

Abstract
Cellular protein folding is challenged by environmental stress and aging, which lead to aberrant protein conformations and aggregation. One way to antagonize the detrimental consequences of protein misfolding is to reactivate vital proteins from aggregates. In the yeast Saccharomyces cerevisiae, Hsp104 facilitates disaggregation and reactivates aggregated proteins with assistance from Hsp70 (Ssa1) and Hsp40 (Ydj1). The small heat shock proteins, Hsp26 and Hsp42, also function in the recovery of misfolded proteins and prevent aggregation in vitro, but their in vivo roles in protein homeostasis remain elusive. We observed that after a sublethal heat shock, a majority of Hsp26 becomes insoluble. Its return to the soluble state during recovery depends on the presence of Hsp104. Further, cells lacking Hsp26 are impaired in the disaggregation of an easily assayed heat-aggregated reporter protein, luciferase. In vitro, Hsp104, Ssa1, and Ydj1 reactivate luciferase:Hsp26 co-aggregates 20-fold more efficiently than luciferase aggregates alone. Small Hsps also facilitate the Hsp104-mediated solubilization of polyglutamine in yeast. Thus, Hsp26 renders aggregates more accessible to Hsp104/Ssa1/Ydj1. Small Hsps partially suppress toxicity, even in the absence of Hsp104, potentially by sequestering polyglutamine from toxic interactions with other proteins. Hence, Hsp26 plays an important role in pathways that defend cells against environmental stress and the types of protein misfolding seen in neurode-generative disease.
Members of the sHsp1 family protect cells from a variety of environmental conditions such as heat and oxidative stress by antagonizing protein aggregation (1–3). Most of them form dynamic oligomeric structures ranging from 9 to 50 subunits (4). sHsps share a conserved α-crystallin domain of 80–100 amino acids at their C terminus (4), whereas their N-terminal regions are highly variable in sequence and length (5).
In the yeast, Saccharomyces cerevisiae, there are two sHsps, Hsp26 and Hsp42. Both proteins are highly expressed when yeast cells enter respiratory growth in preparation for transition to stationary phase or when subjected to moderate heat stress (6). Although yeast cells deleted for HSP26 or HSP42 show no overt heat sensitivity or thermotolerance defects, they do accumulate protein aggregates (6, 7). The aggregated proteins in both deletion mutants overlap substantially, suggesting at least partially overlapping roles of Hsp26 and Hsp42 (6).
In vitro, Hsp26 and Hsp42 form distinct oligomeric complexes and assemble into large co-complexes with aggregation-prone proteins upon heat shock (6, 8–10). It has been proposed that sHsps aid in the refolding of denatured proteins by holding them in a reactivation-competent state (4, 11). Reactivation of aggregated proteins in yeast is carried out by Hsp104 with assistance from the Hsp70/Hsp40 chaperone system both in vivo and in vitro (12). Hsp104 is a member of the AAA+ superfamily of proteins that is generally associated with structural remodeling of proteins and protein complexes. Recent work demonstrates that the prokaryotic homolog of Hsp104, ClpB, achieves disaggregation by active unfolding of protein aggregates followed by refolding by Hsp70/Hsp40. Also, the prokaryotic sHsps, IbpA/B, were shown to assist in the reactivation of aggregated substrates by ClpB, Hsp70, and Hsp40 (13, 14).
Here, we demonstrate that Hsp26 facilitates the disaggregation of aggregated proteins after heat shock by Hsp104 in vivo and in vitro. This is achieved by the co-assembly of Hsp26 with misfolded proteins that bolster access of the disaggregation machinery composed of Hsp104/Hsp70 and Hsp40. As an example of sHsp action relevant to protein misfolding diseases in humans, we demonstrate that sHsps together with Hsp104 antagonize polyglutamine aggregation and toxicity.
MATERIALS AND METHODS
General Yeast Methods, Strains, and Plasmids
Yeast media were prepared using complete supplemental mixtures from Q-BIOGene, Irvine, CA. All strains were constructed in W303 background and are listed in supplemental Table I. Gene deletion strains were constructed using the short flanking homology method using kanamycin or hygromycin resistance cassettes as described (15). Primers used for gene deletions are listed in supplemental Table II. Transformation of yeast was performed as described in Ito et al. (16). Yeast integrating plasmids for the expression of huntingtin exon I-CFP fusions (25Q and 72Q in pRS303 plasmid backbone,2 were linearized by restriction with BstXI prior to transformation. Plasmids for the overexpression of Hsp104, Hsp26, and Hsp42 are described in supplemental Table III.
For growth assays, required strains were grown overnight in selective media. Cell densities were equalized and spotted in 5-fold dilutions on appropriate selective medium using a spotter (Frogger, V&P Scientific, Inc). The plates were documented after 2–3 days of incubation at 30 °C.
Filter Retardation Assay
Protein extractions for immunoblotting were performed essentially as described previously (17) and filter retardation assays of aggregated material were performed essentially as described (18). Dot blots were performed exactly like the filter retardation assays except using polyvinylidene difluoride membranes instead of cellulose acetate. For the induction of expression of polyglutamine huntingtin fragment in yeast, cultures were grown at 30 °C in raffinose containing liquid medium (2%) and transferred to galactose containing medium. After 8 h of growth in galactose, cells were harvested for protein extraction and filter retardation assays.
