

Abstract
Mast cell activation through the high affinity IgE receptor (FcεRI) is a critical component of atopic inflammation. The cytokine TGF-β1 has been shown to inhibit IgE-dependent mast cell activation, possibly serving to dampen mast cell-mediated inflammatory responses. We present proof that TGF-β1 inhibits mast cell FcεRI expression through a reversible pathway that diminishes protein, but not mRNA, expression of the FcεRI subunit proteins α, β, and γ. The stability of the expressed proteins and the assembled cell surface complex was unaltered by TGF-β1 treatment. However, TGF-β1 decreased the rate of FcεRI β-chain synthesis, arguing that this inhibitory cytokine exerts its effects at the level of mRNA translation. TGF-β1 consistently diminished FcεRI expression on cultured human or mouse mast cells as well as freshly isolated peritoneal mast cells. The related cytokines, TGF-β2 and TGF-β3, had similar effects. We propose that TGF-β1 acts as a negative regulator of mast cell function, in part by decreasing FcεRI expression.
The extensively studied TGF-β superfamily is a large group of structurally related, highly conserved growth factors that includes TGF-β1, TGF-β2, TGF-β3, activins, and bone morphogenetic proteins. TGF-β1, the prototypic family member, is synthesized as a precursor polypeptide and secreted in a latent form by most tissues and cell types. The biologically active protein is a disulfide-linked homodimer (1). Signaling by TGF-β family members occurs through interaction with serine/threonine kinase receptors termed type I and type II that, when activated, phosphorylate Smad proteins. Smads form complexes and translocate to the nucleus, where they regulate transcription of target genes together with other cofactors (reviewed in Refs. 2 and 3). TGF-β family members have stimulatory or inhibitory effects in regulating a wide variety of cellular functions, including apoptosis, differentiation, and proliferation. A vast number of studies have implicated TGF-β proteins in several different physiological processes, including inflammation, fibrosis, and angiogenesis. Importantly, TGF-β proteins have been linked to autoimmune disease, atherosclerosis, fibrotic disease, and cancer in humans (4, 5). A number of studies showing the inhibitory effects of TGF-β1 on immune cell function (reviewed in Ref. 5) support the role of TGF-β1 as a suppressor of immunity and inflammation. Underscoring the importance of TGF-mediated immunosuppression is the observation that TGF-β1-null mice develop severe inflammation, wasting syndrome, and organ failure, leading to death by 3 wk of age (6, 7).
Mast cell activation by cross-linking of the heterotetrameric high affinity receptor for IgE (FcεRI) results in the release of pre-formed vasoactive amines and de novo synthesized cytokines, chemokines, and PGs (8). FcεRI is composed of a ligand-binding α-chain, a signal-augmenting β-chain, and a signal-transducing γ-chain dimer. The FcεRI β and γ subunits contain ITAMs that, upon receptor aggregation, bind Src homology domain 2-containing signaling molecules to promote a signaling cascade causing mediator release (8). Mast cells are responsible for immediate hypersensitivity responses. Their widespread distribution in the skin and respiratory tract suggests a role as for these cells as a first-line defense against invading pathogens. However, unregulated activation can result in deleterious effects. In fact, mast cells are often associated with human allergic disease and are implicated in mouse models of the autoimmune diseases multiple sclerosis and, recently, rheumatoid arthritis (9, 10).
Given the role of mast cells in allergic and inflammatory disease and the inhibitory effect of TGF-β on immune cells, the possibility exists that TGF-β is a paracrine or autocrine inhibitor of mast cell function. It has been shown in separate studies that TGF-β1 inhibited IL-3-dependent proliferation of mouse peritoneal mast cells and bone marrow-derived mast cells (BMMCs)3 (11, 12). In addition, TGF-β1 inhibited the rescue of BMMCs from IL-3 deprivation-induced apoptosis by stem cell factor (SCF) (13). Inhibitory effects of TGF-β1 on mast cell effector functions have also been documented. Bissonette et al. (14) showed that Ag-induced histamine and TNF-α production from rat peritoneal mast cells were inhibited by TGF-β1, whereas Meade et al. (15) showed that in vivo treatment with TGF-β1 inhibited an IgE-mediated, mast cell-dependent, immediate hypersensitivity response in mice. Despite these studies supporting the idea that TGF-β1 inhibits mast cell function, contradicting evidence exists about the ability of TGF-β1 to directly inhibit mast cell degranulation. This includes a report by Kim and Lee (16) that TGF-β1 potentiates the IgE-dependent anaphylaxis reaction. Moreover, these studies have not revealed the mechanism of action by which TGF-β acts on mast cells. This study was undertaken to assess a direct effect of TGF-β1 on mast cell IgE receptor expression as a means of dampening mast cell effector responses.
Materials and Methods
Cytokines and reagents
DNP-conjugated human serum albumin and cycloheximide were purchased from Sigma-Aldrich. Murine IL-3, SCF, and TGF-β1 were purchased from R&D Systems. 2.4G2 (rat anti-mouse FcγRII/RIII), mouse IgE, and FITC-conjugated rat anti-mouse kit were purchased from BD Pharmingen. FITC-conjugated rat anti-mouse IgE was purchased from Southern Biotechnology Associates. The anti-human FcεRI-α mAb, 3B4, was a gift from G. Mackay (University of Melbourne, Melbourne, Australia). Nonspecific human IgG (clone MOPC) was obtained from Sigma-Aldrich.
