidate regions to examine expression is quite useful in testing for maternal-specific expression; however, Ube3a failed to demonstrate reduced or absent expression in the UPD samples. It remains possible that imprinting takes place in a particular subset of cells within the brain or other tissue or at a particular time during development. To address the latter, a polymorphism could be identified for analysis of allele-specific expression in normal human fetal tissues or strain-specific sequence variations identified for analysis of F1 mice from reciprocal interspecific matings. The coat color markers used to identify the paternal duplication mice also affect eye color, which allows the identification of UPD mice from ∼12 days gestation.
The localization of UBE3A within a narrowed AS critical region and identification of de novo truncating mutations in AS cases expected to have intragenic mutations confirms that UBE3A is the AS gene (Kishino et al. 1997; Matsuura et al. 1997). Issues of how the biology of the UBE3A protein and the ubiquitin system may relate to the AS phenotype have been discussed elsewhere (Matsuura et al. 1997). Given the genetics of AS, there is a strong argument for the existence of a maternal-specific product from this locus. Although this putative imprinted product has not been identified as yet, ongoing studies of the UBE3A gene may provide insights into some possible mechanisms. We and others have independently identified additional upstream exons for UBE3A (Yamamato et al. 1997; T. Matsuura, J.S. Sutcliffe, and Y.-H. Jiang, unpubl.; GenBank accession no. X98034–98037). Observations by Yamamato and colleagues have suggested that multiple protein isoforms of UBE3A may be encoded by this locus and may be related to alternative splicing at the 5′ end of the gene. It is possible that in addition to tissue or developmental-specific imprinting, a maternal-specific product from this locus could involve a particular isoform of UBE3A, possibly transcribed from an imprinted promoter. Whereas most of the gene structure of UBE3A, including >10 exons, has been determined (Yamamoto et al. 1997; T. Matsuura, unpubl.; GenBank accession nos. X98021–X98030), further upstream exons remain to be characterized. The exon corresponding to the CpG island has been termed exon 0 and has been observed in several cDNA clones. Given the observations of hypermethylation of the CpG island on maternal chromosomes at the imprinted SNRPN locus, it is interesting to speculate whether the UBE3A CpG island might be differentially methylated in some tissue or at a particular developmental stage.
In considering the potential significance of UBE3A transcripts with alternative exon configurations at the 5′ end and whether this is related to the putative maternally expressed product of this locus, it will be interesting to identify the mutation in the familial AS case in which two affected siblings differed in alleles at D15S122 (Greger et al. 1994). This observation suggests a recombination event between the marker locus and the disease-causing mutation in this family and that the mutation should be telomeric to D15S122. Our studies indicate that this marker maps just centromeric to the CpG island and exon 0. Although the precise location of D15S122 relative to all of the 5′ exons is not fully defined, it is clear that this marker maps transcriptionally upstream of exon U1, indicating that the mutation must affect an exon or exons upstream of exon U1. It is possible that some AS patients might have mutations that affect only the putative imprinted, but not biallelically expressed, UBE3A transcript.
Although data presented here provide useful information concerning expression of Ube3a in adult mouse tissues, many questions remain concerning the imprinting of this locus and how mutations in this gene give rise to AS. Additionally, the physical mapping data provide a framework for more detailed physical and transcriptional characterization of the IC, PWS, and AS candidate regions.
METHODS
Screening of the PAC Library
The human PAC library was screened by two rounds of STS PCR screening of plate pools, followed by a final hybridization of a specific probe to a high-density array containing 384 clones representing four plates. STS primer sequences and PCR conditions for PW71, SNRPN, PAR-5, PAR-1, UBE3A, D15S10, and D15S122 have been described previously (Mutirangura et al. 1993; Wagstaff et al. 1993; Greger et al. 1994; Nakao et al. 1994; Sutcliffe et al. 1994). Primers for IPW were forward (5′-ACTCTTCTGGGAGTGAATGTTATC-3′, exon 2) and reverse (5′-GCATGTAGTTCACTTAATAGAGG-3′, exon 3), corresponding to nucleotide positions 989–1013 and 1576–1554 in the published cDNA sequence (Wevrick et al. 1994). PCR was carried out in 10 mm Tris (pH 8.3), 1.5 mm MgCl2, 50 mm KCl, 250 μm dNTPs, and 1 μm primers with cycling conditions of 94°C for 1 min, 58°C for 1 min, and 72°C for 1 min for 35 cycles. Primers for D15S1506 were forward (5′-TTGGCAACTCGATGTTCTTG-3′) and reverse (5′-TTTTATTTTGAACCAACCACCC-3′), with cycling conditions of 94°C for 0.5 min, 55°C for 0.5 min, and 72°C for 0.5 min for 35 cycles. Southern blot hybridization was carried out in 0.125 m NaPO4 (pH 7.0), 0.25 m NaCl, 1 mm EDTA, 10% polyethylene glycol (PEG-8000), 7% SDS, and 1% BSA at 65°C overnight, followed by washing to a final stringency of 0.2× SSC/0.1% SDS at 65°C and autoradiography at −80°C.