Proteins
Hsp104, Ssa1, and Ydj1 were purified as described earlier (19). Purified Hsp26 and antibodies to Hsp26 and Hsp42 were kind gifts of Professor Johannes Buchner and Dr. Martin Haslbeck at Technische Universität München, Garching, Germany. Firefly luciferase and the assay kit were purchased from Promega Corporation.
Heat Shock and Protein Extraction
Starter cultures were grown overnight in YPD medium (yeast extract, peptone, and dextrose) at 30 °C. 70 ml of YPD/strain was inoculated using the starter cultures at a density of 0.05 absorbance units at 600 nm (A600). They were allowed to grow at 30 °C till they reached an A600 value of 0.5 (mid-log phase).Cultures were subjected to 37 °C for 30 min (pretreatment) followed by 46 °C for 30 or 60 min (sublethal heat shock). The cultures were allowed to recover at 30 °C after addition of cycloheximide (10 μg/ml) to block protein synthesis. 10-ml samples were removed at various stages as indicated. The cell pellets were washed with water and frozen at −80 °C until use.
Protein samples were prepared by resuspending cell pellets in 220 μl of lysis buffer (20 mm sodium phosphate (pH 6.9), 10 mm dithiothreitol, 0.1% Tween 20, 0.2 mg/ml zymolyase (Seikagaku) and 25 units of Benzonasetm nuclease (Novagen) containing 1 tablet of Completetm protease inhibitor mixture in 5 ml) incubated for 20 min at 25 °C, followed by sonication in a cup horn sonicator (Virsonic) with 6 × 5-s pulses of 50 watts. Cell ghosts were removed by centrifugation at 2000 rpm for 1 min. 200 μl of supernatant were collected as the total protein extract. The protein concentrations in all samples were equalized. 100 μl of the total extract was subjected to centrifugation at 14,000 × g for 30 min to separate the soluble and aggregated proteins. 100-μl supernatants were removed into fresh tubes, and the pellets were resuspended in 100 μl of lysis buffer. Two-dimensional PAGE was performed following manufacturer’s instructions (Amersham Biosciences) using the DryS-trip (pH 4–7) for first dimension and ExcelGel SDS 12–14% for second dimension with the Multiphor II system. Each spot was dried in a 1.5-ml tube before submitting them for protein identification at the Howard Hughes Medical Institute Biopolymers Facility at the University of Texas South Western. For analysis by SDS-PAGE, all samples were boiled for 10 min after the addition of SDS-sample loading buffer. For Western blotting, 10 μl of each sample was separated by SDS-PAGE, transferred to polyvinylidene difluoride membrane, and probed with antibodies against Hsp26 or Hsp42.
For thermotolerance assays, cells were pretreated at 37 °C for 30 min followed by 48 or 50 °C (lethal heat shock) for varying lengths of time. Samples after each treatment were chilled on ice. The survival of cells was assayed by spotting 5 μl of 5-fold serial dilutions on to YPD plates.
Hsp104-dependent Reactivation Assays
In vivo protein disaggregation assays were performed as described earlier (12). In vitro assays for reactivation of aggregated firefly luciferase (FFL) were performed essentially as described in Glover et al. (19) with the following changes. Denaturation of FFL was carried out by heating at 45 °C for 15 min in the absence or presence of indicated concentrations of Hsp26. Heat-treated FFL samples were added to reactions with indicated chaperones. ATP regeneration system comprising creatine phosphate (15 mm) (Roche Diagnostics) and creatine kinase (0.5 μm) was included. Reactivated FFL activity was measured using the luciferase assay system (Promega)
Dynamic Light Scattering
Hsp26 oligomer (0.4 μm) and firefly luciferase (0.1 or 1 μm) individually or together were centrifuged at 14,000 × g for 1 min to clear large particles. Diffusion coefficients were measured by monitoring intensity of scattered laser light (589 nm) using a Protein Solutions DynaPro instrument equipped with a temperature controlled microsampler. Samples were heated to 45 °C for 5 min followed by cooling to 25 °C. Fifty data points were acquired for each measurement. The viscosity of the buffer at 25 °C of 0.891 centipoise was used for calculations.
RESULTS
Solubility of Hsp26 in Response to Heat Shock
To identify substrates and cofactors of Hsp104-mediated protein disaggregation in yeast, we analyzed proteins that accumulate in aggregates after heat shock and depend on Hsp104 for resolubilization. Using two-dimensional PAGE, we compared proteins from the soluble and the insoluble fractions of wild-type (WT) and Δhsp104 cells. Proteins were examined: 1. before a non-lethal heat shock, 2. immediately thereafter and 3. after recovery (Fig. 1 and data not shown). Several proteins became insoluble after heat shock and were resolubilized in WT but not in Δhsp104 cells. These proteins were identified by mass spectrometry. Among them, Hsp26 was very abundant and was identified with high confidence (supplemental Table IV).
Fig. 1. Two-dimensional PAGE analysis of soluble and insoluble proteins after recovery from heat shock.