Mast cell cultures
BMMC were derived from C57BL/6 or BL6 × 129 mice by culture of bone marrow cells in complete RPMI 1640 medium (cRPMI), supplemented with 25% WEHI-3 cell-conditioned medium or with IL-3 and SCF (30 ng/ml each). After 3–4 wk in culture, >95% of these cells were mast cells, as judged by morphology and surface expression of FcεRI and Kit (data not shown). These cells were used within 3 mo of their maturation. Peritoneal cells were harvested from euthanized animals by lavage of the peritoneum using 5 ml of cRPMI injected into the peritoneal cavity. These cells were cultured in cRPMI/IL-3 and SCF. Human umbilical cord blood-derived mast cell populations were derived as described previously (17).
Tissue culture conditions and flow cytometry to measure inhibition of FcεRI expression
Cells were washed and replated at 3 × 105 cells/ml, 200 μl/well, in 96-well, flat-bottom plates. IL-3 was added to 5 ng/ml, followed by the indicated concentrations of TGF-β1 or mouse IgE. The use of SCF (30 ng/ml) in some experiments did not alter the results. Cultures were incubated for the indicated times. Every 4 days half the media and cytokines or IgE were replaced. Mast cell surface Ag expression was assessed by flow cytometry as described previously (18). The mean fluorescence intensity of IgE staining was used to determine the percent inhibition of FcεRI expression caused by the addition of TGF-β1.
Mast cell activation assays
To assess β-hexosaminidase release or cytokine secretion, BMMC were cultured in IL-3 with or without TGF-β1 as described above, then activated with mouse IgE (10 μg/ml, 45 min, 4°C) and the indicated concentrations of DNP-BSA for 1 h (β-hexosaminidase release) or 9 h (cytokine secretion). After activation, β-hexosaminidase release was measured as previously described (19). The percent β-hexosaminidase release was determined by dividing the amount of β-hexosaminidase in the supernatant by the total amount detected in the supernatant and cell pellet. TNF-α or IL-6 secretion was measured by standard ELISA (R&D Systems).
RNase protection assay (RPA)
RPA assays were performed using the RiboQuant system (BD Pharmingen) according to the manufacturer’s instructions. Pixel intensities of individual bands were obtained using a Typhoon PhosphorImager (Molecular Dynamics). The ratio of the pixel intensity for each band of interest to the sum of the pixel intensities for the housekeeping genes (L32 and GAPDH) in that lane were determined. Calculations of percent change in expression relative to control conditions were determined by comparing these ratios.
35 S labeling and Western blot analysis
FcεRIα, -β, and -γ subunits as well as actin were detected by immunoprecipitation and Western blotting as described previously (20). For immunoprecipitation of FcεRIα, lysates from 30 × 106 cells were incubated with 50 μl of protein A-Sepharose conjugated to 25 μg of rabbit anti-mouse IgG (Jackson ImmunoResearch Laboratories) and 5 μg of anti-FcεRIα (clone 5.14; gift from Z. Eshhar, Weizmann Institute, Rehovat, Israel). Immunoprecipitates were analyzed by Western blotting with 1 μg/ml anti-FcεRIα (clone TW; gift from B. Baird and D. Holowka, Cornell University, Ithaca, NY). Although both the 5.14 and TW Ab clones are specific for rat FcεRI α-chain, they cross-react with mouse α-chain. Anti-α-chain immunoprecipitates were subjected to electrophoresis without reducing agents to prevent cross-reactivity of the detection Ab with the precipitating H chain. FcεRIβ and FcεRIγ were detected by Western blotting of total cell lysates using 1.2 μg/ml anti-FcεRIβ (clone JRK) or a 1/10,000 dilution of anti-γ-chain (clone 06–727; Upstate Cell Signaling Solutions). For 35S labeling, BMMC were incubated in cysteine- and methionine-free RPMI 1640 with 5% FCS for 2.5 h, then pulsed for 20 min with 800 mCi/ml 35S-labeled cysteine and methionine. Lysates were sequentially immunoprecipitated with anti-FcεRI β and anti-actin. Immunoprecipitates were resolved by gel electrophoresis and subjected to phosphorimaging. The ratio of β-chain to actin bands was used to compare samples treated or not treated with TGF-β1.
Results
TGF-β inhibits FcεRI expression on mast cells
TGF-β1 has been shown to inhibit FcεRI-dependent mast cell degranulation and TNF-α production (14), an effect confirmed in our experiments. Mouse BMMC cultured for 3 days with or without TGF-β1 were stimulated by IgE and Ag. TGF-β1 reduced β-hexosaminidase release 50–60% over a range of Ag concentrations and decreased the IgE-mediated production of both TNF-α and IL-6 (Fig. 1). This potent inhibition of IgE-mediated signaling led us to question the effects of TGF-β1 on FcεRI expression.
FIGURE 1.
