

Abstract
Cardiovascular diseases are among the major targets for gene therapy. Initially, clinical experiments of gene transfer of vascular endothelial growth factor (VEGF) improved vascularization and prevented the amputation in patients with critical leg ischemia. However, the majority of trials did not provide conclusive results and therefore further preclinical studies are required. Importantly, data indicate the necessity of regulated expression of angiogenic factors, particularly VEGF, to obtain the therapeutic effect. It is also suggested that the combined delivery of two or more genes may improve the formation of mature vasculature and therefore may be more effective in the amelioration of ischemia. Moreover, experimental approaches in animal models displayed the promise of gene transfer modulating the inflammatory processes and oxidant status of the cells. Particularly, the concept of preemptive gene therapy has been tested, and recent studies have demonstrated that overexpression of heme oxygenase-1 or extracellular superoxide dismutase can prevent heart injury by myocardial infarction induced several weeks after gene instillation. The combination of a preemptive strategy with regulated gene expression, using the vectors in which the therapeutic transgene is driven by exogenously or endogenously controllable promoter, offers another modality. However, we hypothesize that regulatable gene therapy, dependent on the activity of endogenous factors, might be prone to limitations owing to the potential disturbance in the expression of endogenous genes. Here, we demonstrated some indications of these drawbacks. Therefore, the final acceptance of these promising strategies for clinical trials requires careful validation in animal experiments.
Index Entries: Gene therapy, angiogenesis, vascular endothelial growth factor, heme oxygenase, hypoxia inducible factor-1, nitric-oxide synthase, superoxide dismutase
INTRODUCTION
The progress in gene therapy is dependent on the understanding of the molecular basis of diseases as well as on the development of efficient tools allowing delivery of therapeutic nucleic acid. The vast amount of knowledge, which has been accumulated in the last few years, offers countless possibilities of different therapeutic strategies even in the same type of disease. It is therefore impossible in a short review to discuss all crucial aspects of gene therapy, including even the newest strategies for a given type of disease. Therefore, here we focus only on some examples of recent approaches in the development of delivery tools as well as on the exploitation of new therapeutic genes with potential application in cardiovascular diseases.
Delivery of therapeutic nucleic acids is accomplished by vectors. They belong to two categories: nonviral and viral (reviewed in ref. 1). Nonviral vehicles comprise plasmids or short-strand nucleic acids (anti-sense, DNA decoys, and small interfering RNA), which are delivered to the cells either in a so-called naked form or with the help of various chemical or physical methods. The advantages of nonviral vehicles are the simplicity of their construction, relatively easy and cheap method of production in the large quantities necessary for in vivo gene therapy, and overall safety compared with viral vectors. The important limitations are the low efficiency of transfection and transient way of expression of the introduced genes. Thus, delivery of plasmid vectors to the vascular wall by instillation with intravascular catheters results in transfection efficiency that is usually considerably below 1% (2). There are, however, some exceptions to these general features. Naked DNA can transfect skeletal muscles or the heart with the efficiency reaching several percent of cells in the region of the injection, and the level of expression of introduced genes is relatively high for several weeks or even months (3–5). This response occurs despite the episomal localization of the plasmid and is probably because of the structure of the muscles, which possess a well-developed system of endoplasmic reticulum. Therefore this approach, particularly when applied to genes that code for secretory proteins, creates an opportunity for the simple, cheap, and effective gene therapy to be applied.
SELECTION OF DELIVERY VECTORS FOR GENE THERAPY
Viral Vectors
Viral vectors can be integrating and nonintegrating (reviewed in refs. 1, 6). Retroviral vectors and, to a certain extent, adeno-associated vectors (AAVs) are integrating viral vectors. Two groups of retroviral vectors are being used. The first one is derived from animal oncoretroviruses, such as the murine Moloney leukemia virus. These vehicles were applied in the first trials of gene therapy and still constitute the larger proportions of vectors used in clinical trials. It is a consequence of a stable integration of retroviral vectors into the genome of transduced cells that allows for the permanent expression of a transgene. The limitation of oncoretroviral vectors is their inability to transduce nondividing cells. Nevertheless, they can efficiently deliver genes to proliferating progenitor cells; in addition, this type of modification has enabled the treatment of severe combined immunodeficiency (SCID) in children lacking the gene for common γc chain of cytokine receptor (7–9). Lentiviral vectors can transduce nondividing cells. Therefore, those carriers found application in delivery to hematopoietic stem cells and neural cells (10), but they also may be efficient in the transduction of tumor cells (11) and are particularly considered for gene therapy in relation to HIV infections (12).
However, both categories of retroviral vectors create the risk of the insertional mutagenesis owing to random integration into the cellular genome. Although such events have been rarely observed in experimental trials, three cases of leukemia in SCID patients treated with gene therapy highlight the important concerns of the safety of this strategy (7,13). Additionally, the risk of lentiviral-induced oncogenesis has recently been highlighted (14) in relation to potential pro-oncogenic effect of woodchuck post-transcriptional regulatory element (15), which is widely used in many retroviral, adenoviral and AAV vectors.
Adenoviral Vectors
AAV vectors are created from small, nonpathogenic single-stranded DNA viruses, which do not induce any human diseases. Eight serotypes of AAV have been discovered so far, and AAV-2 is the prototype of the AAV vectors that have been the most commonly used in gene therapy. Wild-type AAVs specifically integrate into the clearly defined site on chromosome 19 (16). This integration requires Rep proteins encoded by one of two genes of wild-type AAV. The removal of both rep and cap genes is, however, necessary during AAV vectors synthesis, and, as a consequence, the AAV vectors lost the propensity for the specific and efficient integration. Nevertheless, recombinant AAV vectors can randomly integrate in low proportions into the chromosomal DNA, although the majority of AAV vectors remain in episomal forms. The nonpathogenic nature of AAV decreases the risk of an inflammatory response (17). Moreover, AAV-2 can transduce the majority of quiescent cells with a high efficacy, and long-term expression can be achieved (17). However, some cells are refractory to transduction with AAV and endothelial cells are among them, with efficacy as low as 2% in case of the AAV-2 serotype (18,19), whereas efficacy can be as high as 20–40% for vascular smooth muscle cells (17,20,21). Modifications of the viral capsids (22) as well as the exploitation of the different tropism of other AAV serotypes (23) offer new possibilities to match AAV to vascular cells.
Limitation of Viral Vectors
Inflammatory response is a major concern in the application of adenoviral vectors. They are very efficient in transducing various cell types irrespective of the stage of their cell cycle; and in endothelial cells, a transduction efficiency up to 75% or more in vivo has been reported (17). However, the first generation of adenoviral vectors, which have been the most commonly used besides retroviruses in clinical trials (24), still possess a significant proportion of adenoviral genes. Therefore, in a short time after transduction, the immune response develops against viral proteins. This response may limit the time and level of expression of therapeutic genes. Nevertheless, it seems that these vectors, when carefully delivered, still hold promise of being a potent therapeutic tool, as is suggested by progress in the application of the p53 gene in patients with head and neck cancer (25). Moreover, the construction of the new class of adenoviral vectors, or so-called helper-dependent vectors, creates new, exciting possibilities (26).