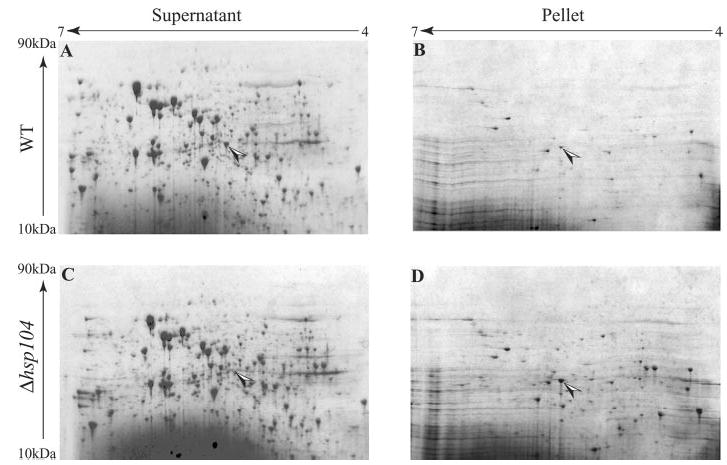
Wild-type and Δhsp104 cells were pretreated at 37 °C for 30 min to induce the expression of heat shock proteins followed by a sub-lethal heat shock at 46 °C for 30 min. Protein synthesis was blocked by the addition of cycloheximide (10 μg/ml). The cells were allowed to recover at 25 °C for 2 h. Soluble and insoluble proteins were analyzed by two-dimensional PAGE. The spot corresponding to Hsp26 is marked with an arrow.
The results were confirmed by Western blotting using antibodies against Hsp26 (Fig. 2A). Heat shock induced the expression of Hsp26 severalfold over untreated levels. Immediately after the heat treatment, most Hsp26 had become insoluble in both the WT and Δhsp104 cells. After recovery at 25 °C for 1 h, the majority of Hsp26 was resolubilized in WT cells but remained insoluble in Δhsp104 cells.
Fig. 2. Solubility of Hsp26 and Hsp42 after heat shock in WT and Δhsp104 cells.
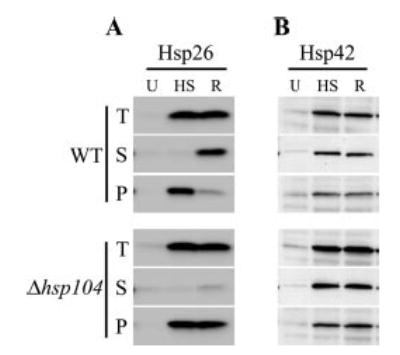
A, Western blot with Hsp26 antibody. B, Western blot with Hsp42 antibody. Samples were untreated (U), subjected to heat shock at 37 °C for 30 min followed by 46 °C for 30 min (HS) and allowed for recovery from heat shock for 1 h (R). Lysates (T, total) were centrifuged to separate supernatant (S) and pellet (P) fractions.
We next tested by Western blot whether the solubility of Hsp42, the other sHsp in yeast, might also depend on the presence of Hsp104 after heat shock. Approximately 50% of Hsp42 was detected in the insoluble fraction regardless of previous heat treatment (Fig. 2B). Upon heat treatment Hsp42 levels increased severalfold, but its solubility in WT and Δhsp104 cells remained similar.
In the accompanying paper, Haslbeck et al. (36) also observed that Hsp26 is the major protein found in the insoluble fraction of yeast cells after heat shock. These observations reveal a functional interaction between Hsp26 (but not necessarily Hsp42) and Hsp104, wherein Hsp26 is sequestered into the insoluble fraction after heat shock and is resolubilized in an Hsp104-dependent manner during recovery.
Thermotolerance of Mutants
To understand the role of sHsps in thermotolerance, we tested the recovery of sHsp deletion strains after a severe heat shock. Previous work demonstrated that neither Hsp26 nor Hsp42 was required for thermotolerance (survival after a severe heat shock) (7, 10). In agreement, we saw no thermotolerance defect in Δhsp26 or Δhsp42 cells (Fig. 3). Even the Δhsp26Δhsp42 double mutant had wild-type levels of thermotolerance. However, Δhsp104 cells have a 5-fold higher rate of survival than Δhsp104 Δhsp26 double deletions after a short (10 min) heat shock at 50 °C. The synthetic effect of the deletions of Hsp104 and Hsp26 in our experiments suggests a functional link between these chaperones in response to heat shock and the ensuing protein disaggregation. Surprisingly, the deletion of Hsp42 in Δhsp104 cells does not show this effect, revealing a non-overlapping function of Hsp26 and Hsp42 in thermotolerance.
Fig. 3. Thermotolerance of various mutants.

Cells were either untreated or given a tolerance-inducing pretreatment at 37 °C for 30 min followed by a 10- or 20 min lethal heat shock at 50 °C. 5 μl of cells were spotted on YPD plates after 5-fold serial dilutions. The strains are as follows: 1, WT; 2, Δhsp104; 3, Δhsp26; 4, Δhsp26Δhsp104; 5, Δhsp42; 6, Δhsp42Δhsp104; 7, Δhsp26Δhsp42; 8, Δhsp26Δhsp42Δhsp104.
In Vivo Effect of Hsp26 Deletion on Hsp104-dependent Reactivation
We hypothesized that Hsp26 binds unfolded proteins after heat shock and thereby enables Hsp104/Ssa1/Ydj1 to resolubilize aggregates more efficiently. To test this, we investigated the Hsp104-dependent disaggregation of a temperature-sensitive protein whose functional state is readily quantified (a previously described luciferase variant (12, 20)). Each strain constitutively expressing luciferase was pretreated at 37 °C for 30 min and then given a sublethal heat shock at 46 °C for 30 min (12). Protein synthesis was inhibited by the addition of cycloheximide after the heat shock to focus on the reactivation of luciferase synthesized prior to heat shock.