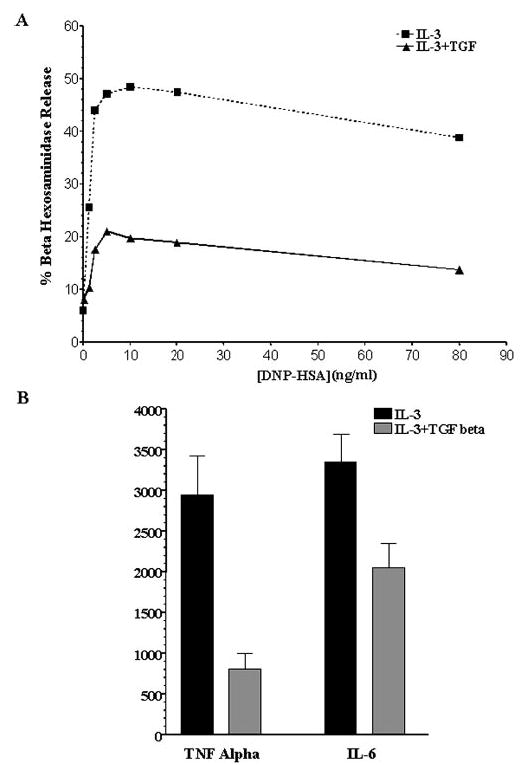
TGF-β1 inhibits IgE-mediated mast cell degranulation and the production of TNF-α and IL-6. BMMC were cultured with or without TGF-β1 (5 ng/ml) for 3 days. Cells were then stimulated with IgE and DNP-HSA. A, BMMC received the indicated concentration of DNP-HSA for 60 min. β-Hexosaminidase release was measured by enzymatic assay as described in Materials and Methods. Data shown are the means of two samples from one of two independent experiments that produced similar results. B, BMMC were stimulated with 50 ng/ml DNP-HSA for 9 h. Culture supernatants were harvested, and cytokine concentrations were determined by ELISA. Data shown are the mean and SE of seven samples from one of four independent experiments that gave similar results.
We first determined the effect of TGF-β1 on mast cell FcεRI surface expression. BMMC were cultured in IL-3 with or without TGF-β1 for 3 days, after which receptor expression was analyzed. Flow cytometric analysis revealed that FcεRI surface expression was markedly reduced on BMMC cultured with TGF-β1 (Fig. 2A). These results were consistent using 15 different BMMC populations derived from individual mice. The kinetics of TGF-β1 effects were measured in time-course and dose-response experiments. We found that TGF-β1 decreased surface FcεRI expression within the first 24 h of culture, and that maximal inhibition (60%) occurred on days 3–4 of culture (Fig. 2B). Importantly, the reduction in FcεRI expression was not due to decreased cell viability. TGF-β1 had no effect on cell death, as judged by propidium iodide or trypan blue staining (data not shown). The inhibitory effect of TGF-β1 showed a concentration dependence, with 50% inhibition of FcεRI expression occurring at ~2 ng/ml TGF-β1 (Fig. 2C). Lastly, the related cytokines, TGF-β2 and TGF-β3, also decreased FcεRI surface expression (Fig. 2D), indicating that this inhibitory effect was consistent among TGF family members.
FIGURE 2.
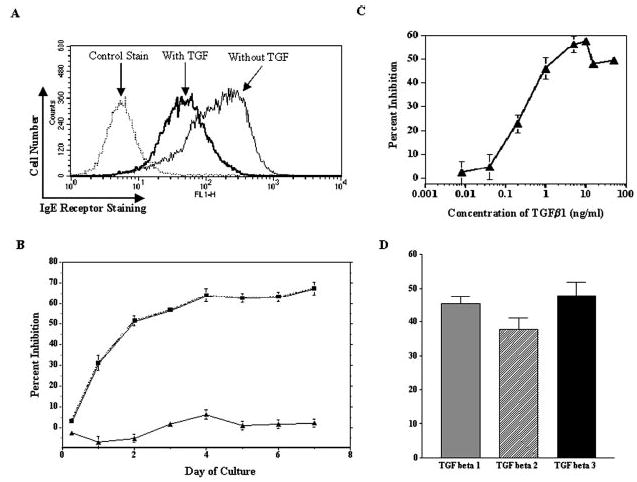
TGF-β1 inhibits mouse mast cell FcεRI expression. A, BMMC were cultured for 3 days in IL-3 and SCF with or without TGF-β1, after which FcεRI surface expression was assessed by flow cytometry. Control staining was with FITC-anti-IgE staining in the absence of IgE. Data shown are a representative histogram from one of 42 experiments using 15 BMMC populations. B, Time course of TGF-β1-mediated inhibition. BMMC were cultured as described in A for the indicated times, after which surface FcεRI expression was assessed by flow cytometry. Percent inhibition was determined by comparing the mean fluorescence intensities of cells cultured in IL-3 with and without TGF-β1. Data shown are the mean and SE of 6–18 samples/point. C, Concentration-dependent effects of TGF-β1 on mouse FcεRI expression. BMMC were cultured as described in A for 3 days. Percent inhibition of FcεRI expression was determined by comparing surface staining of cells cultured in IL-3 with that of cells cultured in IL-3 and TGF-β1. Data shown are the mean and SE from six samples. D, BMMC were cultured for 3 days in IL-3 with or without TGF-β1, TGF-β2, or TGF-β3 (5 ng/ml each). The percent inhibition of surface FcεRI expression was determined as described in B, using six BMMC populations. When comparing the percent inhibition induced by each stimulus, p > 0.05.
To determine whether the inhibitory effect of TGF-β1 on FcεRI expression was restricted to BMMC, we tested other mast cell populations. Peritoneal cells from C57BL6 mice were cultured for 3 days in IL-3 with or without TGF-β1. After loading FcεRI with IgE, peritoneal exudate cells were double-stained for Kit and IgE, then analyzed by flow cytometry. As shown in Fig. 3A, IgE receptor staining intensity on peritoneal mast cells (IgE+Kit+) cultured in IL-3 and TGF-β1 was reduced >70% compared with that on cells cultured in IL-3 alone. These results indicated that the effects of TGF-β1 also were apparent on mature mast cells developed in vivo. Human mast cells also decreased FcεRI expression in response to TGF-β1. As shown in Fig. 3B, human mast cells that were derived from cord blood, then cultured for 3 days in SCF and TGF-β1 exhibited a nearly complete loss of FcεRI expression compared with cells maintained in SCF alone. These data indicate that TGF-β1-mediated inhibition was not a consequence of in vitro development conditions and is consistent in human and mouse models.