Analysis of PAC Clones
Multiple PAC clones were obtained for each locus, and DNA was isolated and purified after banding on a CsCl2 gradient by standard methods. PAC DNA samples were fingerprinted by EcoRI digestion and separation on 0.7% agarose gels in Tris-acetate buffer. STS content was determined by PCR amplification using primers for each of the screening loci as described above, in addition to other markers in the region, and confirmed by Southern blot hybridization to specific probes. Cosmid relationships were determined by hybridization of whole-cosmid inserts to Southern blot filters containing EcoRI-restricted PAC DNA using repeat suppression (see below).
Southern Blotting with PAC, Cosmid, and Individual Fragment Probes
Genomic DNAs from normal individuals, from the mother and affected daughter in the t(14;15) case, and from the mother and affected son in the 15q deletion case were digested with EcoRI or HindIII, separated on 0.7%–0.8% agarose gels, transferred by standard methods to Hybond N+ nylon membrane (Amersham, Arlington Heights, IL), and hybridized as described above to specific probes. For methylation analysis, genomic DNAs from normal, PWS deletion, and AS deletion patients were digested with XbaI and NotI and blotted as described above. Repeat suppression was used for all genomic probes and involved incubation of the denatured 32P-labeled probe in a solution containing ∼6 mg/ml of sheared, denatured, total human placental DNA and 5× SSC at 65°C for 30–60 min. Southern blots were washed and exposed as described above.
cDNA Library Screening
A mouse neonatal brain λ cDNA library (Stratagene cat. no. 937301; La Jolla, CA) was plated, and replica lifts were prepared by standard methods. Approximately 5 × 105 PFU were screened by hybridization with human cDNA clones containing different segments of the coding region. Positive phage were purified, and insert-containing plasmids were isolated after in vivo excision following manufacturer’s protocols.
DNA Sequencing
cDNA clones were subcloned and sequenced using an ABI (Applied Biosystems, Foster City, CA) model 377 automated DNA sequencer using dye terminator chemistry and manufacturer’s protocols. Double-stranded sequence was assembled using the software applications Autoassembler (ABI), and the mouse and human UBE3A proteins were compared using the Wisconsin package (Genetics Computer Group; Madison, WI).
Northern Analysis
Total RNA was isolated from various tissues from UPD mice and littermate controls following homogenization in Ultraspec II (Tel Test; Houston, TX). Total RNA was resolved on a 1.2% agarose gel in 10 mm NaPO4 (pH 6.8) buffer following glyoxal/DMSO denaturation using standard methods. RNA was visualized by ethidium bromide staining and transferred to Hybond N+ membrane. Hybridization and washing conditions were identical to that used for Southern hybridization.
RT–PCR
Single-stranded cDNA was synthesized from total RNA using Superscript II reverse transcriptase (GIBCO-BRL; Gaithersberg, MD) primed with oligo(dT) (Promega, Madison, WI). Primers for PCR amplification of Ube3a were forward (5′-ATATTCCGGAAGTAAAAGGACATTA-3′) and reverse (5′-AACAGGCACAGACAGAGCAC-3′). PCR conditions were the same as above with the exception that annealing was at 58°C.
Acknowledgments
We thank M. Nakao, J. Beuten, X.-Y. Wang, T.-F. Tsai, B. Durtschi, and S. Aradhya for contributions to positional cloning efforts; D. Driscoll and L. Burke, and R. Michaelis for generously providing the t(14;15) and non-AS 15q deletion cell lines, respectively; and P. Howley for providing a preprint of a submitted manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL abeaudet@bcm.tmc.edu; FAX (713) 798-7773.