The Hsp26 deletion did not affect the activity of luciferase at 25 °C or during the conditioning pretreatment at 37 °C. The sublethal heat shock reduced the luciferase activity to <5% in mid-log phase cells (Fig. 4). Cells deleted for Hsp104 did not recover luciferase activity, regardless of the presence of sHsps (Fig. 4 and data not shown).
Fig. 4. In vivo reactivation of aggregated luciferase.
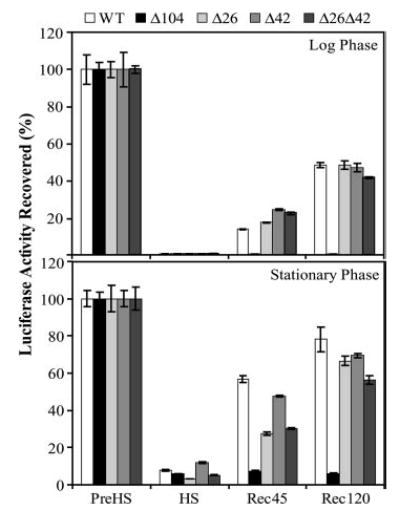
Mutant strains (as indicated) were transformed with plasmid carrying the heat-sensitive luciferase gene. Cells were subjected to pretreatment at 37 °C for 30 min followed by sublethal heat shock at 46 °C for 60 min. Protein synthesis was inhibited by the addition of cycloheximide to a final concentration of 10 μg/ml. The disaggregation of aggregated luciferase was followed post-heat treatment by measuring luciferase activity. Luciferase activity in pretreated cells was set to 100%.
Cells deleted for Hsp26 reactivated luciferase to a similar extent to WT when grown in mid-log phase. In the stationary phase however, Δhsp26 cells reactivated luciferase at a significantly slower rate than WT cells. The increased dependence of stationary phase cells on Hsp26 for disaggregation is also consistent with the role of sHsps in senescence (21–23).
In Vitro Effect of Hsp26 on Hsp104-dependent Reactivation
To investigate mechanistic aspects of the cooperation between Hsp104/Ssa1/Ydj1 and Hsp26 we used purified proteins in aggregation and disaggregation assays. The Hsp104-dependent return of Hsp26 to the soluble state after heat shock likely reflects its ability to capture denatured proteins and keep them in a refolding competent state.
FFL aggregates were prepared by heating FFL at 45 °C in the absence and presence of various concentrations of Hsp26. Heat not only causes the aggregation of FFL but it also activates Hsp26 by disassembling the oligomers (9). Moreover, heat-aggregated FFL on its own is a poor substrate for Hsp104 (19), so we used it to test the effects of Hsp26 (Fig. 5A). Disaggregation reactions were initiated by diluting FFL aggregates 10-fold into reaction mixtures containing no chaperones, Hsp104 alone, Ssa1 and Ydj1, or all three chaperones.
Fig. 5. Hsp26 facilitates reactivation of aggregated FFL by the Hsp104/Ssa1/Ydj1 chaperone machinery.
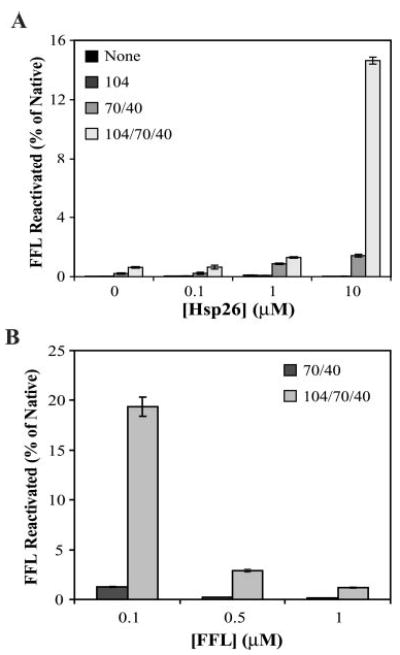
A, titrating Hsp26 (monomer concentrations are indicated). FFL concentration was 0.1 μm. Hsp26-FFL co-complexes were made by heating them at 45 °C for 15 min and diluting 20-fold into chaperone mixtures containing 1 μm each of Hsp104, Ssa1, and Ydj1 and 5 mm ATP. Native FFL activity at the same concentration was set to 100%. B, titrating FFL (concentrations are indicated). Hsp26 monomer concentration was kept constant at 10 μm. Other conditions are as indicated for A.
When Hsp26 was present during the heat denaturation of FFL, it improved resolubilization in a dose-dependent manner (Fig. 5A). Hsp104/Ssa1/Ydj1-mediated FFL reactivation was ~20-fold more efficient when FFL aggregates were prepared in the presence of Hsp26 than in its absence. Replacing Hsp26 with another protein such as bovine serum albumin did not influence FFL reactivation (data not shown). When Hsp26 was added after the aggregation of FFL it did not facilitate disaggregation by Hsp104 (FFL activity was not detectable in all samples; data not shown) indicating that formation of the Hsp26-FFL co-complex was essential for reactivation. These observations provide the first direct evidence for cooperation between Hsp26 and Hsp104/Ssa1/Ydj1 chaperones in the reactivation of aggregated proteins.
Increasing FFL concentration with a constant Hsp26 concentration drastically reduced the efficiency of reactivation (Fig. 5B). These results suggest that a Hsp26 oligomer:FFL ratio of 2:5 was optimal for FFL as a substrate under these conditions. Our results argue that Hsp26 facilitates protein disaggregation by the Hsp104/Ssa1/Ydj1 machinery in a concentration-dependent manner. Importantly, Hsp26 can only perform its solubilizing function when it co-aggregates with the protein substrate and not after substrate aggregation.