CLINICAL TRIALS IN CARDIOVASCULAR DISEASES
Despite the progress in pharmacotherapy, the efficient treatment of the advanced stages of cardiovascular diseases is still lacking. Therefore, great expectations all have been attributed to gene therapy. Particularly, the cloning of vascular endothelial growth factor (VEGF), so the major angiogenic mediator, has raised the hopes for they its application for the stimulation of blood vessels formation in coronary heart diseases or peripheral tissue ischemia. However, despite the initial promising results of small clinical trials in which the delivery of plasmid harboring VEGF cDNA was used for the stimulation of angiogenesis in ischemic legs (27–29), the efficacy of that strategy has not been convincingly confirmed in other experiments (reviewed in ref. 30).
The other clinically tested approach of gene therapy uses the application of DNA decoys for the E2F transcription factor. This strategy relies on the delivery of short, about 20-bp, double-stranded oligonucleotides, containing the sequence to which this transcription factor binds in the promoter of genes, particularly those involved in cell proliferation. In such a way, the expression of several genes, e.g., c-myc, c-myb, or PCNA is prevented. The E2F-based DNA decoy strategy has demonstrated its feasibility in a PREVENT trial and resulted in the attenuation of development of restenosis in arterio-venous bypasses, because of the inhibition of the proliferation of vascular smooth muscle cells (31). Ex vivo, pressure-mediated transfection efficiency of E2F decoys reached approx 90% and resulted in >70% inhibition of PCNA and c-myc expression and bromodeoxyuridine-labeling index (31). However, the approach still requires validation in larger randomized clinical trials. Thus, the progress in cardiovascular gene therapy is slower than expected. Although some clinical end points have been positive in several trials, these did not produce any clear meaningful clinical benefits for the patients (recently reviewed in refs. 30, 32) The reasons are various and include the technical obstacles concerning the delivery of vectors, transfection efficiency, and biological effect of produced factors as well as the selection of patients (reviewed in ref. 30). Therefore, besides solving those problems, it also seems reasonable to look for the other strategies to ameliorate ischemia and to prevent the consequences of myocardial infarction. Some such approaches are discussed in the next section.
RECENT DEVELOPMENTS IN GENE THERAPY FOR CARDIOVASCULAR DISEASES
Modification of Progenitor Cells
Modification of progenitor cells by retroviral vector-mediated delivery of therapeutic gene is being tested for cardiovascular gene therapy. In the study by Iwaguro et al. (33), ex vivo-expanded endothelial progenitor cells (EPCs) were transduced with VEGF before transplantation into an ischemic hindlimb muscle. Such modified EPCs demonstrated a higher capacity to augment neovascularization. In another study, EPCs were transduced with an active subunit of telomerase reverse transcriptase that also enhanced their migratory and proliferative capacity (34).
Recently, Dzau and coworkers have modified rabbit endothelial progenitor cells with retroviral vectors harboring the reporter gene (green fluorescent protein) or with one of two potentially therapeutic genes, namely, endothelial nitric-oxide synthase (eNOS) or heme oxygenase (HO)-1 (35). Such modified cells have then been delivered to the rabbit artery, previously denuded by balloon angioplasty, imitating the procedure performed in atherosclerotic vessels. The instillation of progenitor cells per se enhanced the process of reendothelialization. Moreover, the recovery was faster in animals treated with EPC transduced with the eNOS gene. This study demonstrates the feasibility of the combination of gene with stem cell therapy in cardiovascular diseases and highlights again the important role of nitric oxide (NO) in the endothelial function. Surprisingly, no additional therapeutic effect of HO-1 overexpression in EPSs has been observed in contrast to previous studies showing significant attenuation of neointimal thickening owing to HO-1 overexpression in the damaged vessel wall (36). It is surmised that the beneficial outcome of HO-1 requires a higher level of its expression to reach the sufficiently high production of HO-1 products, carbon monoxide (CO) and/or bilirubin (35).
Modification of EPCs may find application in patients with cardiovascular diseases, particularly in situations when the propensity of their endothelial (and hence also progenitor cells) to generate NO is impaired because of the various underlying factors (37). Such transduced EPCs also may offer better prospects for the seeding of vascular prosthetic grafts, used in patients lacking suitable vessels for a bypass. Also, transduced EPC may be used to cover the stents applied in angioplasty procedure. The limitation of such an approach is, however, the small amount of progenitor cells available for modification from a potential patient.
Regulated Gene Expression Therapy
The level of therapeutic gene expression determines the efficiency of gene therapy. It has been demonstrated that constitutive promoters, the most commonly used in both experimental and clinical gene therapy, can provide sufficient level of expression of genes used in cardiovascular gene therapy. However, it is suggested that the tight regulation of expression of therapeutic genes at least in the case of some genes, might be beneficial (38). Such a strategy would allow for adjusting the production of therapeutic protein to the needs of the modified (or targeted) organ. Regulated gene expression has been particularly considered for the therapeutic application of VEGF. This potent angiogenic factor has been tested for the stimulation of blood vessel formation in ischemic heart and skeletal muscles. Interestingly, some experiments demonstrated that high and unregulated expression of VEGF may lead to harmful effects. Studies indicate that the high overexpression of VEGF, the strong angiogenic but also proinflammatory agent with a potent vascular permeability activity, may result in serious side effects, such as hypotension (39,40) or hemangioma formation (41–43). Therefore, it can be suggested that the application of VEGF in a mode of regulated gene transfer would be a better approach.
Hypoxia Induced Gene Expression Therapy
The expression of genes in hypoxic conditions, including VEGF, is dependent on the activation of a hypoxia inducible factor (HIF)-1 (reviewed in refs. 44, 45). This transcription factor works in the form of a dimer, consisting of HIF-1α and HIF-1β subunits. In normoxia, both proteins are constitutively generated in cells, however, HIF-1α is also permanently degraded, which prevents the dimer formation. Degradation of HIF-1α is dependent on the action of the oxygen-dependent prolyl hydroxylase, which by adding hydroxyl groups to specific proline residues (P402 and P564) present in so-called oxygen-dependent degradation (ODD) domain, creates the signal for von-Hippel-Lindau (VHL) ligase. VHL ubiquitylates HIF-1α, which is targeted for proteasome degradation. This mechanism degradation is abolished in hypoxia, which inhibits prolyl hydroxylase activity, resulting in stabilization of HIF-1α, followed by the dimer formation and its binding to a hypoxia responsive element (HRE), present in the regulatory regions of many hypoxia-dependent genes, e.g., VEGF. As a result, the expression of such genes is enhanced in hypoxic conditions. Hypoxia-regulated gene therapy is an attractive approach that can be applied to specific overexpression of various genes exclusively in the ischemic tissues. The following interesting experimental strategies, applying the so-called vigilant vectors, have been tested (46–49); for reviews of other strategies, see refs. 30, 50 (Fig. 1).
Fig. 1.