REFERENCES
- Buiting K, Dittrich B, Gross S, Greger V, Lalande M, Robinson W, Mutirangura A, Ledbetter DH, Horsthemke B. Molecular definition of the Prader-Willi syndrome chromosome region and orientation of the SNRPN gene. Hum Mol Genet. 1993;2:1991–1994. doi: 10.1093/hmg/2.12.1991. [DOI] [PubMed] [Google Scholar]
- Buiting K, Saitoh S, Gross S, Dittrich B, Schwartz S, Nicholls RD, Horsthemke B. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nature Genet. 1995;9:395–400. doi: 10.1038/ng0495-395. [DOI] [PubMed] [Google Scholar]
- Burke LW, Wiley JE, Glenn CC, Driscoll DJ, Loud KM, Smith AJ, Kushnick T. Familial cryptic translocation resulting in Angelman syndrome: Implications for imprinting or location of the Angelman gene? Am J Hum Genet. 1996;58:777–784. [PMC free article] [PubMed] [Google Scholar]
- Buxion JL, Chan CT, Gilbert H, Clayton-Smith J, Burn J, Pembrey M, Malcolm S. Angelman syndrome associated with a maternal 15q11-13 deletion of less than 200 kb. Hum Mol Genet. 1994;3:1409–1413. doi: 10.1093/hmg/3.8.1409. [DOI] [PubMed] [Google Scholar]
- Cassidy SB, Beaudet AL, Knoll JHM, Ledbetter DH, Nicholls RD, Schwartz S, Butler MG, Watson M. Diagnostic testing for Prader-Willi and Angelman syndromes; Report of the ASHG/ACMG Test and Technology Transfer Committee. Am J Hum Genet. 1996;58:1085–1088. [PMC free article] [PubMed] [Google Scholar]
- Cattanach, B.M., J.A. Barr, C.V. Beechey, J. Martin, J. Noebels, and J. Jones. 1997. A candidate model for Angelman syndrome in the mouse. Mamm. Genome (in press). [DOI] [PubMed]
- Dittrich B, Buiting K, Gross S, Horsthemke B. Characterization of a methylation imprint in the Prader-Willi syndrome chromosome region. Hum Mol Genet. 1993;2:1995–1999. doi: 10.1093/hmg/2.12.1995. [DOI] [PubMed] [Google Scholar]
- Dittrich B, Buiting K, Rickard S, Buxton J, Saitoh S, Nicholls RD, Poustka A, Winterpacht A, Zabel B, Horsthemke B. Imprint switching on human chromosome 15 may involve alternative transcripts of the SNRPN gene. Nature Genet. 1996;14:163–170. doi: 10.1038/ng1096-163. [DOI] [PubMed] [Google Scholar]
- Glenn CC, Nicholls RD, Robinson WP, Saitoh S, Niikawa N, Schinzel A, Horsthemke B, Driscoll DJ. Modification of 15q11-q13 DNA methylation imprints in unique Angelman and Prader-Willi patients. Hum Mol Genet. 1993a;2:1377–1382. doi: 10.1093/hmg/2.9.1377. [DOI] [PubMed] [Google Scholar]
- Glenn CC, Porter KA, Jong MT, Nicholls RD, Driscoll DJ. Functional imprinting and epigenetic modification of the human SNRPN gene. Hum Mol Genet. 1993b;2:2001–2005. doi: 10.1093/hmg/2.12.2001. [DOI] [PubMed] [Google Scholar]
- Glenn CC, Saitoh S, Jong MT, Filbrandt MM, Surti U, Driscoll DJ, Nicholls RD. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am J Hum Genet. 1996;58:335–346. [PMC free article] [PubMed] [Google Scholar]
- Greger V, Woolf E, Lalande M. Cloning of the breakpoints of a submicroscopic deletion in an Angelman syndrome patient. Hum Mol Genet. 1993;2:921–924. doi: 10.1093/hmg/2.7.921. [DOI] [PubMed] [Google Scholar]
- Greger V, Reis A, Lalande M. The critical region for Angelman syndrome lies between D15S122 and D15S113. Am J Med Genet. 1994;53:396–398. doi: 10.1002/ajmg.1320530425. [DOI] [PubMed] [Google Scholar]
- Hamabe J, Kuroki Y, Imaizumi K, Sugimoto T, Fukushima Y, Yamaguchi A, Izumikawa Y, Niikawa N. DNA deletion and its parental origin in Angelman syndrome patients. Am J Med Genet. 1991;41:64–68. doi: 10.1002/ajmg.1320410117. [DOI] [PubMed] [Google Scholar]
- Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY, Greenberg F. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91:398–402. [PMC free article] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Howley PM. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interation of the human papillomavirus E6 oncoprotein with p53. Mol Cell Biol. 1993;13:775–784. doi: 10.1128/mcb.13.2.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nature Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- Ledbetter DH, Ballabio A. Molecular cytogenetics of contiguous gene syndromes: Mechanisms and consequences of gene dosage imbalance. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York, NY: McGraw Hill; 1995. pp. 811–839. [Google Scholar]
