

Abstract
Initiation factors IF2 in bacteria and eIF2 in eukaryotes are GTPases that bind Met-tRNA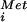 to the small ribosomal subunit. eIF5B, the eukaryotic ortholog of IF2, is a GTPase that promotes ribosomal subunit joining. Here we show that eIF5B GTPase activity is required for protein synthesis. Mutation of the conserved Asp-759 in human eIF5B GTP-binding domain to Asn converts eIF5B to an XTPase and introduces an XTP requirement for subunit joining and translation initiation. Thus, in contrast to bacteria where the single GTPase IF2 is sufficient to catalyze translation initiation, eukaryotic cells require hydrolysis of GTP by both eIF2 and eIF5B to complete translation initiation.
to the small ribosomal subunit. eIF5B, the eukaryotic ortholog of IF2, is a GTPase that promotes ribosomal subunit joining. Here we show that eIF5B GTPase activity is required for protein synthesis. Mutation of the conserved Asp-759 in human eIF5B GTP-binding domain to Asn converts eIF5B to an XTPase and introduces an XTP requirement for subunit joining and translation initiation. Thus, in contrast to bacteria where the single GTPase IF2 is sufficient to catalyze translation initiation, eukaryotic cells require hydrolysis of GTP by both eIF2 and eIF5B to complete translation initiation.
GTP-binding proteins (G proteins) play critical roles in regulating cell growth and differentiation and as components in signal transduction, transport, and protein synthesis pathways. Elongation factors 1A (EF1A/EF-Tu) and 2 (EF2/EF-G) promote, respectively, aminoacyl-tRNA binding to ribosomes and translocation during elongation. Analogous to EF1A-catalyzed binding of aminoacyl-tRNAs to the ribosome during elongation, initiation factor 2 (IF2) in bacteria and eukaryotic IF2 (eIF2) in eukaryotes bind the initiator Met-tRNA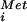 to the small ribosomal subunit in the first step of translation initiation (reviewed in ref. 1). In eukaryotes, the stable eIF2⋅GTP⋅Met-tRNA
to the small ribosomal subunit in the first step of translation initiation (reviewed in ref. 1). In eukaryotes, the stable eIF2⋅GTP⋅Met-tRNA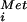 complex associates with the 40S subunit along with additional factors, and the resulting complex then binds an mRNA at the 5′ end and scans to locate the AUG start codon. Base-pairing between the Met-tRNA
complex associates with the 40S subunit along with additional factors, and the resulting complex then binds an mRNA at the 5′ end and scans to locate the AUG start codon. Base-pairing between the Met-tRNA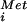 in the ribosomal complex and the AUG codon triggers GTP hydrolysis by eIF2 in a reaction also requiring eIF5 and is followed by release of eIF2 and possibly other IFs. Subsequent joining of the 60S ribosomal subunit completes translation initiation and generates an 80S ribosome that can synthesize proteins. The apparently conserved function of IF2 and eIF2 in the first step of the pathway led to the generally accepted model that only a single GTP molecule was required for translation initiation in all organisms. Consistent with this model, biochemical experiments using purified mammalian IFs provided evidence that a single GTP was needed for 80S complex formation (2). However, a recent kinetic analysis indicated a second GTP-dependent reaction in eukaryotic translation initiation (3).
in the ribosomal complex and the AUG codon triggers GTP hydrolysis by eIF2 in a reaction also requiring eIF5 and is followed by release of eIF2 and possibly other IFs. Subsequent joining of the 60S ribosomal subunit completes translation initiation and generates an 80S ribosome that can synthesize proteins. The apparently conserved function of IF2 and eIF2 in the first step of the pathway led to the generally accepted model that only a single GTP molecule was required for translation initiation in all organisms. Consistent with this model, biochemical experiments using purified mammalian IFs provided evidence that a single GTP was needed for 80S complex formation (2). However, a recent kinetic analysis indicated a second GTP-dependent reaction in eukaryotic translation initiation (3).
Our recent discovery of bacterial IF2 orthologs in archaea and eukaryotes suggested that there was a second GTP requirement in eukaryotic translation initiation (4, 5). The FUN12 gene in yeast encodes a protein now called eIF5B that resembles IF2. Deletion of the FUN12 gene caused a severe slow growth phenotype due to impaired translation initiation, and recombinant eIF5B restored translation in extracts prepared from fun12Δ (ΔeIF5B) strains (6). The discovery that mammalian eIF5B was necessary for subunit joining established a role for this GTPase in eukaryotic translation initiation (7). eIF5B was demonstrated to bind GTP and hydrolyze GTP in the presence of ribosomes [this latter observation was reported originally for the factor IF-M2A (8), which is the same protein as eIF5B]. In addition, when nonhydrolyzable GDPNP was substituted for GTP, eIF5B catalyzed subunit joining; however, the factor was unable to dissociate from the 80S ribosome after subunit joining (7).
To dissect the function of the eIF5B G domain and test the model that two GTP molecules are required in translation initiation, we mutated conserved residues in the eIF5B G domain and tested the function of the mutant proteins in translation initiation. When the nucleotide specificity of eIF5B was switched from GTP to XTP, translation initiation depended on both GTP (eIF2) and XTP (eIF5B). Therefore, there are at least two steps requiring GTP in eukaryotic translation initiation. Finally, experiments in yeast revealed an increase in ribosomes' leaky scanning past AUG start codons in strains lacking eIF5B. Thus, a defect in subunit joining can influence the efficiency of start site selection during translation initiation.