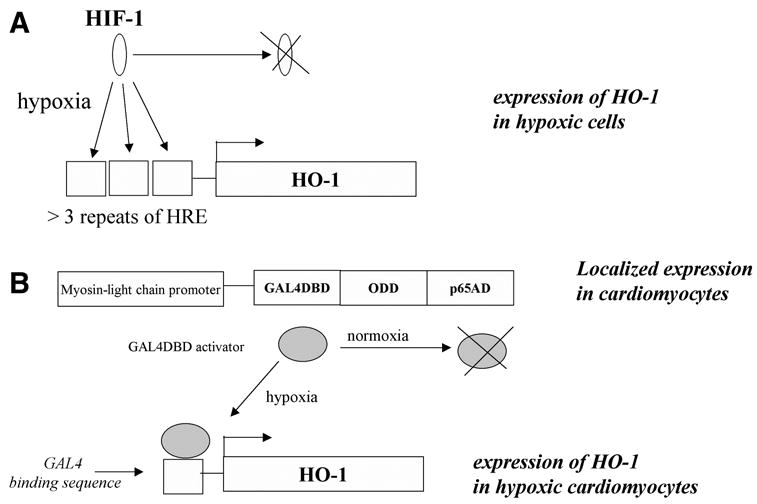
Strategies for hypoxia-dependent regulation of gene expression in gene therapy. (A) Expression of a therapeutic gene, e.g., HO-1, is driven by several repeats of the hypoxia-responsive element. Activation is exerted when HIF-1α produced constitutively in the cells, is stabilized by hypoxia and forms an active dimmer with HIF-1β(B) Expression of a trans-gene is driven by a complex transcription factor, containing the ODD domain of HIF-1α. The amount of complex increases in hypoxia because of stabilization of ODD.
In the first strategy, the DNA sequence, harboring several HRE sequences, is linked to a transgene (Fig. 1A). The classical HIF-binding sequence (HBS), present in the HRE part of the VEGF promoter, consists of six nucleotides (TACGTG). Three to nine HRE sequences containing HBS are essential to achieve the sufficient level of expression during hypoxia (51–53). Because HIF-1α is unstable in the presence of oxygen, the trans-gene should not be expressed in normoxic conditions. Once the oxygen tension decreases, HIF-1α is stabilized, and the HIF-1 transcription factor binds to HRE and induces the expression of therapeutic transgene.
The second tested approach combines hypoxia-dependent gene expression with the organ-specific over-expression of HIF-1α, or its ODD domain (reviewed in refs. 46, 47) (Fig. 1B). To this end, two vectors have to be constructed. The first one harbors the ODD coding sequences under the control of the cell-specific promoter, e.g., myosin light-chain promoter, which drives the expression only in cardiomyocytes. The ODD coding sequence is bound to the fragment encoding the yeast GAL4DBD activation protein. The second vector consists of a therapeutic gene, e.g., HO-1, driven by the GAL4 binding sequence. Both vectors are introduced into the ischemic heart. The expression of ODD occurs only in the cardiomyocytes as a result of the specificity of the myosin light-chain promoter. Hypoxia stabilizes the ODD and results in stabilization of the whole activation factor (GAL4DBD-ODD-p65AD), which by binding to the Gal4 binding sequence, induces locally the expression of a therapeutic gene in the ischemic heart (Fig. 1B).
Another approach to achieve the required level of gene expression is based on promoter sequences regulated by exogenously applied stimuli. The most commonly used are tetracycline-dependent promoters (54), but artificially engineered transcription factors have recently been used (55, 56). To this end, an interesting opportunity is offered by rapamycin. First, it can act as an external regulator of the expression of genes that are driven by the artificial rapamycin-dependent promoter (57). Second, the drug also is used to cover vascular stents as it attenuates restenosis by inhibition of proliferation of vascular smooth muscle cells (58). Third, the therapeutic potential of rapamycin can be exerted by its direct effect on the expression of other genes in vascular cells. Indeed, rapamycin has been proven to increase the expression of HO-1 (59).
Mechanisms of the rapamycin-dependent gene expression rely on the employment of its intracellular targets. Rapamycin binds to the FK-binding protein 12 (FKB12) forming a complex that binds to the mammalian targets of rapamycin (mTOR), inhibiting both DNA and protein synthesis. This effect is responsible for the inhibition of vascular smooth muscle cell proliferation and also is probably involved in the induction of the HO-1 expression (59). The FKB12 protein also is used in the system of rapamycin-dependent artificial promoters. It is the complex of the bipartite transcription factor consisting of two chimeric human peptides. The first one is composed of a domain ZFHD1 complexed to FKB12, whereas the second one is the truncated protein FRAP connected with the nuclear factor-κB p65 protein. In the presence of rapamycin, the ZFHD1-FKB protein binds to the FRAP-p65 chimera to form a complex that activates ZFHD1 sites in a promoter driving the therapeutic gene expression (reviewed in ref. 54).
NEW THERAPEUTIC OPTIONS FOR CARDIOVASCULAR GENE THERAPY
AAV vectors harboring the VEGF cDNA under the control of the HRE sequence have been constructed and delivered into the ischemic rat myocardium (52,53). The expression of VEGF was sufficient to induce the formation of new blood vessels. Thus, the hypoxia-regulated production of VEGF may prevent the negative aspects of high expression of this potent angiogenic factor. Moreover, the combination of HRE-dependent regulation of VEGF expression and the cardiac-specific over-expression of HIF-1 may offer additional benefits, limiting the transgene expression only to the ischemic heart (52). Interestingly, it also is possible that not only the total amount of VEGF should be regulated but also its local concentration. Accordingly, in a recent article, Ozawa and coworkers demonstrated that the formation of hemangiomatous blood vessels was strictly dependent on the amount of locally released VEGF (41). In this study, mice myoblasts were stably transduced with retroviral vector harboring VEGF. Several clones of VEGF-producing cells have been selected based on the level of production of VEGF. When such cells were implanted into the muscles of adult mice, the formation of normal or tumor-type blood vessels was dependent on the VEGF dosage. Clonal myoblasts that secrete low-to-medium levels of VEGF (up to about 70 ng/106 cells/d) induced growth of stable, pericyte-coated capillaries of uniform size that were not porous. In contrast, the clones producing high amounts of VEGF induced hemangiomas. Surprisingly, when only a few myoblasts generating very high amounts of VEGF were mixed with nontrandsuced cells, which did not produce VEGF, the formation of hemangioma still occurred after implantation of such a mixture into the muscle. Therefore, it is suggested that it was not the total amount of VEGF that was produced by the cells in a certain location but the local concentration of VEGF that determined the formation of either normal or tumorigenic blood vessels. If so, even long-term, local continuous delivery of VEGF, when maintained below a threshold microenvironmental level, may lead to normal angiogenesis (41). Thus, a very high local or systemic level of VEGF may not necessarily be the required aim of gene therapy, because it may result in significant edema (28), the formation of hemangioma (42,43), or aggravation of atherosclerotic lesions (60).
In addition to the proper level of VEGF, other angiogenic mediators also are necessary for the formation of fully mature blood vessels (reviewed in refs. 30, 50). The recruitment of mural cells and the formation of an organized vascular wall are required for the development of fully functional veins and arteries. As suggested by Ozawa’s work, it is possible that this can be achieved by tight regulation of local production of VEGF. Another approach is to use the combination of genes of various growth factors. The potential candidates are fibroblast growth factors (FGFs) and angiopoietin-1 as well as the platelet-derived growth factor, which in several experimental settings have been demonstrated to stimulate the growth of mature vasculature (reviewed in refs. 30, 50). It also can be suggested that bicistronic vectors, harboring two therapeutic genes can be used for such purposes. In such an approach, the expression of one gene, e.g., VEGF, is under the control of a constitutive promoter, whereas the expression of a second gene, e.g., FGF-2, is driven by the internal ribosome entry site (IRES) (61). IRES allows the cap-independent translation, and it can permit the protein synthesis also under hypoxic conditions (62). Some experiments demonstrated the feasibility of such an approach (46,64). However, the therapeutic potentials of such vectors in cardiovascular settings remain to be established. Nevertheless, IRES-dependent translation of VEGF from plasmid vector efficiently restored blood flow in a murine hindlimb ischemia model (62), suggests that IRES-based vectors may be exploited for therapeutic purposes.