FIGURE 3.
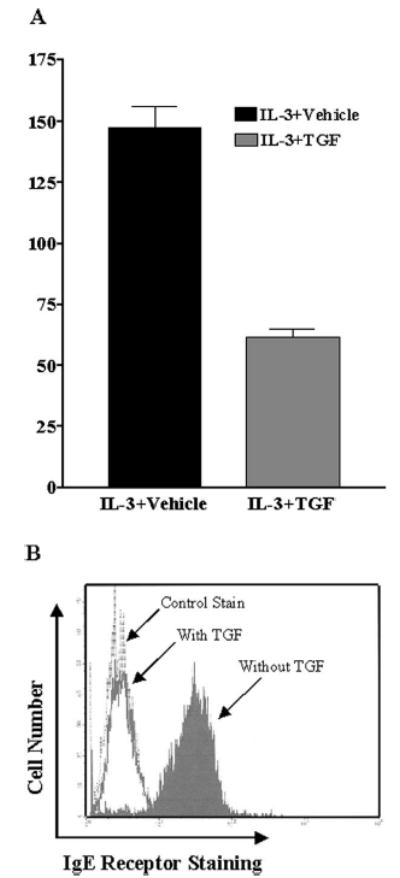
TGF-β1 inhibits FcεRI surface expression on mouse peritoneal mast cells and cultured human mast cells. A, Mouse peritoneal lavage cells were cultured in IL-3 and SCF with or without TGF-β1 for 3 days. Data shown are the average and SD of four separate populations, showing the mean fluorescence intensity of FcεRI staining on Kit-positive cells (mast cells) in the presence or the absence of TGF-β1. B, Human mast cells were cultured in SCF with or without TGF-β1 (15 ng/ml) for 3 days. FcεRI surface expression was determined by flow cytometry using anti-FcεRIα or a control Ab. Results are representative of two different mast cell cultures.
Effect of TGF-β1 on occupied IgE receptors
It has been reported that monomeric IgE increases mast cell FcεRI expression (21–23), an effect that could be critical to IgE-elicited inflammatory responses in atopic disease. To determine whether TGF-β1 could alter the effects of monomeric IgE, we cultured BMMC with or without IgE for 4 days to up-regulate FcεRI expression, then added TGF-β1 for an additional 3 days. As shown in Fig. 4, TGF-β1 reduced FcεRI expression on cells that had previously been cultured with IgE. However, the effects of TGF-β1 appeared to be less potent when IgE was present, because the decrease in FcεRI surface expression averaged 31% compared with 50–60% without IgE (Fig. 2). These data indicate that TGF-β1-mediated down-regulation of FcεRI can occur on occupied receptors, but to a lesser extent than on unoccupied receptors.
FIGURE 4.
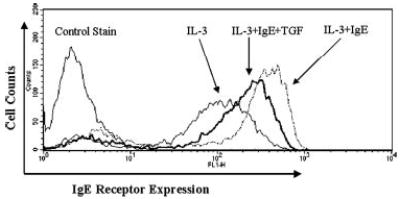
TGF-β1 inhibits IgE-mediated up-regulation of FcεRI surface expression. BMMC were cultured for 4 days in the presence or the absence of IgE (1 μg/ml). On day 4 of culture, cells were fed, and TGF-β1 (5 ng/ml) was added to one set of samples containing IgE for 3 days. Surface FcεRI expression was assessed by flow cytometry. Data shown are one representative histogram from seven BMMC populations that gave similar responses.
Effect of TGF-β on FcεRI subunit mRNA and protein expression
To determine the mechanism by which TGF-β1 negatively regulates expression of the IgE receptor, we assessed mRNA and protein levels of the FcεRIα, -β, and -γ subunits in BMMCs cultured with IL-3 and SCF with or without TGF-β1. No significant differences in mRNA expression were detected by RPA after culture with or without TGF-β1 for 6, 24, or 72 h (Fig. 5). In contrast, TGF-β1 substantially decreased total α, β, and γ protein expression, as shown by Western blotting (Fig. 6). Densitometric analysis showed that the FcεRIα subunit was decreased 34% (n = 2), whereas the β and γ subunits were each inhibited nearly 70% (β, 67.7 ± 11.8% (n = 4); γ, 70.6 ± 14.2% (n = 3)). Collectively, these data indicate that changes in FcεRI transcription or mRNA stability are not the mechanisms by which TGF-β1 decreases IgE receptor expression. Instead, this inhibition appears to be accomplished through a post-transcriptional mechanism that leads to reduced expression of all three FcεRI subunits, particularly the β-and γ-chains.
FIGURE 5.