Physical Nature of Hsp26-FFL Interactions
The simplest explanation for the relative inefficiency of FFL reactivation at low Hsp26:FFL ratios, is that Hsp26 was unable to capture all of the FFL. To investigate this we studied the physical nature of Hsp26-FFL complexes by dynamic light scattering (Fig. 6 and supplemental Table V).
Fig. 6. Complex formation between Hsp26 and FFL monitored by dynamic light scattering.
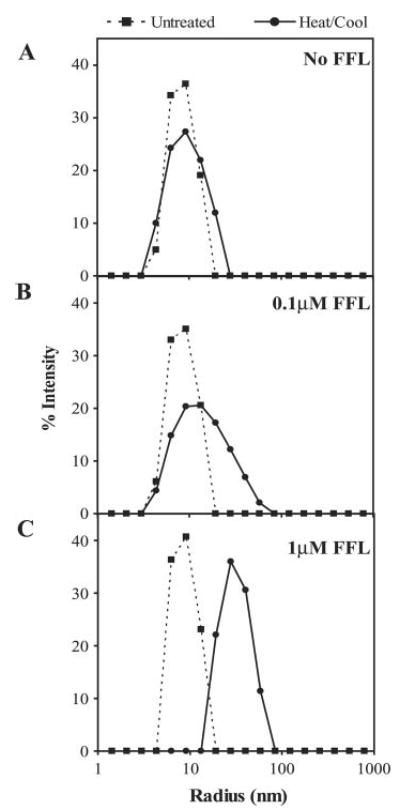
Samples were 0.42 μm Hsp26 oligomer (A), 0.42 μm Hsp26 oligomer with 0.1 μm FFL (B), and 0.42 μm Hsp26 oligomer with 1 μm FFL (C). Dotted line, untreated (25 °C); solid line, heated at 45 °C for 10 s followed by cooling to 25 °C.
In the absence of a misfolded client protein, Hsp26 is an oligomer of ~18 nm in diameter (Fig. 6) and has been shown to disassemble at 45 °C (9). FFL aggregated into very large particles when heated to 45 °C without Hsp26 (>300 nm, data not shown). However, when the two proteins are mixed at a ~4:1 or a ~1:2 ratio (Hsp26 oligomer:FFL) and heated to 45 °C no large aggregates were observed. Instead, the measured particle diameter was ~33 or ~66 nm respectively.
Thus, Hsp26 is able to capture all of the FFL even when Hsp26:FFL ratios were low. However, complexes formed with high Hsp26 concentrations lead to the formation of smaller particles. These are readily resolubilized by Hsp104/Ssa1/Ydj1. At lower Hsp26 concentrations, larger particles are formed, which are more resilient to resolubilization.
sHsps Modulate Polyglutamine Aggregation and Toxicity
We next tested whether the function of Hsp26 and the Hsp104/Ssa1/Ydj1 disaggregation machinery applies to proteins involved in human protein misfolding disease. Previously, our laboratory and others demonstrated that yeast serves as a useful model for studying human neurodegenerative diseases involving protein aggregation such as Huntington disease (24, 25) and Parkinson disease (26). We tested whether deletion or overexpression of Hsp26, Hsp42, Hsp104, or various combinations thereof, would affect the toxicity conferred by expanded polyglutamine region (72Q) of human huntingtin. Yeast cells expressing a normal polyglutamine stretch (25Q) under identical conditions were used as a control.
Deletions of Hsp26 or Hsp42 had almost no effect on the toxicity of 72Q (supplemental Fig. 1). This is not surprising given that both sHsps are normally expressed at very low levels in yeast. Overexpressing Hsp104, Hsp26, or Hsp42 reduced 72Q-induced toxicity (Fig. 7A). However, only overexpressing combinations of Hsp104 together with Hsp26 or Hsp42 strongly reduced the toxicity of 72Q (Fig. 7A). Importantly, the overexpression of these genes does not change the status of the [RNQ+] prion (data not shown), which is crucial for the aggregation and toxicity of 72Q in yeast (25). Notably, the toxicity of α-synuclein in yeast (26) could not be suppressed by the over-expression of Hsp104 and Hsp26 or Hsp42 (supplemental Fig. 2). Thus the ability of these chaperones to suppress toxicity depends on the nature of the aggregated protein.
Fig. 7. sHsps and polyglutamine toxicity and aggregation.