- Matsuura T, Sutcliffe JS, Fang P, Galjaard R-J, Jiang Y-h, Benton C, Rommens JM, Beaudet AL. Mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nature Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- Meijers-Heijboer EJ, Sandkuijl LA, Brunner HG, Smeets HJ, Hoogeboom AJ, Deelen WH, van Hemel JO, Nelen MR, Smeets DF, Niermeijer MF, Halley DJJ. Linkage analysis with chromosome 15q11-13 markers shows genomic imprinting in familial Angelman syndrome. J Med Genet. 1992;29:853–857. doi: 10.1136/jmg.29.12.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis RC, Skinner SA, Lethco BA, Simensen RJ, Donlon TA, Tarleton J, Phelan MC. Deletion involving D15S113 in a mother and son without Angelman syndrome: refinement of the Angelman syndrome critical deletion region. Am J Med Genet. 1995;55:120–126. doi: 10.1002/ajmg.1320550131. [DOI] [PubMed] [Google Scholar]
- Mutirangura A, Jayakumar A, Sutcliffe JS, Nakao M, McKinney MJ, Buiting K, Horsthemke B, Beaudet AL, Chinault AC, Ledbetter DH. A complete YAC contig of the Prader-Willi/Angelman chromosome region (15q11-13) and refined localiztion of the SNRPN gene. Genomics. 1993;18:546–552. doi: 10.1016/s0888-7543(11)80011-x. [DOI] [PubMed] [Google Scholar]
- Nakao M, Sutcliffe JS, Durtschi B, Mutirangura A, Ledbetter DH, Beaudet AL. Imprinting analysis of three genes in the Prader-Willi/Angelman region: SNRPN, E6-associated protein, and PAR-2 (D15S225E) Hum Mol Genet. 1994;3:309–315. doi: 10.1093/hmg/3.2.309. [DOI] [PubMed] [Google Scholar]
- Nicholls RD. Genomic imprinting and candidate genes in the Prader-Willi and Angelman syndromes. Curr Opin Genet Dev. 1993;3:445–456. doi: 10.1016/0959-437x(93)90119-a. [DOI] [PubMed] [Google Scholar]
- Ozcelik T, Leff S, Robinson W, Donlon T, Lalande M, Sanjines E, Schinzel A, Francke U. Small nuclear ribonucleoprotien polypeptide N (SNRPN), an expressed gene in the Prader-Willi syndrome critical region. Nature Genet. 1992;2:265–269. doi: 10.1038/ng1292-265. [DOI] [PubMed] [Google Scholar]
- Reed ML, Leff SE. Maternal imprinting of human SNRPN, a gene deleted in Prader-Willi syndrome. Nature Genet. 1994;6:163–167. doi: 10.1038/ng0294-163. [DOI] [PubMed] [Google Scholar]
- Reis A, Dittrich B, Greger V, Buiting K, Lalande M, Gillessen-Kaesbach G, Anvret M, Horsthemke B. Imprinting mutations suggested by abnormal DNA methylation patterns in familial Angelman and Prader-Willi syndrome. Am J Hum Genet. 1994;54:741–747. [PMC free article] [PubMed] [Google Scholar]
- Saitoh S, Kubota T, Ohta T, Jinno Y, Niikawa N, Sugimoto T, Wagstaff J, Lalande M. Familial Angelman syndrome caused by imprinted submicroscopic deletion encompassing GABAA receptor beta 3-subunit gene. Lancet. 1992;339:366–367. doi: 10.1016/0140-6736(92)91686-3. [DOI] [PubMed] [Google Scholar]
- Sutcliffe JS, Nakao M, Christian S, Orstavik KH, Tommerup N, Ledbetter DH, Beaudet AL. Deletions of a differently methylated CpD island at the SNRPN gene define a putative imprinting control region. Nature Genet. 1994;8:52–58. doi: 10.1038/ng0994-52. [DOI] [PubMed] [Google Scholar]
- Wagstaff J, Shugart YY, Lalande M. Linkage analysis in familial Angelman syndrome. Am J Hum Genet. 1993;53:105–112. [PMC free article] [PubMed] [Google Scholar]
- Wevrick R, Kerns JA, Francke U. Identification of a novel paternally expressed gene in the Prader-Willi syndrome region. Hum Mol Genet. 1994;3:1877–1882. doi: 10.1093/hmg/3.10.1877. [DOI] [PubMed] [Google Scholar]
- Williams CA, Angelman H, Clayton-Smith J, Driscoll DJ, Hendrickson JE, Knoll JH, Magenis RE, Schinzel A, Wagstaff J, Whidden EM, et al. Angelman syndrome: Consensus for diagnostic criteria. Angelman Syndrome Foundation. Am J Med Genet. 1995;56:237–238. doi: 10.1002/ajmg.1320560224. [DOI] [PubMed] [Google Scholar]
- Woodage T, Lindeman R, Deng ZM, Fimmel A, Smith A, Trent RJ. Physical mapping studies at D15S10: Implications for candidate gene identification in the Angelman syndrome/Prader-Willi syndrome chromosome region of 15q11-q13. Genomics. 1994;19:170–172. doi: 10.1006/geno.1994.1031. [DOI] [PubMed] [Google Scholar]
- Yamamoto, Y., J.M. Huibregtse, and P.M. Howley. 1997. The human E6-AP gene (UBE3A) encodes three potential protein isoforms generated by differential splicing. Genomics (in press). [DOI] [PubMed]