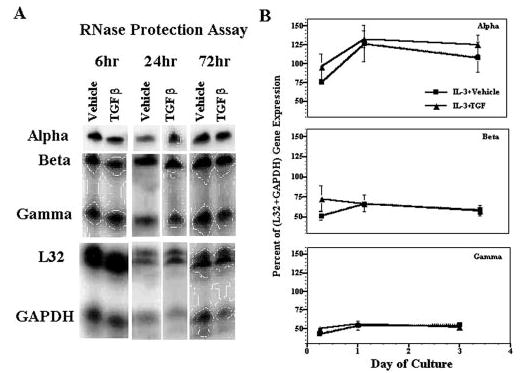
TGF-β1 does not affect FcεRI mRNA expression. A, BMMC were cultured in IL-3 plus vehicle or TGF-β1 (5 ng/ml) for the indicated times. Down-regulation of surface FcεRI expression was confirmed by flow cytometry on day 3 of culture (not shown). The expression of FcεRI subunits and control mRNAs was determined by RPA using total RNA. B, RPA results were quantified by phosphorimaging, using L32 and GAPDH housekeeping gene expression as loading controls. The expression of each FcεRI subunit mRNA relative to the sum of housekeeping gene expression in each lane is shown. Data are the mean and SE of four separate samples.
FIGURE 6.
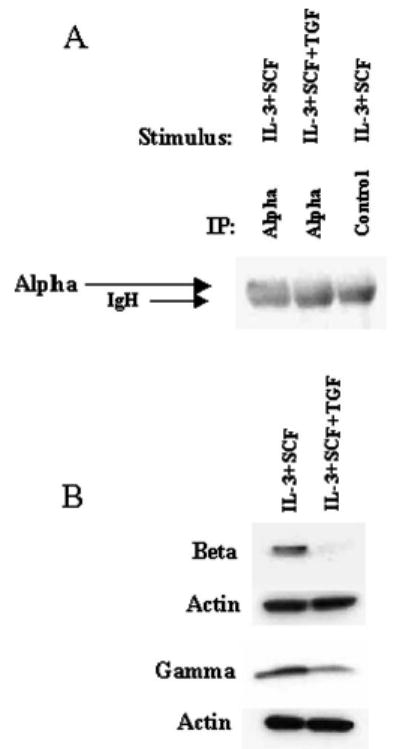
TGF-β1 inhibits the expression of FcεRI subunit proteins. BMMC were cultured in IL-3 and SCF with or without TGF-β1 for 3 days, and down-regulation of surface FcεRI expression was confirmed by flow cytometry. A, Total protein from 30 million cells was immunoprecipitated with anti-FcεRIα or control IgG. Immunoprecipitates were subjected to SDS-PAGE and Western blotting with anti-FcεRI. Data shown are representative of two independent experiments. B, Total protein from ~1 million cells was subjected to SDS-PAGE and Western blotting with anti-FcεRIβ or -γ. The same membranes were stripped and reprobed with anti-actin to demonstrate protein loading. Data shown are representative of three to five independent experiments.
Because post-transcriptional regulation is often used as a means of rapidly controlling expression, we measured the stability of TGF-β1-mediated FcεRI inhibition. In washout experiments, we found that the effects of TGF-β1 were reversible over a 24- to 48-h period. BMMC were cultured for 3 days in the presence of TGF-β1, then washed extensively and replated in IL-3 and SCF with no added TGF-β1. As shown in Fig. 7A, the effect of TGF-β1 was diminished within 24 h, and normal IgE receptor expression was restored within 48 h of TGF-β1 removal. Thus, TGF-β1-mediated inhibition of FcεRI expression was maintained only when this cytokine was present.
FIGURE 7.
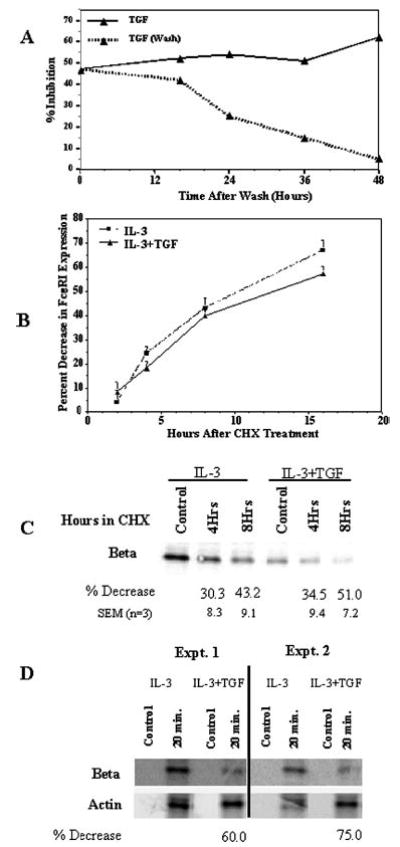
The inhibitory effects of TGF-β1 on surface FcεRI expression are reversible and do not enhance the rate of FcεRI degradation. A, BMMC were cultured for 3 days with or without TGF-β1. Half the TGF-β1-stimulated cells were washed and replated in medium without TGF-β1 on day 3. Over the following 48 h, samples were assessed for surface FcεRI expression by flow cytometry. Percent inhibition of surface FcεRI expression was determined using cultures never receiving TGF-β1, as described in Fig. 1. Data are the mean and SE (too small to be visualized) of eight BMMC populations. B, BMMC were cultured for 3 days in IL-3 and SCF with or without TGF-β1. On day 3, cycloheximide (CHX) or DMSO was added to the cultures for 2–16 h. Using DMSO-treated cells as a control, the CHX-mediated decrease in surface FcεRI expression on cells cultured with or without TGF-β1 was assessed by flow cytometry. Data shown are the mean and SE of 12 samples. C, Cells were cultured and treated with CHX or DMSO as described in B. Total cell lysates from 1 million cells were subjected to SDS-PAGE and Western blotting to detect FcεRI β-chain expression. The percent decrease in expression obtained from three experiments is shown below the respective bands. D, BMMC were cultured in IL-3 with or without TGF-β1 for 3 days. Cells were starved of cysteine and methionine (control), then incubated with 35S-labeled cysteine and methionine for 20 min. FcεRI β-chain and actin were sequentially immunoprecipitated and resolved by gel electrophoresis as described in Materials and Methods.