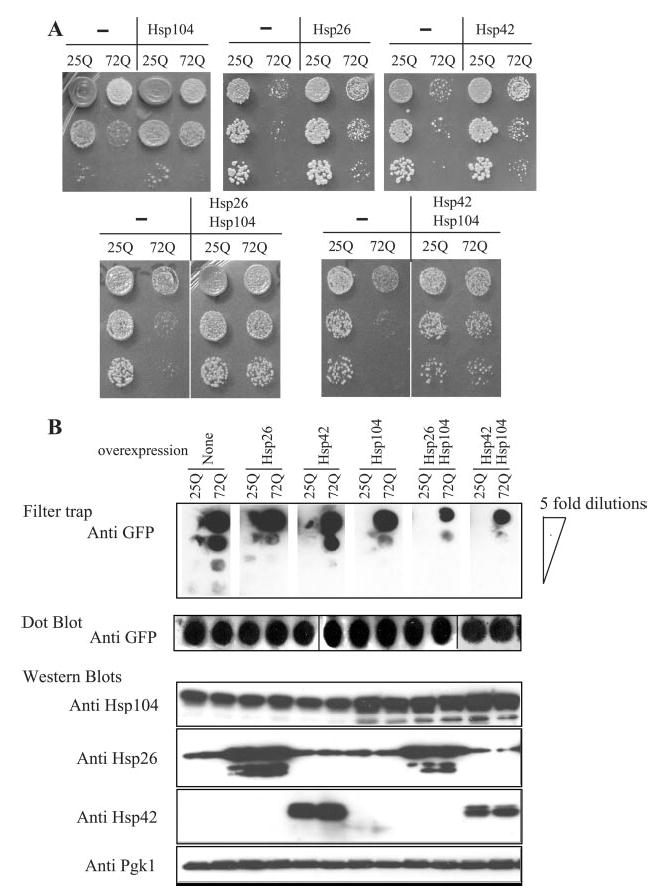
A, growth assays of yeast cells expressing Hsp104 or Hsp26 or the indicated combinations of both in addition to human huntingtin polyglutamine regions exon I containing 25Q or 72Q. B, upper panel, filter-trap assays to analyze aggregation profiles of 25Q and 72Q samples. Lower panel, dot blot and Western blots to determine the expression levels of each of the indicated proteins. Pgk1 was used as loading control. GFP, green fluorescent protein.
To test whether the chaperone-mediated reduction of poly-glutamine-induced toxicity correlates with protein disaggregation activity, we analyzed the polyglutamine aggregation in filter-trap assays (Fig. 7B). The combinations of Hsp104 with Hsp26 or Hsp42 markedly reduced 72Q aggregation. The over-expression of the chaperones was confirmed by Western blotting (Fig. 7B). Notably, the reduction in 72Q aggregation and toxicity was achieved without a reduction of its expression level as demonstrated by the dot blot (Fig. 7B).
In summary, these experiments show that sHsps antagonize the toxicity of a polyglutamine protein in yeast. This reduction of toxicity correlates with the solubilization of the polyglutamine aggregates.
DISCUSSION
Our work illustrates that sHsps are captors of misfolded proteins and modulators of their disaggregation. When proteins misfold, sHsps bind them and, as a consequence, co-aggregate with them. This keeps the entire aggregate in a state that allows efficient disaggregation by the AAA(+) protein Hsp104. In doing so, Hsp104 both liberates sHsps and passes misfolded proteins to the Hsp70/40 machinery. This machinery, in turn, facilitates their reactivation (Fig. 8). Thus, sHsps act at the first step in the pathway that leads to the reactivation of misfolded substrates.
Fig. 8. Chaperone cascade model.
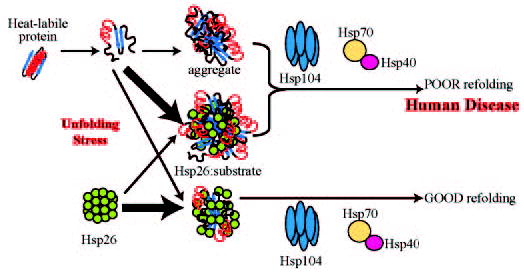
Misfolded and aggregation-prone proteins are trapped by Hsp26 into co-complexes. The nature of the co-complexes depends on the ratio of Hsp26 to the substrate protein. At lower Hsp26 concentrations, larger (and perhaps tighter) complexes formed with misfolded proteins, which are poorly reactivated by the Hsp104/Hsp70/Hsp40 chaperone system. At higher Hsp26 concentrations smaller complexes form with the misfolded proteins, which are efficiently reactivated by the Hsp104/Hsp70/Hsp40 chaperone system. A shift in the delicate balance from the resolvable complexes to the non-resolvable aggregates could result from a reduced efficacy of the chaperone pathway. These complexes may normally serve to resolubilize aggregates, but excessive accumulation of the complexes could result in toxic inclusions.
Similarly, sHsps enable Hsp104 to solubilize polyglutamine aggregates and ameliorate polyglutamine toxicity in yeast. This helps to clarify the role of sHsps in the pathobiology of numerous protein conformation disorders such as Huntington disease. Because sHsps and Hsp104 are likely to act on mis-folded oligomeric intermediates, as well as on larger aggregates, our work does not identify the toxic species. However, the modulation of both the physical state and the toxicity of polyglutamine proteins by sHsps and Hsp104 establishes a direct link between the conformational state of the protein and its toxicity.
sHsps Rescues Polyglutamine Toxicity in Multiple Ways
The human sHsp, HSP27, was identified as a suppressor of polyglutamine-mediated toxicity in a cellular model of Huntington disease (27). In this study, HSP27 suppressed cell death without suppressing polyglutamine aggregation. Instead, sHsps antagonize the effects of polyglutamine-induced oxidative stress and thereby reduce cell death.
Our results suggest that the ability of sHsps to efficiently decrease polyglutamine aggregation by changing its solubility and its toxicity is directly associated with Hsp104-dependent disaggregation. This only takes place efficiently when sHsps and Hsp104 are simultaneously overexpressed. Notably, in accordance with Wyttenbach et al. (27) in our microscopic studies no major changes in the number or morphology of polyglutamine aggregates where detected when sHsps were overexpressed (data not shown).