Materials and Methods
Plasmids.
The low copy number URA3 plasmid pC982 expressing full-length yeast eIF5B was constructed by subcloning the ≈3.9-kb SalI–BamHI fragment from pC479 (6) to pRS316. The yeast eIF5B G domain mutants were created by site-directed mutagenesis and inserted into the same vector. The plasmid pC484 used to express GST-yeast eIF5B396–1002 in bacteria has been described (4). Derivatives of this plasmid were constructed to express yeast eIF5B mutants. Plasmids expressing WT and G domain mutant forms of GST-human eIF5B587–1220 have been described (5). The eIF5B portions from these latter plasmids were subcloned to pET28a, generating plasmids to express hexa-histidine- and T7-tagged (His6) versions of human eIF5B587–1220.
Biochemical Techniques.
Yeast polysome analyses and in vitro translation assays were conducted as described (6). Subunit joining or 80S complex assembly assays, methionyl-puromycin (MP) synthesis assays, and GTP hydrolysis assays were conducted as described (7). For subunit joining assays 5 pmol 40S subunits, 5 pmol 60S subunits, 5 pmol [35S]Met-tRNA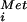 , 0.5 μg of eIF1, 0.5 μg of eIF1A, 3 μg of eIF2, 8 μg of eIF3, 2 μg of eIF4A, 0.5 μg of eIF4B, 2 μg of eIF4F, 0.3 μg of eIF5, and 1.5 μg of eIF5B were incubated in a final volume of 100 μl. For MP synthesis assays 2 pmol 40S subunits, 2.5 pmol 60S subunits, 3 pmol [35S]Met-tRNA
, 0.5 μg of eIF1, 0.5 μg of eIF1A, 3 μg of eIF2, 8 μg of eIF3, 2 μg of eIF4A, 0.5 μg of eIF4B, 2 μg of eIF4F, 0.3 μg of eIF5, and 1.5 μg of eIF5B were incubated in a final volume of 100 μl. For MP synthesis assays 2 pmol 40S subunits, 2.5 pmol 60S subunits, 3 pmol [35S]Met-tRNA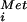 , 1 nmol AUG trinucleotide, 0.5 μg of eIF1, 0.5 μg of eIF1A, 2 μg of eIF2, 5 μg of eIF3, 0.5 μg of eIF5, and 1 μg of eIF5B were incubated in a final volume of 40 μl. [γ-32P]XTP was synthesized by Perkin–Elmer Life Sciences.
, 1 nmol AUG trinucleotide, 0.5 μg of eIF1, 0.5 μg of eIF1A, 2 μg of eIF2, 5 μg of eIF3, 0.5 μg of eIF5, and 1 μg of eIF5B were incubated in a final volume of 40 μl. [γ-32P]XTP was synthesized by Perkin–Elmer Life Sciences.
Results and Discussion
Mutations in the Conserved G Domain of eIF5B Disrupt Translational Activity and Impair Cell Growth.
All G proteins contain five consensus sequence motifs (Fig. 1A) involved in nucleotide binding (9). Conserved amino acids in the G1 and G3 motifs, which interact with the phosphates of GTP, and the G4 motif, which interacts with the guanine base, were mutated in yeast and human eIF5B. Yeast lacking eIF5B (ΔeIF5B) exhibit a severe slow-growth phenotype, and the eIF5B mutants failed to restore WT growth when introduced into the ΔeIF5B strain (Fig. 1B). Yeast expressing the G1 (V414G) and G3 (H480E) mutants of eIF5B grew worse than strains lacking eIF5B. Strains expressing the G4 (K531E) mutant grew like the deletion strain, consistent with the observation that the corresponding mutation in other G proteins blocks nucleotide binding. Thus, in the absence of GTP or GDP binding eIF5B cannot function to promote growth. Finally, expression of the H480Q and G4 (D533N) mutants partially rescued the slow growth of the ΔeIF5B strain, indicating that these factors were able to stimulate translation in vivo. Immunoblot analysis confirmed that the eIF5B mutants were expressed at levels equivalent to WT eIF5B (data not shown).
Fig 1.

Mutational analysis of the G domain of yeast eIF5B. (A) Amino acid sequences of the G1, G3, and G4 sequence motifs in yeast and human eIF5B. The residues in uppercase are conserved in all G proteins; the residues in lowercase are conserved in human and yeast eIF5B. The residues mutated in these studies are underlined and the position numbers in human and yeast eIF5B are indicated. (B) Growth rate analysis of yeast expressing eIF5B mutants. An empty vector (ΔeIF5B) or plasmids expressing the indicated WT or mutant forms of yeast eIF5B were introduced into the ΔeIF5B strain J111 (Matα ura3–52 leu2–3 leu2–112 fun12Δ). Transformants were streaked on synthetic minimal medium and incubated for 7 days at 30°C. (C) In vitro translation assay. The indicated GST or GST-eIF5B396–1002 fusion proteins were purified from bacteria and added along with a luciferase mRNA to translation extracts prepared from isogenic WT or ΔeIF5B strains as indicated. Translational activity in reactions (15 μl final volume) containing 0–800 ng GST or GST-eIF5B was determined by measuring luminescence. Results are representative of at least two independent experiments.