Functional improvement of the vascular network also can be accomplished indirectly by the overexpression of genes, whose products stimulate VEGF synthesis (Fig. 2). Among genes regulating VEGF production are those of which cardiovascular protective effect is well established. Accordingly, we have shown recently that NO enhances VEGF expression and that this effect can be achieved by the transfer of NOS III or NOS II genes (65,66). Furthermore, we demonstrated that HO-1, the enzyme generating CO, iron, and biliverdin from a substrate heme could be a mediator of VEGF synthesis (67). Accordingly, the gene transfer of HO-1 augmented the VEGF expression and angiogenic activity of endothelial cells (67,68). It also is possible to increase the VEGF synthesis by gene transfer of superoxide dismutase-1 (SOD1), an enzyme-dismutating superoxide radical to hydrogen peroxide (69). Interestingly, the stimulation of VEGF by reactive oxygen species, including SOD1-derived H2O2 also involves the enhancement of the HO-1 expression (70).
Fig. 2.
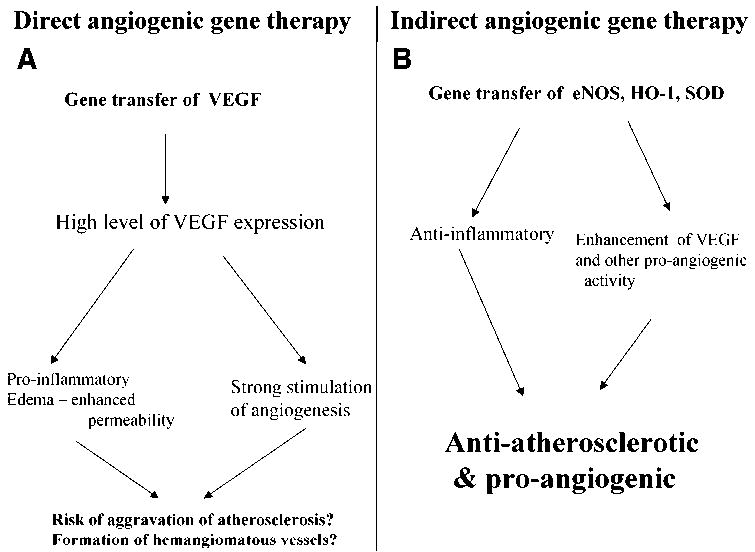
Strategies for direct and indirect stimulation of angiogenesis. (A) In a direct strategy, the formation of new blood vessels is enhanced by overexpression of the genes of angiogenic factors. Because of the other activities of those factors, the strategy may be connected with the risk of side effects, such as edema, inflammation, or hemangioma formation. In the setting of atherosclerosis, the risk of disease aggravation has to be considered (60). (B) In the indirect approach, the synthesis of proangiogenic factors is enhanced by overexpression of the vasculoprotective genes, such as eNOS, HO-1, or SOD1. Besides stimulation of the synthesis of growth factors, the transfer can have an anti-inflammatory effect.
Our data obtained from in vitro experiments have recently been corroborated in animal studies, which showed the stimulation of angiogenesis in ischemic tissues by gene transfer of NOS III (71) or the HO-1 gene (72). Moreover, overexpression of MnSOD (SOD2) or eNOS has been demonstrated to promote wound healing, the process being also dependent on the proper vasculature formation (73). The indirect stimulation of angiogenesis by overexpression of anti-inflammatory genes offers another possibility of stimulation of the growth of vasculature in ways that may prevent the unwanted effect of high expression of VEGF (Fig. 2). However, this hypothesis requires further verification in animal experiments.
THERAPEUTIC POTENTIAL OF GENE TRANSFER OF HO-1
The absence of HO-1 exacerbates atherosclerosis (74). Accordingly, HO-1 overexpression prevented the development of atherosclerosis in several animal models (75–78). The beneficial effect of HO-1 gene transfer on prevention of restenosis after balloon angioplasty also has been shown (36,79). These advantageous effects are the result of the modulation of the several crucial process, such as augmentation of the endothelial cells resistance to proapoptotic stimuli, inhibition of the vascular smooth muscle cell proliferation, and inhibition of the synthesis of proinflammatory mediators (protective actions of HO-1 reviewed in ref. 80).
The application of HO-1 for stimulation of angiogenesis in ischemic tissues provides an interesting option. However, the animal data on this approach are still limited to one study, in which Suzuki et al. (72) showed that the adenoviral transduction of HO-1 enhanced VEGF expression in rat ischemic hindlimbs, which in turn induced angiogenesis and improved blood flow. It can be suggested that similar beneficial effects can be applied for the prevention of ischemia reperfusion injury in myocardial infarction (46,81). Addressing the concept of preventive gene therapy with HO-1, Tang and coworkers used two plasmid vectors, one of which harbored the ODD coding sequence of HIF-1α and the second the HO-1gene under the control of Gal4 sequence (Fig. 1). Plasmids were injected into the mouse heart subjected to myocardial ischemia. The overexpression of hypoxia-induced HO-1 attenuated the formation of fibrotic scar, the effect probably being related to the down-regulation of proapoptotic Bax, Bak, and caspase 3 levels and upregulation of antiapoptotic Bcl-2 protein (46).
Another, preemptive strategy also has been tested by Pachori and coworkers (81) who used AAV vector containing human HO-1 cDNA, driven by an erythropoietin gene-derived HRE sequence. The single administration of such a vector into the rat heart prevented the ischemia/reperfusion injury by myocardial infarction, which was induced 8 wk later. Ischemia in such pre-treated hearts resulted in approximately a 50-fold induction in human HO-1 over endogenous rat HO-1, whereas the rise of the latter was delayed by about 12 h and did not reach the level attained from the transgene. There also was an immediate 5-fold (sustained for 24 h) increase in total HO-1 protein levels in human HO-1-transduced animals after 1 h of ischemia.
Furthermore, Agrawal and coworkers (82) have evaluated the preemptive gene therapy using extracellular SOD (EC-SOD and SOD3). AAV-mediated delivery of EC-SOD, driven by a constitutive promoter, prevented the injury caused by myocardial infarction induced 6 wk later. Importantly, analysis performed 16 mo after gene transfer demonstrated the persistent increase of functional SOD in the EC-SOD-treated ventricles (82). Thus, the proof of the concept of a preemptive gene therapy strategy has been demonstrated in several recent studies (46,48,49). This approach may be used for patients at high risk of acute coronary ischemia, and, as pointed out by Pachori and coworkers (81), may be administered in cardiac catheterization laboratory during a revascularization procedure or in the operating room during cardiac surgery.
POTENTIAL DRAWBACKS OF HYPOXIA-REGULATED GENE THERAPY
Endogenous regulation of delivered genes by changes in the microenvironment of the targeted organ offers the possibility of obtaining the expression of therapeutic transgene only in the conditions when it is really required. Therefore, the idea of hypoxia-dependent regulation of transduced genes has gained so much attention. Interestingly, so far the effect of such a strategy on the expression of endogenous genes, regulated by the same transcription factor that drives the transgene expression, has not been investigated. However, one may presume that delivery of large amounts of plasmid or viral vector molecules may result in the trapping of the transcription factor by large amounts of the regulatory sequences delivered to the cells. We have tested the hypothesis in the following experiments.