Because the inhibitory effect of TGF-β1 occurred post-transcriptionally, it was possible that diminished FcεRI expression was mediated by degradation of the receptor subunits. To address this issue, we treated BMMCs that had been cultured for 3 days in the presence or the absence of TGF-β1 with the protein synthesis inhibitor cycloheximide and assessed surface FcεRI expression by flow cytometry as well as total FcεRIβ subunit expression by Western blotting. The day 3 time point was chosen because it demonstrated maximal effects of TGF-β1 that were maintained through at least day 7 (Fig. 2B). As shown in Fig. 7, B and C, cycloheximide treatment decreased the expression of surface FcεRI and total FcεRIβ subunit at rates that were not affected by the presence of TGF-β1. These data indicated that TGF-β1 inhibits FcεRI expression by a reversible post-transcriptional mechanism that does not involve enhanced receptor endocytosis or protein degradation.
One explanation for reduced FcεRI expression in the absence of altered endocytosis or degradation could be that TGF-β1 reduces the rate of FcεRI protein synthesis. We determined the effect of TGF-β1 on the rate of FcεRI β-chain synthesis. BMMC cultured for 3 days in IL-3 and TGF-β1 before incubation with 35S-labeled methionine and cysteine showed a significant reduction in FcεRI β-chain synthesis compared with cells cultured in IL-3 alone (Fig. 7D). Using actin production as a reference, TGF-β1-treated cells showed a 60–75% reduction in FcεRI β-chain protein production. These data argue that TGF-β1 controls expression of the mast cell IgE receptor by inhibiting FcεRI protein synthesis, leading to reduced surface expression and function.
Discussion
IgE-mediated mast cell activation is a central tenet in atopic diseases such as allergic asthma. The incidence of these diseases has risen dramatically in recent years, emphasizing the importance of understanding and controlling mast cell function. Our efforts have focused on mast cell homeostasis: the regulation of mast cell development, function, and survival. Because mast cells provide critical resistance to bacterial and parasitic infections, but also can elicit inflammatory cascades related to atopy, arthritis, multiple sclerosis, and heart disease, this cellular homeostasis is paramount in balancing health and disease.
Mast cells are responsive to many cytokines that can provide homeostatic control. We have recently found that IL-4 and IL-10 exert such effects partly through control of FcεRI expression. To gain a complete view of how cytokines control mast cell activity, we have sought other factors capable of similar regulatory function. Recently, Tridandapani et al. (24) demonstrated that TGF-β1 could inhibit FcR γ-chain expression on myeloid cells and monocytes. Because this subunit is shared among several FcRs, including FcεRI, it appeared possible that TGF-β1 might regulate mast cell FcεRI expression. Furthermore, TGF-β1 diminishes IgE-mediated histamine release and TNF-α production in vitro (14) and inhibits in vivo mast cell responses (15). TGF-β1 has also been shown to inhibit IL-3-, IL-4-, and SCF-mediated signaling in mast cells, decreasing survival and proliferation (11–13). In contrast, TGF-β1 has been reported to elicit mast cell migration (25, 26). Hence, like IL-4 and IL-10, TGF-β1 appears to have both positive and negative effects on mast cell function. These effects might be mediated by mast cells in an autocrine fashion, because mast cells produce IL-4, IL-10, and TGF-β1. Furthermore, mast cell proteases are reportedly capable of cleaving latent TGF-β1 (27, 28). It is also interesting to note that polymorphisms in the TGF-β1 promoter region have been shown to segregate with asthmatic families that have elevated serum IgE (29). These data encouraged our interest in TGF-β1 as a mediator of mast cell homeostasis.
We show in the current work that TGF-β1 inhibits mast cell FcεRI surface expression, with peak effects after 3 days of culture. This inhibition was consistently found in cultured mouse mast cells, freshly isolated peritoneal mouse mast cells, and cultured human mast cells, making it unlikely that our data are related to culture artifacts or species differences. The effects of TGF-β1 were reproduced using the related cytokines TGF-β2 and TGF-β3, which, like TGF-β1, signal through TGF-β receptors I and II (1). This inhibitory signaling was sensitive, occurring at a concentration of 1 ng/ml. Mast cells express >100,000 FcεRI molecules/cell, a fraction of which need to be aggregated to elicit signaling (8). Therefore, it is interesting to note that TGF-β1 reduced surface FcεRI expression ~60%, leaving many more receptors than needed for activation. Consistent with other studies, we found that the reduction in FcεRI expression correlated with significantly reduced IgE-mediated activation. Specifically, TGF-β1 inhibited IgE-mediated mast cell degranulation and TNF-α production, as shown by Bissonette et al. (14). We also found that production of the inflammatory cytokine IL-6 was decreased. Furthermore, the ability of monomeric IgE to increase FcεRI expression was dampened by TGF-β1, an effect that could be critical in patients with chronically elevated serum IgE. Thus, the effects of TGF-β1 were consistent among distinct mast cell populations and were functionally relevant.