Consistent with the inability of the H480E mutant to promote cell growth, recombinant eIF5B-H480E failed to restore translational activity in extracts from a ΔeIF5B strain (Fig. 1C). The severe slow-growth phenotype in yeast expressing eIF5B-H480E indicated that this mutant factor interfered with an eIF5B-independent pathway of translation initiation operative in ΔeIF5B strains. The H480E mutation in yeast eIF5B corresponds to an H706E mutation in human eIF5B (Fig. 1A). To further analyze the effects of this G3 motif mutation, we examined the mutant and WT factors in several biochemical assays for eIF5B function. For all in vitro experiments an N-terminally truncated form of human eIF5B587–1220 was used (see ref. 5). Whereas recombinant human eIF5B possessed robust ribosome-dependent GTPase activity, the H706E mutation reduced this activity to below background levels (Fig. 2A). The subunit joining activity of WT or mutant forms of human eIF5B was assessed by adding 60S subunits, eIF5, and eIF5B to 48S complexes assembled with [35S]Met-tRNA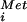 . In the absence of eIF5B, the labeled Met-tRNA
. In the absence of eIF5B, the labeled Met-tRNA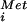 was bound to 48S complexes, but not 80S complexes (Fig. 2B). Addition of WT eIF5B promoted subunit joining, as revealed by the transfer of the [35S]Met-tRNA
was bound to 48S complexes, but not 80S complexes (Fig. 2B). Addition of WT eIF5B promoted subunit joining, as revealed by the transfer of the [35S]Met-tRNA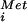 from the 48S complexes to 80S complexes (Fig. 2B). The H706E mutation in human eIF5B severely impaired 80S complex formation (Fig. 2B). This impaired activity may reflect a defect in GTP binding or the structural integrity of the recombinant mutant factor. Previously, we showed that inclusion of nonhydrolyzable GDPNP in place of GTP in the subunit-joining assay blocked release of eIF5B from 80S complexes after subunit joining (7). Thus, GTP hydrolysis by eIF5B is required for its release from the ribosome. As the H706E mutation impaired the GTPase activity of eIF5B, we monitored the association of eIF5B and eIF5B-H706E with the 80S complexes formed in the subunit joining assay. A small proportion of eIF5B-H706E, but not WT eIF5B, was readily detected in the 80S fraction from the subunit joining assays (Fig. 2D). As the amount of 80S complex formed by eIF5B-H706E was substantially less than that formed by WT eIF5B, the detection of eIF5B-H706E on the 80S ribosomes implies a stable association of the mutant factor with the ribosome. This apparent defect in eIF5B-H706E release from the 80S ribosome after subunit joining is consistent with the dominant-negative growth defect observed in yeast overexpressing eIF5B-H480E (data not shown).
from the 48S complexes to 80S complexes (Fig. 2B). The H706E mutation in human eIF5B severely impaired 80S complex formation (Fig. 2B). This impaired activity may reflect a defect in GTP binding or the structural integrity of the recombinant mutant factor. Previously, we showed that inclusion of nonhydrolyzable GDPNP in place of GTP in the subunit-joining assay blocked release of eIF5B from 80S complexes after subunit joining (7). Thus, GTP hydrolysis by eIF5B is required for its release from the ribosome. As the H706E mutation impaired the GTPase activity of eIF5B, we monitored the association of eIF5B and eIF5B-H706E with the 80S complexes formed in the subunit joining assay. A small proportion of eIF5B-H706E, but not WT eIF5B, was readily detected in the 80S fraction from the subunit joining assays (Fig. 2D). As the amount of 80S complex formed by eIF5B-H706E was substantially less than that formed by WT eIF5B, the detection of eIF5B-H706E on the 80S ribosomes implies a stable association of the mutant factor with the ribosome. This apparent defect in eIF5B-H706E release from the 80S ribosome after subunit joining is consistent with the dominant-negative growth defect observed in yeast overexpressing eIF5B-H480E (data not shown).
Fig 2.

Biochemical analysis of human eIF5B G domain mutants. (A) Ribosome-dependent GTP or XTP hydrolysis assay. Recombinant WT, or the indicated mutant, His6-human eIF5B587–1220 protein was incubated with [γ-32P]GTP or [γ-32P]XTP in the presence or absence of purified mammalian ribosomes, as indicated. TLC was used to separate [32P]Pi from [32P]NTP as indicated. (B and C) Subunit joining assay. Sucrose density gradient centrifugation of 80S complexes assembled with 60S subunits, eIF5, GTP (and XTP where indicated), and the indicated His-6-eIF5B587–1220 fusion added to reaction mixtures containing 48S complexes preassembled with β-globin mRNA, 40S subunits, GTP, [35S]Met-tRNA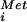 , eIF1, eIF1A, eIF2, eIF3, eIF4A, eIF4B, and eIF4F. Fractions were collected and monitored for radioactivity, and the peaks representing 48S complexes (40S subunits with bound factors, Met-tRNA
, eIF1, eIF1A, eIF2, eIF3, eIF4A, eIF4B, and eIF4F. Fractions were collected and monitored for radioactivity, and the peaks representing 48S complexes (40S subunits with bound factors, Met-tRNA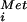 , and mRNA) and 80S ribosomes are labeled. (D) Immunoblot analysis of eIF5B in ribosomal complexes assembled with His6-human eIF5B587–1220 and His6-human eIF5B587–1220-H706E fusion proteins as indicated. Fractions from 48S, 80S, and the top of the sucrose density gradients in B were subjected to immunoblot analysis with anti-T7-tag antibodies; the arrow marks the position of eIF5B.