Human microvascular endothelial cells (HMEC)-1 have been transfected in vitro with a plasmid vector harboring the HRE of human VEGF promoter coupled to a reporter luciferase gene. The other cells have been transfected by a plasmid harboring an HRE-related NPAS2:BML1 response element (NRE sequence) (83), which is not activated in hypoxia (84). Cells were exposed to 1% oxygen. After 24 h of hypoxia, the activity of the reporter gene was determined, and enzyme-linked immunosorbent assay (ELISA) assessed the VEGF release into the media. As expected, hypoxia strongly induced the production of VEGF (Fig. 3). The synthesis of VEGF in cells transfected with NRE-luc plasmid was similar to those obtained in cells not transfected. However, the synthesis of VEGF in cells transfected with HRE-luc plasmid was approx 3 times lower (Fig. 3). Comparable data have been obtained in several independent experiments. A similar effect also has been noted in other sets of experiments. HMEC-1 cells were transduced with plasmids harboring HO-1 cDNA under the control of the HRE sequence (p-HRE-HO-1) or plasmid harboring HO-1 cDNA under the control of the cytomegalovirus (CMV) promoter (p-CMV-HO-1). Cells were treated with cobalt chloride to mimic hypoxia conditions (85). As expected, CoCl2 induced the HO-1 expression driven by the HRE sequence. CoCl2 also enhanced the production of VEGF, although it was less potent in cells transfected with pHRE-HO-1 vector in comparison with cells transfected with pCMV-HO-1 (not shown).
Fig. 3.
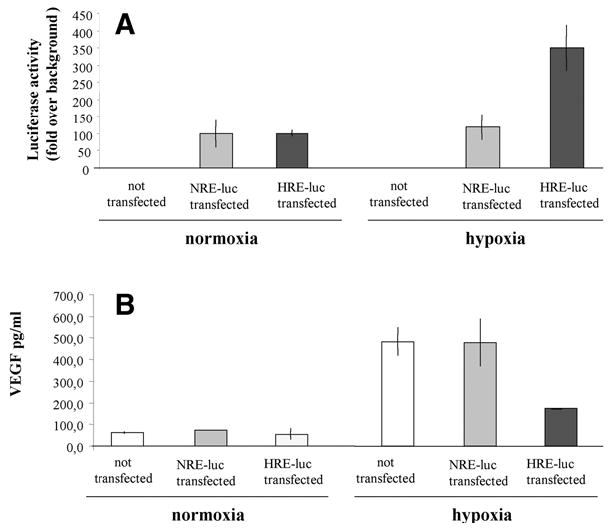
Effect of transfer of plasmid vector harboring HRE sequence on the expression of endogenous VEGF in human microvascular endothelial cells. (A) HMEC-1 were seeded onto a 24-well plate and were transfected with 250 ng HRE or NRE (mutated HRE site) luciferase reporter vectors by using Superfect (QIAGEN, Valencia, CA). Twenty-four hours after transfection, cells were placed in hypoxic (1% O2) conditions for the next 24 h. The efficiency of transfection and response to hypoxia were measured by luciferase activity. (B) In nontransfected cells, luciferase was not detected. Hypoxia-mediated induction of HRE-driven luciferase expression was paralleled by attenuated induction of endogenous HRE-dependent protein VEGF as measured by ELISA.
The detailed mechanisms of such changes will require further investigation. However, one can presume that the trapping of the HIF-1 transcription factor by HRE sequences present in the introduced plasmids, analogically to the effect exerted by DNA decoys, may be responsible for the less effective stimulation of VEGF production. The effect on other hypoxia-dependent genes also should be determined. Investigators in studies performed to date by using the hypoxia-regulated gene expression have not noted such changes. However, it is reasonable to suppose that the amount of transcription factor available in the cells may be limited. The outcome of the enhanced expression of one therapeutic gene (e.g., HO-1) together with the concomitant decrease in the endogenous production of other important genes, such as VEGF, remains to be elucidated before the application of this type of therapy in clinical conditions.
CONCLUSIONS
Gene therapy with proangiogenic factors did not fulfill the initial promise. In such a situation, other potentially therapeutic genes are tested. Among them, HO-1 emerges as an important target for interventions in various diseases. Perhaps the anti-inflammatory and proangiogenic effects of HO-1 can be exploited for treatment of cardiovascular diseases, which will require the augmentation of protection against oxidative stress and enhancement of blood supply to the ischemic tissues (Fig. 2). Additionally, redox-induced genes are promising targets for therapeutic applications not only in atherosclerosis but also in other diseases, particularly in cancers. The coming years should provide more data demonstrating the feasibility of such an approach in animal models and first clinical trials.
The concept of preemptive gene therapy, as exemplified here by experiments with AAV-EC-SOD or AAV-HO-1 expression is a novel approach offering exciting possibilities. It is therefore suggested that in future preemptive myocardial gene therapy may find its utility in selected high-risk patients undergoing interventional or a surgical revascularization procedure (82). However, further studies are required to elucidate whether the application of vectors regulated by induced transcription factors, such as HIF-1, will not result in the attenuation of the expression of endogenous genes dependent on the same activation pathway. It remains to be tested whether the combined strategy, namely the overexpression of the transcriptional activator (ODD domain of HIF-1), together with the target gene (e.g., HO-1 gene) could overcome such an inhibitory effect as observed and described for in vitro experiments herein. The improvement in gene transfer technology and in modes of regulation of therapeutic gene expression is thought to increase the effectiveness of clinical trials. It is necessary to follow this road with the aim of transforming gene therapy from a purely experimental to a more therapeutic approach.
Acknowledgments
Research on cardiovascular gene therapy was supported by KBN grant 096/P04/2004 from the Ministry of Science and Informatics Technology. A.J. is the Wellcome Trust International Research Senior Fellow in Biomedical Science.