The timing of TGF-β1-mediated inhibitory effects was reminiscent of our previous work with IL-4 and IL-10, both of which diminished FcεRI expression after 3 days of culture (18, 20). We have postulated that this delay in inhibitory signaling may frame an inflammatory window during which mast cell responses elicit inflammation to control infection. IL-4, IL-10, and TGF-β1 would be produced during the T cell- and mast cell-mediated responses that characterize inflammation elicited by IgE. It is intriguing that these cytokines may initially offer survival or migration signals to mast cells, but subsequently inhibit mast cell function and survival. Both the timing and the effects of this cytokine-mediated response fit a description of homeostasis.
The similarities among IL-4, IL-10, and TGF-β1 were partly conserved in their mechanism of action. Like IL-4 and IL-10, TGF-β1 inhibited FcεRI expression without significantly altering the expression of the mRNAs encoding the three FcεRI subunits. Instead, the protein expression of all three subunits was decreased after TGF-β1 treatment and occurred without any notable change in protein stability. Like the study by Tridandapani et al. (24), we noted a decrease in FcR γ-chain expression. We also noted a similar decrease in FcεRI β-chain expression. This is an interesting point of regulation, because IL-4 and IL-10 also reduced FcεRI β expression. As shown by several studies, FcεRIβ is a central regulator of FcεRI expression and function, serving as an amplifier of the IgE response (30–33). β-Chain targeting by these cytokines emphasizes the importance of this subunit as a point of control in IgE receptor function.
The inhibition of FcεRI expression by TGF-β1 may be mediated at the level of protein synthesis. Thus, TGF-β1-stimulated BMMC synthesized FcεRIβ at a rate at least 60% slower than that in control cells. Our data provide a model in which TGF-β1 reduces IgE receptor expression by controlling the rate of translation without altering mRNA expression or the stability of the proteins produced. This model is supported by a recent paper demonstrating the phosphorylation of eukaryotic initiation factor 2α by TGF-β1 receptor (34), which could decrease the synthesis of some proteins. Post-transcriptional control is typically used for flexible, rapid changes in expression, consistent with the reversibility of the TGF-β1 effects we observed. In fact, the effects of TGF-β1 were fairly transient, with a loss of inhibition 36–48 h after TGF-β1 was removed from the culture medium. Hence, the reversible nature of the effects of TGF-β1 fits our theory of how this cytokine alters IgE receptor expression.
These data support the theory that TGF-β1 produced by mast cells or other cells involved in the inflammatory cascade provides a delayed and reversible inhibition of IgE-mediated mast cell inflammatory function. The delay in its effects is postulated to allow protective mast cell responses, which are dampened after 3 days to avert chronic inflammation. These effects are exerted at the level of protein production, creating a nimble homeostatic system to control the potent effector functions of mast cells.
Footnotes
This work was supported in part by grants to the Ryan laboratory from the National Institutes of Health (1RO1AI43433 and 1R01CA91839).
Abbreviations used in this paper: BMMC, bone marrow-derived mast cell; cRPMI, complete RPMI 1640 medium; RPA, RNase protection assay; SCF, stem cell factor.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Massague J. The transforming growth factor-β family. Annu Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- 2.Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 3.ten Dijke P, Hill CS. New insights into TGF-β SMAD signaling. Trends Biochem Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor β in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 5.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-β. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 6.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor β1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rivera J. Molecular adapters in FcεRI signaling and the allergic response. Curr Opin Immunol. 2002;14:688–693. doi: 10.1016/s0952-7915(02)00396-5. [DOI] [PubMed] [Google Scholar]
- 9.Secor VH, Secor WE, Gutekunst CA, Brown MA. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med. 2000;191:813–821. doi: 10.1084/jem.191.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297:1689–1692. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- 11.Toyota N, Hasimoto Y, Matsuo S, Izuka H. Transforming growth factor β1 inhibits IL-3- and IL-4-dependent mouse connective tissue-type mast cell proliferation. Arch Dermatol Res. 1995;287:198–201. doi: 10.1007/BF01262332. [DOI] [PubMed] [Google Scholar]
- 12.Broide DH, Wasserman SI, Alvaro-Gracia J, Zvaifler NJ, Firestein G. Transforming growth factor-β1 selectively inhibits IL-3-dependent mast cell proliferation without affecting mast cell function or differentiation. J Immunol. 1989;143:1591–1597. [PubMed] [Google Scholar]
- 13.Mekori YA, Metcalfe DD. Transforming growth factor-β prevents stem cell factor-mediated rescue of mast cells from apoptosis after IL-3 deprivation. J Immunol. 1994;153:2194–2203. [PubMed] [Google Scholar]