, and mRNA) and 80S ribosomes are labeled. (D) Immunoblot analysis of eIF5B in ribosomal complexes assembled with His6-human eIF5B587–1220 and His6-human eIF5B587–1220-H706E fusion proteins as indicated. Fractions from 48S, 80S, and the top of the sucrose density gradients in B were subjected to immunoblot analysis with anti-T7-tag antibodies; the arrow marks the position of eIF5B.
The affect of the H706E mutation on eIF5B translational activity was further assessed by using a MP synthesis assay, a model assay for formation of the first peptide bond. 48S complexes assembled by using eIF2, GTP, [35S]Met-tRNA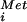 , 40S subunits, and AUG triplet were incubated with eIF5, eIF5B, 60S subunits, GTP, and puromycin. Recombinant WT human eIF5B587–1220 was as effective as native rabbit eIF5B (residues 1–1,220) in stimulating MP synthesis (Fig. 3A). Consistent with its defect in GTPase and subunit joining assays, human eIF5B-H706E failed to stimulate MP synthesis (Fig. 3A). After subunit joining and GTP hydrolysis by eIF5B, we propose that eIF5B is released and the A site is vacant to accept the first elongating tRNA (or puromycin in the MP synthesis assay). Addition of human eIF5B-H706E to sucrose density gradient-purified 80S ribosomes that had been preassembled by using WT eIF5B in the presence of GTP caused a 50% inhibition of MP synthesis (Fig. 3B). This inhibitory effect of eIF5B-H706E was observed in the presence of either GTP or GDPNP. In contrast, addition of WT eIF5B in the presence of GTP had no affect on MP synthesis; however, when added in the presence of GDPNP WT eIF5B strongly inhibited MP synthesis (Fig. 3B). Thus, blockage of eIF5B GTPase activity by addition of nonhydrolyzable GDPNP to WT eIF5B or by the H706E mutation in the G domain locks eIF5B on the 80S ribosome and prevents MP synthesis. The more significant inhibition observed with WT eIF5B in the presence of GDPNP versus eIF5B-H706E in the presence of GTP or GDPNP probably reflects reduced ability of the mutant factor to bind GTP and adopt the conformation required for binding the ribosome. Finally, as eIF5B is thought to bind the ribosome near the A site, overlapping the binding sites for EF1A and EF2 (10), the inhibitory effect of eIF5B-H706E on MP synthesis by preformed 80S complexes may reflect eIF5B competition with elongating tRNAs (or puromycin) for access to the A site.
, 40S subunits, and AUG triplet were incubated with eIF5, eIF5B, 60S subunits, GTP, and puromycin. Recombinant WT human eIF5B587–1220 was as effective as native rabbit eIF5B (residues 1–1,220) in stimulating MP synthesis (Fig. 3A). Consistent with its defect in GTPase and subunit joining assays, human eIF5B-H706E failed to stimulate MP synthesis (Fig. 3A). After subunit joining and GTP hydrolysis by eIF5B, we propose that eIF5B is released and the A site is vacant to accept the first elongating tRNA (or puromycin in the MP synthesis assay). Addition of human eIF5B-H706E to sucrose density gradient-purified 80S ribosomes that had been preassembled by using WT eIF5B in the presence of GTP caused a 50% inhibition of MP synthesis (Fig. 3B). This inhibitory effect of eIF5B-H706E was observed in the presence of either GTP or GDPNP. In contrast, addition of WT eIF5B in the presence of GTP had no affect on MP synthesis; however, when added in the presence of GDPNP WT eIF5B strongly inhibited MP synthesis (Fig. 3B). Thus, blockage of eIF5B GTPase activity by addition of nonhydrolyzable GDPNP to WT eIF5B or by the H706E mutation in the G domain locks eIF5B on the 80S ribosome and prevents MP synthesis. The more significant inhibition observed with WT eIF5B in the presence of GDPNP versus eIF5B-H706E in the presence of GTP or GDPNP probably reflects reduced ability of the mutant factor to bind GTP and adopt the conformation required for binding the ribosome. Finally, as eIF5B is thought to bind the ribosome near the A site, overlapping the binding sites for EF1A and EF2 (10), the inhibitory effect of eIF5B-H706E on MP synthesis by preformed 80S complexes may reflect eIF5B competition with elongating tRNAs (or puromycin) for access to the A site.
Fig 3.

MP synthesis assay of human eIF5B G domain mutants. (A) MP synthesis by active 80S ribosomes assembled with 48S complexes, eIF5, 60S subunits, GTP or GTP plus XTP or no nucleotide, and native human eIF5B (native) or recombinant WT or mutant forms of His6-eIF5B587–1220, as indicated. 48S complexes were assembled with 40S subunits, GTP, [35S]Met-tRNA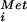 , AUG trinucleotide, eIF1, eIF1A, eIF2, and eIF3. Each value was corrected for background, determined with 48S complexes alone. (B) MP synthesis by 80S complexes assembled by WT His6-eIF5B587–1220 in the presence of GTP, purified from sucrose gradients, and incubated with 0.2 mM GTP, 0.2 mM GDPNP, and 0.3 μg of eIF5B587–1220 or eIF5B587–1220-H706E as indicated.