References
- 1.Gruchala M, Roy H, Bhardwaj S, Yla-Herttuala S. Gene therapy for cardiovascular diseases. Curr Pharm Des. 2004;10:407–423. doi: 10.2174/1381612043453379. [DOI] [PubMed] [Google Scholar]
- 2.Flugelman MY, Jaklitsch MT, Newman KD, Casscells W, Bratthauer GL, Dichek DA. Low level in vivo gene transfer into the arterial wall through a perforated balloon catheter. Circulation. 1992;85:1110–1117. doi: 10.1161/01.cir.85.3.1110. [DOI] [PubMed] [Google Scholar]
- 3.Lu QL, Bou-Ghario G, Partridge TA. Nonviral gene delivery in skeletal muscle: a protein factory. Gene Ther. 2003;10:131–142. doi: 10.1038/sj.gt.3301874. [DOI] [PubMed] [Google Scholar]
- 4.Danko I, Wolff JA. Direct gene transfer into muscle. Vaccine. 1994;12:1499–1502. doi: 10.1016/0264-410x(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 5.Rissanen TT, Rutanen J, Yla-Herttuala S. Gene transfer for therapeutic vascular growth in myocardial and peripheral ischemia. Adv Genet. 2004;52:117–164. doi: 10.1016/S0065-2660(04)52004-7. [DOI] [PubMed] [Google Scholar]
- 6.St George JA. Gene therapy progress and prospects: adenoviral vectors. Gene Ther. 2003;10:1135–1141. doi: 10.1038/sj.gt.3302071. [DOI] [PubMed] [Google Scholar]
- 7.Cavazzana-Calvo M, Lagresle C, Hacein-Bey-Abina S, Fischer A. Gene therapy for severe combined immunodeficiency. Annu Rev Med. 2005;56:585–602. doi: 10.1146/annurev.med.56.090203.104142. [DOI] [PubMed] [Google Scholar]
- 8.Cavazzana-Calvo M, Fischer A. Efficacy of gene therapy for SCID is being confirmed. Lancet. 2004;364:2155–2156. doi: 10.1016/S0140-6736(04)17603-4. [DOI] [PubMed] [Google Scholar]
- 9.Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 10.Wiznerowicz M, Trono D. Harnessing HIV for therapy, basic research and biotechnology. Trends Biotechnol. 2005;23:42–47. doi: 10.1016/j.tibtech.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 11.Pellinen R, Hakkarainen T, Wahlfors T, et al. Cancer cells as targets for lentivirus-mediated gene transfer and gene therapy. Int J Oncol. 2004;25:1753–1762. [PubMed] [Google Scholar]
- 12.Morris KV, Rossi JJ. Anti-HIV-1 gene expressing lentiviral vectors as an adjunctive therapy for HIV-1 infection. Curr HIV Res. 2004;2:185–191. doi: 10.2174/1570162043484906. [DOI] [PubMed] [Google Scholar]
- 13.Couzin J, Kaiser J. Gene therapy. As Gelsinger case ends, gene therapy suffers another blow. Science. 2005;307:1028. doi: 10.1126/science.307.5712.1028b. [DOI] [PubMed] [Google Scholar]
- 14.Read ML, Spice R, Parker AL, Mir S, Logan A. 12th Annual congress of the European society of gene therapy. Expert Opin Biol Ther. 2005;5:137–141. doi: 10.1517/14712598.5.1.137. [DOI] [PubMed] [Google Scholar]
- 15.Kingsman SM, Mitrophanous K, Olsen JC. Potential oncogene activity of the woodchuck hepatitis post-transcriptional regulatory element (WPRE) Gene Ther. 2005;12:3–4. doi: 10.1038/sj.gt.3302417. [DOI] [PubMed] [Google Scholar]
- 16.McCarty DM, Young SM, Jr, Samulski RJ. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu Rev Genet. 2004;38:819–845. doi: 10.1146/annurev.genet.37.110801.143717. [DOI] [PubMed] [Google Scholar]
- 17.Gruchala M, Bhardwaj S, Pajusola K, et al. Gene transfer into rabbit arteries with adeno-associated virus and adenovirus vectors. J Gene Med. 2004;6:545–554. doi: 10.1002/jgm.535. [DOI] [PubMed] [Google Scholar]
- 18.Vassalli G, Bueler H, Dudler J, von Segesser LK, Kappenberger L. Adeno-associated virus (AAV) vectors achieve prolonged transgene expression in mouse myocardium and arteries in vivo: a comparative study with adenovirus vectors. Int J Cardiol. 2003;90:229–238. doi: 10.1016/s0167-5273(02)00554-5. [DOI] [PubMed] [Google Scholar]
- 19.White SJ, Nicklin SA, Buning H, et al. Targeted gene delivery to vascular tissue in vivo by tropism-modified adeno-associated virus vectors. Circulation. 2004;109:513–519. doi: 10.1161/01.CIR.0000109697.68832.5D. [DOI] [PubMed] [Google Scholar]
- 20.Richter M, Iwata A, Nyhuis J, et al. Adeno-associated virus vector transduction of vascular smooth muscle cells in vivo. Physiol Genomics. 2000;2:117–127. doi: 10.1152/physiolgenomics.2000.2.3.117. [DOI] [PubMed] [Google Scholar]
- 21.Buning H, Nicklin SA, Perabo L, Hallek M, Baker AH. AAV-based gene transfer. Curr Opin Mol Ther. 2003;5:367–375. [PubMed] [Google Scholar]
- 22.Nicklin SA, Baker AH. Tropism-modified adenoviral and adeno-associated viral vectors for gene therapy. Curr Gene Ther. 2002;2:273–293. doi: 10.2174/1566523023347797. [DOI] [PubMed] [Google Scholar]
- 23.Chen S, Kapturczak M, Loiler SA, et al. Efficient transduction of vascular endothelial cells with recombinant adeno-associated virus serotype 1 and 5 vectors. Hum Gene Ther. 2005;16:235–247. doi: 10.1089/hum.2005.16.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edelstein ML, Abedi MR, Wixon J, Edelstein RM. Gene therapy clinical trials worldwide 1989–2004 – an overview. J Gene Med. 2004;6:597–602. doi: 10.1002/jgm.619. [DOI] [PubMed] [Google Scholar]
- 25.No authors listed. INGN 201: Ad-p53, Ad5CMV-p53, Adenoviral p53, INGN 101, p53 gene therapy—Introgen, RPR/INGN 201. BioDrugs. 2003;17:216–222. doi: 10.2165/00063030-200317030-00010. [DOI] [PubMed] [Google Scholar]
- 26.Kim IH, Jozkowicz A, Piedra PA, Oka K, Chan L. Lifetime correction of genetic deficiency in mice with a single injection of helper-dependent adenoviral vector. Proc Natl Acad Sci USA. 2001;98:13,282–13,287. doi: 10.1073/pnas.241506298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isner JM, Pieczek A, Schainfeld R, et al. Clinical evidence of angiogenesis after arterial gene transfer of phVEGF165 in patient with ischaemic limb. Lancet. 1996;348:370–374. doi: 10.1016/s0140-6736(96)03361-2. [DOI] [PubMed] [Google Scholar]
- 28.Baumgartner I, Pieczek A, Manor O, et al. Constitutive expression of phVEGF165 after intramuscular gene transfer promotes collateral vessel development in patients with critical limb ischemia. Circulation. 1998;97:1114–1123. doi: 10.1161/01.cir.97.12.1114. [DOI] [PubMed] [Google Scholar]
- 29.Shyu KG, Chang H, Wang BW, Kuan P. Intramuscular vascular endothelial growth factor gene therapy in patients with chronic critical leg ischemia. Am J Med. 2003;114:85–92. doi: 10.1016/s0002-9343(02)01392-x. [DOI] [PubMed] [Google Scholar]