- 14.Bissonnette EY, Enciso JA, Befus AD. TGF-β1 inhibits the release of histamine and tumor necrosis factor-α from mast cells through an autocrine pathway. Am J Respir Cell Mol Biol. 1997;16:275–282. doi: 10.1165/ajrcmb.16.3.9070612. [DOI] [PubMed] [Google Scholar]
- 15.Meade R, Askenase PW, Geba GP, Neddermann K, Jacoby RO, Pasternak RD. Transforming growth factor-β1 inhibits murine immediate and delayed type hypersensitivity. J Immnuol. 1992;149:521–528. [PubMed] [Google Scholar]
- 16.Kim HM, Lee YM. Role of TGF-β1 on the IgE-dependent anaphylaxis reaction. J Immnuol. 1999;162:4960–4965. [PubMed] [Google Scholar]
- 17.Kepley CL, Taghavi S, Mackay G, Zhu D, Morel PA, Zhang K, Ryan JJ, Satin LS, Zhang M, Pandolfi PP, P P, et al. Co-aggregation of FcγRII with FcεRI on human mast cells inhibits antigen-induced secretion and involves SHIP-Grb2-Dok complexes. J Biol Chem. 2004;279:35139–35149. doi: 10.1074/jbc.M404318200. [DOI] [PubMed] [Google Scholar]
- 18.Ryan JJ, Desimone S, Klisch G, Shelburne C, McReynolds LJ, Han K, Kovacs R, Mirmonsef P, Huff TF. IL-4 inhibits mouse mast cell FcεRI expression through a STAT6-dependent mechanism. J Immunol. 1998;161:6915. [PubMed] [Google Scholar]
- 19.Schwartz LB, Austen K, Wasserman SI. Immunological release of β-hexosminidase and β-glucuronidase from purified rat serosal mast cells. J Immunol. 1979;123:1445–1450. [PubMed] [Google Scholar]
- 20.Gillespie SR, DeMartino RR, Zhu J, Chong HJ, Ramirez C, Shelburne CP, Bouton LA, Bailey DP, Gharse A, Mirmonsef P, et al. IL-10 inhibits FcεRI expression in mouse mast cells. J Immunol. 2004;172:3181–318. doi: 10.4049/jimmunol.172.5.3181. [DOI] [PubMed] [Google Scholar]
- 21.Malveaux FJ, Conroy MC, Adkinson NF, Jr, Lichtenstein LM. IgE receptors on human basophils: relationship to serum IgE concentration. J Clin Invest. 1978;62:176–181. doi: 10.1172/JCI109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furuichi K, Rivera J, Isersky C. The receptor for immunoglobulin E on rat basophilic leukemia cells: effect of ligand binding on receptor expression. Proc Natl Acad Sci USA. 1985;82:1522–1525. doi: 10.1073/pnas.82.5.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamaguchi M, Lantz CS, Oettgen HC, Katona IM, Fleming T, Miyajima I, Kinet JP, Galli SJ. IgE enhances mouse mast cell FcεRI expression in vitro and in vivo: evidence for a novel amplification mechanism in IgE-dependent reactions. J Exp Med. 1997;185:663. doi: 10.1084/jem.185.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tridandapani S, Wardrop R, Baran CP, Wang Y, Opalek JM, Caligiuri MA, Marsh CB. TGF-β1 suppresses myeloid Fcγ receptor function by regulating the expression and function of the common γ-subunit. J Immunol. 2003;170:4572–457. doi: 10.4049/jimmunol.170.9.4572. [DOI] [PubMed] [Google Scholar]
- 25.Olsson N, Piek E, Sundstrom M, ten Dijke P, Nilsson G. Transforming growth factor-β-mediated mast cell migration depends on mitogen-activated protein kinase activity. Cell Signal. 2001;13:483–490. doi: 10.1016/s0898-6568(01)00176-0. [DOI] [PubMed] [Google Scholar]
- 26.Olsson N, Piek E, ten Dijke P, Nilsson G. Human mast cell migration in response to members of the transforming growth factor-β family. J Leukocyte Biol. 2000;67:350–356. doi: 10.1002/jlb.67.3.350. [DOI] [PubMed] [Google Scholar]
- 27.Taipale J, Lohi J, Saarinen J, Kovanen PT, Keski-Oja J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-β1 from the extracellular matrix of cultured human epithelial and endothelial cells. J Biol Chem. 1995;270:4689–4696. doi: 10.1074/jbc.270.9.4689. [DOI] [PubMed] [Google Scholar]
- 28.Lindstedt KA, Wang Y, Shiota N, Saarinen J, Hyytiainen M, Kokkonen JO, Keski-Oja J, Kovanen PT. Activation of paracrine TGF-β1 signaling upon stimulation and degranulation of rat serosal mast cells: a novel function for chymase. FASEB J. 2001;15:1377–1388. doi: 10.1096/fj.00-0273com. [DOI] [PubMed] [Google Scholar]
- 29.Hobbs K, Negri J, Klinnert M, Rosenwasser LJ, Borish L. Interleukin-10 and transforming growth factor-β promoter polymorphisms in allergies and asthma. Am J Respir Crit Care Med. 1998;158:1958–1962. doi: 10.1164/ajrccm.158.6.9804011. [DOI] [PubMed] [Google Scholar]
- 30.Takai R, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR γchain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 31.Lin S, Cicala C, Scharenberg AM, Kinet JP. The FcεRIβ subunit functions as an amplifier of FcεRI mediated cell activation signals. Cell. 1996;85:22076. doi: 10.1016/s0092-8674(00)81300-8. [DOI] [PubMed] [Google Scholar]
- 32.Dombrowicz D, Lin S, Flamand V, Brini A, Koller B, Kinet JP. Allergy-associated FcRβ is a molecular amplifier of IgE- and IgG-mediated in vivo responses. Immunity. 1998;8:517–529. doi: 10.1016/s1074-7613(00)80556-7. [DOI] [PubMed] [Google Scholar]
- 33.Donnadieu D, Jouvin MH, Kinet JP. A second amplifier function for the allergy-associated FcεRI-β subunit. Immunity. 2000;12:515. doi: 10.1016/s1074-7613(00)80203-4. [DOI] [PubMed] [Google Scholar]
- 34.McGonigle S, Beall MJ, Pearce EJ. Eukaryotic initiation factor 2α subunit associates with TGFβ receptors and 14-3-3ε and acts as a modulator of the TGFβ response. Biochemistry. 2002;41:579–587. doi: 10.1021/bi011407z. [DOI] [PubMed] [Google Scholar]