, AUG trinucleotide, eIF1, eIF1A, eIF2, and eIF3. Each value was corrected for background, determined with 48S complexes alone. (B) MP synthesis by 80S complexes assembled by WT His6-eIF5B587–1220 in the presence of GTP, purified from sucrose gradients, and incubated with 0.2 mM GTP, 0.2 mM GDPNP, and 0.3 μg of eIF5B587–1220 or eIF5B587–1220-H706E as indicated.
The Human eIF5B-D759N Mutation Alters the Nucleotide Specificity of the Factor to XTP and Introduces an XTP Requirement for Protein Synthesis.
The G4 motif is responsible for the guanine specificity of G proteins. The conserved Asp residue in G4 interacts via a hydrogen bond with the 2-amino group in GTP. Substitution of Asn in place of this Asp residue in a number of G proteins including EF1A (11) and eEF1A (12) alters the nucleotide specificity from GTP to XTP. The Asn side chain cannot form a hydrogen bond with the 2-amino group of GTP, but it can form a hydrogen bond with the 2-keto group present in XTP. The corresponding D759N mutation in the G4 motif of human eIF5B impaired the ribosome-dependent GTPase activity of the factor as predicted (Fig. 2A). In addition, whereas WT eIF5B only weakly hydrolyzed XTP, eIF5B-D759N possessed robust ribosome-dependent XTPase activity (Fig. 2A). Thus, the D759N mutation changed the nucleotide specificity of human eIF5B from GTP to XTP.
We used the human eIF5B-D759N mutant to further examine the nucleotide requirements for translation initiation. In the presence of GTP, human eIF5B-D759N weakly stimulated 80S complex formation (Fig. 2C), consistent with the decreased ability of the mutant factor to bind or hydrolyze GTP (Fig. 2A and data not shown). However, substitution of XTP in place of GTP restored 80S complex formation in assays containing eIF5B-D759N (Fig. 2C). Likewise, in the MP synthesis assay eIF5B-D759N was nonfunctional in reactions containing only GTP (Fig. 3A). Addition of XTP to reactions containing WT eIF5B reduced MP synthesis, possibly because XTP competed with GTP for binding to eIF5B (Fig. 3A). In contrast, addition of XTP to assays containing eIF5B-D759N strongly stimulated MP synthesis (Fig. 3A). In both the subunit joining and MP synthesis assays 48S complexes were assembled in the presence of GTP, which was essential for eIF2-dependent binding of Met-tRNA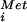 to the 40S subunit. Thus, in reactions containing eIF5B-D759N both GTP (eIF2) and XTP (eIF5B) were required for subunit joining and MP synthesis, indicating the presence of at least two GTP-dependent steps in eukaryotic translation initiation.
to the 40S subunit. Thus, in reactions containing eIF5B-D759N both GTP (eIF2) and XTP (eIF5B) were required for subunit joining and MP synthesis, indicating the presence of at least two GTP-dependent steps in eukaryotic translation initiation.
Model for Two GTP-Dependent Steps in Eukaryotic Translation Initiation.
Models depicting the GTP-dependent steps in prokaryotic and eukaryotic translation initiation are depicted in Fig. 4A. In prokaryotes, a preinitiation complex containing the 30S ribosomal subunit, fMet-tRNA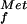 , and IF1, IF2⋅GTP, and IF3 assembles at the AUG codon of an mRNA (reviewed in ref. 1). The selection of the AUG start codon is mediated in part by base-pairing contacts between the 3′ end of the 16S rRNA and the Shine–Dalgarno sequence located 5–7 nt upstream of the AUG codon. Joining of the 50S ribosomal subunit triggers GTP hydrolysis by IF2 and release of the factor, resulting in a 70S ribosome competent to begin translation elongation. This model is supported by the subunit association activity of IF2 (13) and the analysis of IF2 G domain mutants (14); however, a recent kinetic analysis of bacterial translation initiation challenged the importance of IF2 GTP binding and hydrolysis (15). In eukaryotes, a 43S preinitiation complex composed of the 40S ribosomal subunit, Met-tRNA
, and IF1, IF2⋅GTP, and IF3 assembles at the AUG codon of an mRNA (reviewed in ref. 1). The selection of the AUG start codon is mediated in part by base-pairing contacts between the 3′ end of the 16S rRNA and the Shine–Dalgarno sequence located 5–7 nt upstream of the AUG codon. Joining of the 50S ribosomal subunit triggers GTP hydrolysis by IF2 and release of the factor, resulting in a 70S ribosome competent to begin translation elongation. This model is supported by the subunit association activity of IF2 (13) and the analysis of IF2 G domain mutants (14); however, a recent kinetic analysis of bacterial translation initiation challenged the importance of IF2 GTP binding and hydrolysis (15). In eukaryotes, a 43S preinitiation complex composed of the 40S ribosomal subunit, Met-tRNA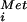 , and eIF1, eIF1A, eIF2⋅GTP, eIF3, and likely eIF5 binds to an mRNA near the 5′ end and scans down the mRNA searching for an AUG start codon. Base-pairing between the anticodon of the Met-tRNA
, and eIF1, eIF1A, eIF2⋅GTP, eIF3, and likely eIF5 binds to an mRNA near the 5′ end and scans down the mRNA searching for an AUG start codon. Base-pairing between the anticodon of the Met-tRNA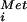 in the 43S complex and the AUG codon in the mRNA stalls the scanning ribosome and triggers GTP hydrolysis by eIF2 and the release of eIF2 in a complex with GDP. In this way GTP hydrolysis by eIF2 is a checkpoint to ensure proper start site selection (16). We propose that eIF5B in complex with GTP binds to the 40S subunit mediated in part by contacts with eIF1A (4) bound in the ribosomal A site and stabilizes the Met-tRNA