- 30.Markkanen JE, Rissanen TT, Kivela A, Yla-Herttuala S. Growth factor-induced therapeutic angiogenesis and arteriogenesis in the heart-gene therapy. Cardiovasc Res. 2005;65:656–664. doi: 10.1016/j.cardiores.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 31.Mann MJ, Whittemore AD, Donaldson MC, et al. Ex-vivo gene therapy of human vascular bypass grafts with E2F decoy: the PREVENT single-centre, randomised, controlled trial. Lancet. 1999;354:1493–1498. doi: 10.1016/S0140-6736(99)09405-2. [DOI] [PubMed] [Google Scholar]
- 32.Yla-Herttuala S, Markkanen JE, Rissanen TT. Gene therapy for ischemic cardiovascular diseases: some lessons learned from the first clinical trials. Trends Cardiovasc Med. 2004;14:295–300. doi: 10.1016/j.tcm.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 33.Iwaguro H, Yamaguchi J, Kalka C, et al. Endothelial progenitor cell vascular endothelial growth factor gene transfer for vascular regeneration. Circulation. 2002;105:732–738. doi: 10.1161/hc0602.103673. [DOI] [PubMed] [Google Scholar]
- 34.Murasawa S, Llevadot J, Silver M, Isner JM, Losordo DW, Asahara T. Constitutive human telomerase reverse transcriptase expression enhances regenerative properties of endothelial progenitor cells. Circulation. 2002;106:1133–1139. doi: 10.1161/01.cir.0000027584.85865.b4. [DOI] [PubMed] [Google Scholar]
- 35.Kong D, Melo LG, Mangi AA, et al. Enhanced inhibition of neointimal hyperplasia by genetically engineered endothelial progenitor cells. Circulation. 2004;109:1769–1775. doi: 10.1161/01.CIR.0000121732.85572.6F. [DOI] [PubMed] [Google Scholar]
- 36.Tulis DA, Durante W, Liu X, Evans AJ, Peyton KJ, Schafer AI. Adenovirus-mediated heme oxygenase-1 gene delivery inhibits injury-induced vascular neointima formation. Circulation. 2001;104:2710–2715. doi: 10.1161/hc4701.099585. [DOI] [PubMed] [Google Scholar]
- 37.Cooke JP, Losordo DW. Nitric oxide and angiogenesis. Circulation. 2002;105:2133–2135. doi: 10.1161/01.cir.0000014928.45119.73. [DOI] [PubMed] [Google Scholar]
- 38.Carmeliet P. VEGF gene therapy: stimulating angiogenesis or angioma-genesis? Nat Med. 2000;6:1102–1103. doi: 10.1038/80430. [DOI] [PubMed] [Google Scholar]
- 39.Hariawala MD, Horowitz JR, Esakof D, et al. VEGF improves myocardial blood flow but produces EDRF-mediated hypotension in porcine hearts. J Surg Res. 1996;63:77–82. doi: 10.1006/jsre.1996.0226. [DOI] [PubMed] [Google Scholar]
- 40.Sato K, Wu T, Laham RJ, et al. Efficacy of intra-coronary or intravenous VEGF165 in a pig model of chronic myocardial ischemia. J Am Coll Cardiol. 2001;37:616–623. doi: 10.1016/s0735-1097(00)01144-x. [DOI] [PubMed] [Google Scholar]
- 41.Ozawa CR, Banfi A, Glazer NL, et al. Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J Clin Investig. 2004;113:516–527. doi: 10.1172/JCI18420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee RJ, Springer ML, Blanco-Bose WE, Shaw R, Ursell PC, Blau HM. VEGF gene delivery to myocardium: deleterious effects of unregulated expression. Circulation. 2000;102:898–901. doi: 10.1161/01.cir.102.8.898. [DOI] [PubMed] [Google Scholar]
- 43.Springer ML, Chen AS, Kraft PE, Bednarski M, Blau HM. VEGF gene delivery to muscle: potential role for vasculogenesis in adults. Mol Cell. 1998;2:549–558. doi: 10.1016/s1097-2765(00)80154-9. [DOI] [PubMed] [Google Scholar]
- 44.Zagorska A, Dulak J. HIF-1: the knowns and unknowns of hypoxia sensing. Acta Biochim Pol. 2004;51:563–585. [PubMed] [Google Scholar]
- 45.Maxwell PH, Ratcliffe PJ. Oxygen sensors and angiogenesis. Semin Cell Dev Biol. 2002;13:29–37. doi: 10.1006/scdb.2001.0287. [DOI] [PubMed] [Google Scholar]
- 46.Tang YL, Tang Y, Zhang YC, Qian K, Shen L, Phillips MI. Protection from ischemic heart injury by a vigilant heme oxygenase-1 plasmid system. Hypertension. 2004;43:746–751. doi: 10.1161/01.HYP.0000120152.27263.87. [DOI] [PubMed] [Google Scholar]
- 47.Tang Y, Schmitt-Ott K, Qian K, Kagiyama S, Phillips MI. Vigilant vectors: adeno-associated virus with a biosensor to switch on amplified therapeutic genes in specific tissues in life-threatening diseases. Methods. 2002;28:259–266. doi: 10.1016/s1046-2023(02)00231-1. [DOI] [PubMed] [Google Scholar]
- 48.Tang Y, Jackson M, Qian K, Phillips MI. Hypoxia inducible double plasmid system for myocardial ischemia gene therapy. Hypertension. 2002;39:695–698. doi: 10.1161/hy0202.103784. [DOI] [PubMed] [Google Scholar]
- 49.Phillips MI, Tang Y, Schmidt-Ott K, Qian K, Kagiyama S. Vigilant vector: heart-specific promoter in an adeno-associated virus vector for cardioprotection. Hypertension. 2002;39:651–655. doi: 10.1161/hy0202.103472. [DOI] [PubMed] [Google Scholar]
- 50.Blau HM, Banfi A. The well-tempered vessel. Nat Med. 2001;7:532–534. doi: 10.1038/87850. [DOI] [PubMed] [Google Scholar]
- 51.Binley K, Askham Z, Martin L, et al. Hypoxia-mediated tumour targeting. Gene Ther. 2003;10:540–549. doi: 10.1038/sj.gt.3301944. [DOI] [PubMed] [Google Scholar]
- 52.Su H, Joho S, Huang Y, et al. Adeno-associated viral vector delivers cardiac-specific and hypoxia-inducible VEGF expression in ischemic mouse hearts. Proc Natl Acad Sci USA. 2004;101:16,280–16,285. doi: 10.1073/pnas.0407449101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su H, Arakawa-Hoyt J, Kan YW. Adeno-associated viral vector-mediated hypoxia response element-regulated gene expression in mouse ischemic heart model. Proc Natl Acad Sci USA. 2002;99:9480–9485. doi: 10.1073/pnas.132275299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Agha-Mohammadi S, Lotze MT. Regulatable systems: applications in gene therapy and replicating viruses. J Clin Investig. 2000;105:1177–1183. doi: 10.1172/JCI10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Snowden AW, Zhang L, Urnov F, et al. Repression of vascular endothelial growth factor A in glioblastoma cells using engineered zinc finger transcription factors. Cancer Res. 2003;63:8968–8976. [PubMed] [Google Scholar]
- 56.Rebar EJ, Huang Y, Hickey R, et al. Induction of angiogenesis in a mouse model using engineered transcription factors. Nat Med. 2002;8:1427–1432. doi: 10.1038/nm1202-795. [DOI] [PubMed] [Google Scholar]
- 57.Weber W, Fussenegger M. Artificial mammalian gene regulation networks-novel approaches for gene therapy and bioengineering. J Biotechnol. 2002;98:161–187. doi: 10.1016/s0168-1656(02)00130-x. [DOI] [PubMed] [Google Scholar]
- 58.Easton JB, Houghton PJ. Therapeutic potential of target of rapamycin inhibitors. Expert Opin Ther Targets. 2004;8:551–564. doi: 10.1517/14728222.8.6.551. [DOI] [PubMed] [Google Scholar]
- 59.Visner GA, Lu F, Zhou H, Liu J, Kazemfar K, Agarwal A. Rapamycin induces heme oxygenase-1 in human pulmonary vascular cells: implications in the antiproliferative response to rapamycin. Circulation. 2003;107:911–916. doi: 10.1161/01.cir.0000048191.75585.60. [DOI] [PubMed] [Google Scholar]