We also observed a significant reduction of polyglutamine toxicity when sHsps were overexpressed alone. This indicates an additional mode of action of sHsps, antagonizing polyglutamine-induced toxicity by sequestration. We suggest that when sHsps bind polyglutamine-containing proteins, it prevents them from interacting with other proteins in a toxic manner. Among other things, this would be expected to reduce oxidative stress. Finally, for those proteins that are oxidatively damaged or misfolded through interactions with polyglutamines, sHsp binding might facilitate their degradation and/or reactivation.
In contrast to polyglutamine we did not observe any effect of the overexpression of sHsps on the toxicity of α-synuclein in yeast, even in the presence of overexpressed Hsp104 (supplemental Fig. 2). We speculated that this difference can be explained by the diverse nature of α-synuclein and polyglutamine aggregates. Although sHsps interact closely with the highly insoluble polyglutamine they may not recognize the more soluble misfolded α-synuclein. This result might also demonstrate a certain degree of substrate specificity of the disaggregating function of sHsps.
Requirement for Hsp104/ClpB and sHsps in Protein Disaggregation
sHsps of different organisms are only moderately conserved in sequence, but their mechanism of action seems very similar. For example, denatured proteins bound by bacterial (28), plant (29), or mammalian (11, 30) sHsps can be reactivated by the chaperones Hsp70/Hsp40. Bacterial ClpB/DnaK/DnaJ/GrpE chaperones can reactivate proteins bound to Hsp16.6 from Synechocystis sp., Hsp18.1 from Pisum sativum, or IbpA/IbpB from Escherichia coli (13, 14). Such observations establish that the interaction between sHsps, Hsp104/ClpB, and Hsp70/Hsp40 have been evolutionarily conserved. But even though multiple sHsps have been identified in humans (31), no close Hsp104/ClpB homologues have been found in mammalian cells. Is there a functional analogue of Hsp104/ClpB in mammals that interacts with sHsps in protein disaggregation? It has been speculated that the proteins p97 and torsin A, which are also members of the AAA+ family of protein remodeling factors, are able to perform an Hsp104/ClpB-like function in mammalian cells (32). However, a recent study by Glover and colleagues (33) suggests that there may not be an Hsp104/ClpB-like activity in mammalian cells, even though Hsp104 is able to perform its disaggregating activity in mammalian cells. Considering the strong dependence of sHsps on Hsp104/ClpB-mediated protein disaggregation in other systems it will be key to elucidate to what degree this mechanism is conserved in mammals.
Functional Specificity of sHsps
Although previous work demonstrated that Hsp26 and Hsp42 are capable of binding highly overlapping groups of proteins (6), our results suggest a functional distinction between the two sHsps. Although the majority of Hsp26 is found associated with aggregated proteins only after heat shock, ~50% of Hsp42 is always found in the insoluble protein fraction. In addition, the deletion of Hsp104 does not result in any measurable differences in Hsp42 solubility. The two sHsps may differ in their modes of action or require specific cellular conditions to function. Despite these differences both sHsps show virtually the same effects on suppression of polyglutamine aggregation and toxicity.
Do sHsps Act as Adaptors for Hsp104/ClpB?
Members of the AAA(+) protein family use adaptor proteins to bind their substrates. For example, in E. coli, SsrA-tagged proteins are delivered to the AAA+ protein ClpXP for degradation by its adaptor SspB (34). Likewise, ubiquitinated proteins in the endoplasmic reticulum of eukaryotic cells are recognized by the adaptor proteins Npl4 and Ufd1 before they interact with the AAA+ protein p97/Cdc48 (35).
In contrast to the direct interaction of these AAA(+) proteins and their adaptors, a direct binding of Hsp104 to sHsps has not been established, despite attempts to find it.3 Also, no modification of the client proteins like the SsrA tag or ubiquitylation in the before-mentioned examples has been observed. How then are the misfolded client proteins recognized by Hsp104? We suggest that rather than binding Hsp104 directly and recruiting it to aggregates, sHsps keep the client proteins in a state that allows fruitful interaction with Hsp104. To this end sHsps have to form a co-complex with client proteins during their aggregation. It then can promote disaggregation post facto. This mode of action allows Hsp104 to disaggregate a wide range of clients, including stress-damaged proteins or misfolding polyglutamine huntingtin without any further modifications.
Supplementary Material
Acknowledgments
We thank Professor Johannes Buchner, Dr. Martin Haslbeck, and Dr. Thusnelda Stromer for providing re-agents for this study and for sharing unpublished results. We thank the members of the Lindquist laboratory for helpful comments.
Footnotes
The on-line version of this article (available at http://www.jbc.org) contains supplemental figures and tables.
The abbreviations used are: sHsp, small heat shock proteins; WT, wild-type; FFL, firefly luciferase; AAA, ATPases associated with various activities.