in the 43S complex and the AUG codon in the mRNA stalls the scanning ribosome and triggers GTP hydrolysis by eIF2 and the release of eIF2 in a complex with GDP. In this way GTP hydrolysis by eIF2 is a checkpoint to ensure proper start site selection (16). We propose that eIF5B in complex with GTP binds to the 40S subunit mediated in part by contacts with eIF1A (4) bound in the ribosomal A site and stabilizes the Met-tRNA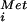 in the P site. Joining of the 60S ribosomal subunit triggers GTP hydrolysis by eIF5B and release of the factor, resulting in an 80S ribosome competent to enter the elongation phase of protein synthesis. As illustrated in Fig. 4A, aside from the different mechanisms used for selection of AUG start codons, the initiation pathways in prokaryotes and eukaryotes are highly conserved with IF2 and eIF5B performing analogous roles coupling GTP hydrolysis with formation of a ribosome that is competent for translation elongation.
in the P site. Joining of the 60S ribosomal subunit triggers GTP hydrolysis by eIF5B and release of the factor, resulting in an 80S ribosome competent to enter the elongation phase of protein synthesis. As illustrated in Fig. 4A, aside from the different mechanisms used for selection of AUG start codons, the initiation pathways in prokaryotes and eukaryotes are highly conserved with IF2 and eIF5B performing analogous roles coupling GTP hydrolysis with formation of a ribosome that is competent for translation elongation.
Fig 4.

Model for two GTP-dependent steps in eukaryotic translation initiation. (A) In prokaryotes (Left), the 30S ribosomal complex containing fMet-tRNA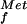 and IF1, IF2⋅GTP, and IF3 binds to an mRNA as directed by base-pairing interactions between 16S rRNA and the Shine–Dalgarno sequence upstream of the AUG start codon. Joining of the 50S subunit triggers GTP hydrolysis by IF2, release of the factor, and assembly of the translationally competent 70S ribosome. In eukaryotes (Right), the 40S ribosomal complex containing Met-tRNA
and IF1, IF2⋅GTP, and IF3 binds to an mRNA as directed by base-pairing interactions between 16S rRNA and the Shine–Dalgarno sequence upstream of the AUG start codon. Joining of the 50S subunit triggers GTP hydrolysis by IF2, release of the factor, and assembly of the translationally competent 70S ribosome. In eukaryotes (Right), the 40S ribosomal complex containing Met-tRNA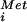 and eIF1, eIF1A, eIF2⋅GTP, eIF3, and eIF5 scans down the mRNA. Base-pairing contacts between the AUG codon on the mRNA and the anticodon of the Met-tRNA
and eIF1, eIF1A, eIF2⋅GTP, eIF3, and eIF5 scans down the mRNA. Base-pairing contacts between the AUG codon on the mRNA and the anticodon of the Met-tRNA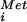 stalls scanning and triggers GTP hydrolysis by eIF2 and release of eIF2⋅GDP and possibly other factors. We propose that eIF1A remains bound in the ribosomal A site (10) and provides a docking site for eIF5B. Binding of eIF5B⋅GTP promotes subunit joining, which in turn induces GTP hydrolysis by eIF5B and release of the factor. The 80S ribosome is then poised to enter the elongation phase of protein synthesis. (B) Model for halfmer polysomes. (Upper) A 40S complex has scanned to the AUG codon of an mRNA that also has an 80S ribosome elongating in the ORF. (Lower) After GTP hydrolysis by eIF2, factor release, and binding of eIF5B, the 40S complex is poised at the AUG codon waiting for joining of the 60S ribosomal subunit. In rpl16bΔ strains, the abundance of 60S subunits is reduced and subunit joining is slowed. The mRNA complexes represented by the lower schematic accumulate and resolve as a shoulder or extra peak sedimenting slightly heavier than the 80S monosome peak in a sucrose density gradient. We propose that the 40S subunit bound at the AUG codon in the halfmer complex is unstable in the absence of eIF5B.
stalls scanning and triggers GTP hydrolysis by eIF2 and release of eIF2⋅GDP and possibly other factors. We propose that eIF1A remains bound in the ribosomal A site (10) and provides a docking site for eIF5B. Binding of eIF5B⋅GTP promotes subunit joining, which in turn induces GTP hydrolysis by eIF5B and release of the factor. The 80S ribosome is then poised to enter the elongation phase of protein synthesis. (B) Model for halfmer polysomes. (Upper) A 40S complex has scanned to the AUG codon of an mRNA that also has an 80S ribosome elongating in the ORF. (Lower) After GTP hydrolysis by eIF2, factor release, and binding of eIF5B, the 40S complex is poised at the AUG codon waiting for joining of the 60S ribosomal subunit. In rpl16bΔ strains, the abundance of 60S subunits is reduced and subunit joining is slowed. The mRNA complexes represented by the lower schematic accumulate and resolve as a shoulder or extra peak sedimenting slightly heavier than the 80S monosome peak in a sucrose density gradient. We propose that the 40S subunit bound at the AUG codon in the halfmer complex is unstable in the absence of eIF5B.
eIF5B Is Required for 48S Complex Stability and Efficient Subunit Joining at AUG Codons in Vivo.