- 60.Celletti FL, Waugh JM, Amabile PG, Brendolan A, Hilfiker PR, Dake MD. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat Med. 2001;7:425–429. doi: 10.1038/86490. [DOI] [PubMed] [Google Scholar]
- 61.Malecki M, Przybyszewska M, Janik P. Construction of a bicistronic proangiogenic expression vector and its application in experimental angiogenesis in vivo. Acta Biochim Pol. 2003;50:875–882. [PubMed] [Google Scholar]
- 62.Roguin A, Avivi A, Nitecki S, et al. Restoration of blood flow by using continuous perimuscular infiltration of plasmid DNA encoding subterranean mole rat Spalax ehrenbergi VEGF. Proc Natl Acad Sci USA. 2003;100:4644–4648. doi: 10.1073/pnas.0330833100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kapturczak M, Zolotukhin S, Cross J, et al. Transduction of human and mouse pancreatic islet cells using a bicistronic recombinant adeno-associated viral vector. Mol Ther. 2002;5:154–160. doi: 10.1006/mthe.2002.0522. [DOI] [PubMed] [Google Scholar]
- 64.Fan L, Drew J, Dunckley MG, Owen JS, Dickson G. Efficient coexpression and secretion of anti-atherogenic human apolipoprotein AI and lecithin-cholesterol acyltransferase by cultured muscle cells using adeno-associated virus plasmid vectors. Gene Ther. 1998;5:1434–1440. doi: 10.1038/sj.gt.3300746. [DOI] [PubMed] [Google Scholar]
- 65.Dulak J, Jozkowicz A, Dembinska-Kiec A, et al. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:659–666. doi: 10.1161/01.atv.20.3.659. [DOI] [PubMed] [Google Scholar]
- 66.Jozkowicz A, Cooke JP, Guevara I, et al. Genetic augmentation of nitric oxide synthase increases the vascular generation of VEGF. Cardiovasc Res. 2001;51:773–783. doi: 10.1016/s0008-6363(01)00344-3. [DOI] [PubMed] [Google Scholar]
- 67.Dulak J, Jozkowicz A, Foresti R, et al. Heme oxygenase activity modulates vascular endothelial growth factor synthesis in vascular smooth muscle cells. Antioxid Redox Signal. 2002;4:229–240. doi: 10.1089/152308602753666280. [DOI] [PubMed] [Google Scholar]
- 68.Jozkowicz A, Huk I, Nigisch A, et al. Heme oxygenase and angiogenic activity of endothelial cells: stimulation by carbon monoxide and inhibition by tin proto-porphyrin-IX. Antioxid Redox Signal. 2003;5:155–162. doi: 10.1089/152308603764816514. [DOI] [PubMed] [Google Scholar]
- 69.Grzenkowicz-Wydra J, Cisowski J, Nakonieczna J, et al. Gene transfer of CuZn superoxide dismutase enhances the synthesis of vascular endothelial growth factor. Mol Cell Biochem. 2004;264:169–181. doi: 10.1023/b:mcbi.0000044386.45054.70. [DOI] [PubMed] [Google Scholar]
- 70.Cisowski J, Loboda A, Jozkowicz A, Chen S, Agarwal A, Dulak J. Role of heme oxygenase-1 in hydrogen peroxide-induced VEGF synthesis: effect of HO-1 knockout. Biochem Biophys Res Commun. 2005;326:670–676. doi: 10.1016/j.bbrc.2004.11.083. [DOI] [PubMed] [Google Scholar]
- 71.Namba T, Koike H, Murakami K, et al. Angiogenesis induced by endothelial nitric oxide synthase gene through vascular endothelial growth factor expression in a rat hindlimb ischemia model. Circulation. 2003;108:2250–2257. doi: 10.1161/01.CIR.0000093190.53478.78. [DOI] [PubMed] [Google Scholar]
- 72.Suzuki M, Iso-o N, Takeshita S, et al. Facilitated angiogenesis induced by heme oxygenase-1 gene transfer in a rat model of hindlimb ischemia. Biochem Biophys Res Commun. 2003;302:138–143. doi: 10.1016/s0006-291x(03)00114-1. [DOI] [PubMed] [Google Scholar]
- 73.Luo JD, Wang YY, Fu WL, Wu J, Chen AF. Gene therapy of endothelial nitric oxide synthase and manganese superoxide dismutase restores delayed wound healing in type 1 diabetic mice. Circulation. 2004;110:2484–2493. doi: 10.1161/01.CIR.0000137969.87365.05. [DOI] [PubMed] [Google Scholar]
- 74.Yet SF, Layne MD, Liu X, et al. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003;17:1759–1761. doi: 10.1096/fj.03-0187fje. [DOI] [PubMed] [Google Scholar]
- 75.Sabaawy HE, Zhang F, Nguyen X, et al. Human heme oxygenase-1 gene transfer lowers blood pressure and promotes growth in spontaneously hypertensive rats. Hypertension. 2001;38:210–215. doi: 10.1161/01.hyp.38.2.210. [DOI] [PubMed] [Google Scholar]
- 76.Juan SH, Lee TS, Tseng KW, et al. Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2001;104:1519–1525. doi: 10.1161/hc3801.095663. [DOI] [PubMed] [Google Scholar]
- 77.Liu XM, Chapman GB, Wang H, Durante W. Adenovirus-mediated heme oxygenase-1 gene expression stimulates apoptosis in vascular smooth muscle cells. Circulation. 2002;105:79–84. doi: 10.1161/hc0102.101369. [DOI] [PubMed] [Google Scholar]
- 78.Bouche D, Chauveau C, Roussel JC, et al. Inhibition of graft arteriosclerosis development in rat aortas following heme oxygenase-1 gene transfer. Transpl Immunol. 2002;9:235–238. doi: 10.1016/s0966-3274(02)00037-0. [DOI] [PubMed] [Google Scholar]
- 79.Tulis DA, Durante W, Peyton KJ, Evans AJ, Schafer AI. Heme oxygenase-1 attenuates vascular remodeling following balloon injury in rat carotid arteries. Atherosclerosis. 2001;155:113–122. doi: 10.1016/s0021-9150(00)00552-9. [DOI] [PubMed] [Google Scholar]
- 80.Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–455. doi: 10.1016/s1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 81.Pachori AS, Melo LG, Hart ML, et al. Hypoxia-regulated therapeutic gene as a preemptive treatment strategy against ischemia/reperfusion tissue injury. Proc Natl Acad Sci USA. 2004;101:12,282–12,287. doi: 10.1073/pnas.0404616101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Agrawal RS, Muangman S, Layne MD, et al. Pre-emptive gene therapy using recombinant adeno-associated virus delivery of extracellular superoxide dismutase protects heart against ischemic reperfusion injury, improves ventricular function and prolongs survival. Gene Ther. 2004;11:962–969. doi: 10.1038/sj.gt.3302250. [DOI] [PubMed] [Google Scholar]
- 83.Reick M, Garcia JA, Dudley C, McKnight SL. NPAS2: an analog of clock operative in the mammalian forebrain. Science. 2001;293:506–509. doi: 10.1126/science.1060699. [DOI] [PubMed] [Google Scholar]
- 84.Jozkowicz A, Nigisch A, Wegrzyn J, Weigel G, Huk I, Dulak J. Opposite effects of prostaglandin-J2 on VEGF in normoxia and hypoxia: role of HIF-1. Biochem Biophys Res Commun. 2004;314:31–38. doi: 10.1016/j.bbrc.2003.12.059. [DOI] [PubMed] [Google Scholar]
- 85.Wang GL, Semenza GL. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: implications for models of hypoxia signal transduction. Blood. 1993;82:3610–3615. [PubMed] [Google Scholar]