M. Duennwald, S. Jagadish, F. Giorgini, S. Willingham, S. L. Lindquist, and P. J. Muchowski, manuscript in preparation.
A. G. Cashikar and S. L. Lindquist, unpublished data.
References
- 1.Jakob U, Gaestel M, Engel K, Buchner J. J Biol Chem. 1993;268:1517–1520. [PubMed] [Google Scholar]
- 2.Welsh MJ, Gaestel M. Ann N Y Acad Sci. 1998;851:28–35. doi: 10.1111/j.1749-6632.1998.tb08973.x. [DOI] [PubMed] [Google Scholar]
- 3.Narberhaus F. Microbiol Mol Biol Rev. 2002;66:64–93. doi: 10.1128/MMBR.66.1.64-93.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haslbeck M. Cell Mol Life Sci. 2002;59:1649–1657. doi: 10.1007/PL00012492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haslbeck M, Ignatiou A, Saibil H, Helmich S, Frenzl E, Stromer T, Buchner J. J Mol Biol. 2004;343:445–455. doi: 10.1016/j.jmb.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 6.Haslbeck M, Braun N, Stromer T, Richter B, Model N, Weinkauf S, Buchner J. EMBO J. 2004;23:638–649. doi: 10.1038/sj.emboj.7600080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petko L, Lindquist S. Cell. 1986;45:885–894. doi: 10.1016/0092-8674(86)90563-5. [DOI] [PubMed] [Google Scholar]
- 8.Susek RE, Lindquist SL. Mol Cell Biol. 1989;9:5265–5271. doi: 10.1128/mcb.9.11.5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haslbeck M, Walke S, Stromer T, Ehrnsperger M, White HE, Chen S, Saibil HR, Buchner J. EMBO J. 1999;18:6744–6751. doi: 10.1093/emboj/18.23.6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wotton D, Freeman K, Shore D. J Biol Chem. 1996;271:2717–2723. doi: 10.1074/jbc.271.5.2717. [DOI] [PubMed] [Google Scholar]
- 11.Ehrnsperger M, Graber S, Gaestel M, Buchner J. EMBO J. 1997;16:221–229. doi: 10.1093/emboj/16.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parsell DA, Kowal AS, Singer MA, Lindquist S. Nature. 1994;372:475–478. doi: 10.1038/372475a0. [DOI] [PubMed] [Google Scholar]
- 13.Mogk A, Deuerling E, Vorderwulbecke S, Vierling E, Bukau B. Mol Microbiol. 2003;50:585–595. doi: 10.1046/j.1365-2958.2003.03710.x. [DOI] [PubMed] [Google Scholar]
- 14.Mogk A, Schlieker C, Friedrich KL, Schonfeld HJ, Vierling E, Bukau B. J Biol Chem. 2003;278:31033–31042. doi: 10.1074/jbc.M303587200. [DOI] [PubMed] [Google Scholar]
- 15.Wach A, Brachat A, Pohlmann R, Philippsen P. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- 16.Ito H, Fukuda Y, Murata K, Kimura A. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnsson N, Varshavsky A. EMBO J. 1994;13:2686–2698. doi: 10.1002/j.1460-2075.1994.tb06559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Proc Natl Acad Sci U S A. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glover JR, Lindquist S. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 20.Escher A, O’Kane DJ, Lee J, Szalay AA. Proc Natl Acad Sci U S A. 1989;86:6528–6532. doi: 10.1073/pnas.86.17.6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrow G, Tanguay RM. Semin Cell Dev Biol. 2003;14:291–299. doi: 10.1016/j.semcdb.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 22.Hsu AL, Murphy CT, Kenyon C. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- 23.Walker GA, White TM, McColl G, Jenkins NL, Babich S, Candido EP, Johnson TE, Lithgow GJ. J Gerontol A Biol Sci Med Sci. 2001;56:281–287. doi: 10.1093/gerona/56.7.b281. [DOI] [PubMed] [Google Scholar]
- 24.Krobitsch S, Lindquist S. Proc Natl Acad Sci U S A. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meriin AB, Zhang X, He X, Newman GP, Chernoff YO, Sherman MY. J Cell Biol. 2002;157:997–1004. doi: 10.1083/jcb.200112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Outeiro TF, Lindquist S. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wyttenbach A, Sauvageot O, Carmichael J, Diaz-Latoud C, Arrigo AP, Rubinsztein DC. Hum Mol Genet. 2002;11:1137–1151. doi: 10.1093/hmg/11.9.1137. [DOI] [PubMed] [Google Scholar]
- 28.Veinger L, Diamant S, Buchner J, Goloubinoff P. J Biol Chem. 1998;273:11032–11037. doi: 10.1074/jbc.273.18.11032. [DOI] [PubMed] [Google Scholar]
- 29.Lee GJ, Vierling E. Plant Physiol. 2000;122:189–198. doi: 10.1104/pp.122.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stege GJ, Brunsting JF, Kampinga HH, Konings AW. J Cell Physiol. 1995;164:579–586. doi: 10.1002/jcp.1041640316. [DOI] [PubMed] [Google Scholar]
- 31.Kappe G, Franck E, Verschuure P, Boelens WC, Leunissen JA, de Jong WW. Cell Stress Chaperones. 2003;8:53–61. doi: 10.1379/1466-1268(2003)8<53:thgecs>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weibezahn J, Bukau B, Mogk A. Microb Cell Fact 16. 2004;3:1–12. doi: 10.1186/1475-2859-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mosser DD, Ho S, Glover JR. Biochemistry. 2004;42:8107–8115. doi: 10.1021/bi0493766. [DOI] [PubMed] [Google Scholar]
- 34.Wah DA, Levchenko I, Baker TA, Sauer RT. Chem Biol. 2002;9:1237–1245. doi: 10.1016/s1074-5521(02)00268-5. [DOI] [PubMed] [Google Scholar]
- 35.Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G. EMBO J. 2000;19:2181–2192. doi: 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haslbeck M, Miess A, Stromer T, Water S, Buchner J. J Biol Chem. 2005 April 20;280:23861–23868. doi: 10.1074/jbc.M502697200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.