With the in vitro evidence in mammalian and yeast systems that eIF5B promotes the subunit joining step of translation initiation (7, 17), we tested whether deleting the eIF5B gene altered subunit joining in vivo. The classic evidence for a subunit joining defect is the presence of halfmer polysomes containing shoulders or discreet peaks sedimenting slightly faster than the polysome peaks on sucrose gradients. Halfmer polysomes are most easily detected as shoulders on the 80S and disome peaks in polysome profiles from strains with reduced amounts of 60S subunits, such as strains lacking a 60S ribosomal subunit protein like L16B (see Fig. 5A, rpl16bΔ strain). In the rpl16bΔ strain the 80S peak represents an mRNA with a single translating ribosome, whereas the halfmer peak is thought to contain a translating 80S and a second 40S subunit stalled at the AUG codon awaiting 60S subunit joining (Fig. 4B). If eIF5B is essential for subunit joining, then we would expect to see halfmer polysomes in the ΔeIF5B strain. However, as shown in Fig. 5A, polysome profiles from ΔeIF5B strains revealed only a reduction of the conventional polysome peaks with a corresponding increase in the 80S peak, but no halfmer peaks. As our model in Fig. 4B predicted that eIF5B would be a component in the halfmer 40S subunit, it is possible that the halfmer ribosome is unstable in the absence of eIF5B. To test this possibility, we deleted the eIF5B gene in the rpl16bΔ strain. Polysome analysis revealed that the loss of eIF5B in the rpl16bΔ ΔeIF5B double mutant strain resulted in a 25–50% reduction in the abundance of the halfmer polysomes compared with the rpl16bΔ strain (Fig. 5A). This result is consistent with the notion that eIF5B is required for the stability of the halfmer ribosomes, and it explains the lack of halfmer peaks in the polysome profiles from ΔeIF5B strains. We propose that eIF5B stabilizes the 40S initiation complexes by stabilizing Met-tRNA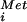 binding after release of eIF2. In this way eIF5B and bacterial IF2 perform analogous roles promoting the binding of Met-tRNA
binding after release of eIF2. In this way eIF5B and bacterial IF2 perform analogous roles promoting the binding of Met-tRNA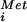 or fMet-tRNA
or fMet-tRNA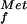 , respectively, to the small ribosomal subunit.
, respectively, to the small ribosomal subunit.
Fig 5.

Altered polysome profiles and impaired GCN4 translation in eIF5B mutant strains. (A) Polysome analysis of the indicated isogenic WT (J115), ΔeIF5B (J116F), rpl16bΔ (J113), and double mutant [ΔeIF5B, rpl16bΔ (J113F)] strains. Whole-cell extracts were centrifuged on 7–47% sucrose gradients and fractionated while monitoring absorbance at 254 nm. Positions of 40S, 60S, 80S, disome (two 80S ribosomes on one mRNA), and polysome peaks are indicated. The arrows with asterisks denote the position of the halfmer polysomes. The halfmer ratios were calculated by measuring the area under the halfmer peak and the adjacent 80S or disome peak. (B and C) Analysis of GCN4-lacZ expression in eIF5B mutant strains. The yeast strain J111 carrying an empty low copy number LEU2 vector (ΔeIF5B) or the same vector containing WT or mutant alleles of eIF5B, as indicated, was transformed with URA3 plasmids containing a WT GCN4-lacZ allele (B) or GCN4-lacZ alleles with the indicated altered 5′ leader (C), that have been described (21–23). Transformants were grown under nonstarvation conditions (C and R column in B) or under starvation conditions imposed the histidine analog 3-amino- triazole (DR in B), and cells were harvested and β-galactosidase activities were determined as described (22). Results shown are the averages of at least two independent transformants, and individual measurements deviated from the average by 25% or less.
In previous work we demonstrated that yeast strains lacking eIF5B failed to derepress GCN4 expression under amino acid starvation conditions (6). In WT strains GCN4 expression is low on nutrient-rich medium and increases ≈7- to 10-fold when cells are starved for amino acids [Fig. 5B, eIF5B (WT)]. GCN4 expression in yeast expressing the various eIF5B G domain mutants correlated closely with the growth rates of the strains. For example, yeast expressing the eIF5B-V414G and eIF5B-H480E mutants grew most slowly (Fig. 1B) and these strains yielded the lowest GCN4 expression under starvation conditions (Fig. 5B). Thus, the eIF5B mutations had similar affects on general and GCN4-specific translation.
The 5′ leader of the GCN4 mRNA contains four short ORFs; however, the first and fourth ORFs are sufficient for normal regulation (reviewed in ref. 18). Nearly all of the ribosomes that bind to the 5′ end of the GCN4 mRNA are thought to scan down and translate the first upstream ORF (ORF1). After translation of ORF1 roughly 50% of the ribosomes resume scanning. Under nutrient-rich conditions translation reinitiation is efficient and ribosomes translate one of the subsequent upstream ORFs 2–4. In contrast to ORF1, ribosomes are thought to disengage from the GCN4 mRNA after translation of ORF2, ORF3, or ORF4; and GCN4 is not expressed. Under amino acid starvation conditions the kinase GCN2 phosphorylates eIF2, thereby limiting the availability of eIF2⋅GTP⋅Met-tRNA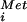 ternary complexes, which are necessary for scanning 40S subunits to recognize an AUG codon. Accordingly, in starvation conditions ribosomes translate ORF1 and then scan a longer time and further distance, bypassing the inhibitory ORFs 2–4 before reinitiating translation at the GCN4 start codon. Deletion of ORF1 blocks GCN4 expression under all conditions, indicating that ribosomes must translate ORF1 and then encounter ORFs 2–4 in the reinitiation mode to bypass these inhibitory elements under starvation conditions.
ternary complexes, which are necessary for scanning 40S subunits to recognize an AUG codon. Accordingly, in starvation conditions ribosomes translate ORF1 and then scan a longer time and further distance, bypassing the inhibitory ORFs 2–4 before reinitiating translation at the GCN4 start codon. Deletion of ORF1 blocks GCN4 expression under all conditions, indicating that ribosomes must translate ORF1 and then encounter ORFs 2–4 in the reinitiation mode to bypass these inhibitory elements under starvation conditions.
To gain insight into how the lack of eIF5B impairs GCN4 expression, we used a series of constructs with altered mRNA leaders to examine GCN4 expression in strains containing or lacking eIF5B. As shown in Fig. 5C (construct 1), ORFs 1 and 4 are sufficient to maintain the low level of expression of GCN4 in nutrient-rich conditions. Shortening the distance between ORFs 1 and 4, while maintaining the normal distance between ORF1 and the GCN4 AUG start site, increased GCN4 expression in the WT strain, but not in the strain lacking eIF5B (Fig. 5C, construct 2). Presumably, in the WT strain the shorter distance between ORFs 1 and 4 did not provide ribosomes scanning down the GCN4 leader after translation of ORF1 with sufficient time to reacquire a ternary complex before encountering the ORF4 start site. These ribosomes thus bypass ORF4 and translate GCN4 instead. The failure to increase GCN4 expression from this construct in the ΔeIF5B strain may indicate that ribosomes become competent for reinitiation faster in strains lacking eIF5B, or that a greater number of ribosomes bypass ORF1 and then translate the inhibitory ORF4 in this mutant strain.
To further examine how the interplay between ORF1 and the presence or absence of eIF5B affects GCN4 expression, we used a series of constructs containing only ORF1 at different distances before the GCN4 start site. Using the construct with WT (350 nt) spacing between ORF1 and the GCN4 ORF, high levels of GCN4 were expressed in both the WT and ΔeIF5B strains (Fig. 5C, construct 3). Reducing the spacing between ORF1 and the GCN4 start site progressively lowered GCN4 expression in both strains; however, GCN4 expression was ≈2-fold higher in the strain lacking eIF5B (Fig. 5C, constructs 4 and 5). These results are consistent with both the faster reinitiation and the ORF1-skipping models to explain the effects of eIF5B deletion on GCN4 expression. To definitively test whether the lack of eIF5B resulted in scanning ribosomes skipping the ORF1 AUG codon (leaky scanning, ref. 19) and initiating instead at the GCN4 start site, we used a construct (Fig. 5C, construct 6) in which ORF1 was elongated such that it terminated after the GCN4 start site in an alternative reading frame. As ribosomes that translate ORF1 in this construct would have to scan in a 3′ to 5′ direction 130 nt and bypass four AUG codons to access the GCN4 start site (20, 21), it is likely that only ribosomes that bypass the ORF1 start site will express GCN4. As expected, GCN4 expression from this construct was very low in WT strains; however, GCN4 expression increased roughly 10-fold in strains lacking eIF5B (Fig. 5C, construct 6). This result indicates a 10-fold increase in the number of ribosomes that leaky scan past the ORF1 AUG start codon without initiating translation in strains lacking eIF5B. Presumably, impaired subunit joining or unstable 40S initiation complexes (or Met-tRNA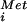 binding) after release of eIF2 (halfmers) in the ΔeIF5B strain results in inefficient translation initiation at the ORF1 AUG codon. On the WT GCN4 leader this leaky scanning of the ORF1 AUG codon will result in ribosomes translating one of the inhibitory ORFs, and thus likely contributes to the poor expression of GCN4 in the ΔeIF5B strain.
binding) after release of eIF2 (halfmers) in the ΔeIF5B strain results in inefficient translation initiation at the ORF1 AUG codon. On the WT GCN4 leader this leaky scanning of the ORF1 AUG codon will result in ribosomes translating one of the inhibitory ORFs, and thus likely contributes to the poor expression of GCN4 in the ΔeIF5B strain.
The impaired stability of halfmer ribosomes and the increase in leaky scanning in yeast lacking eIF5B provide in vivo support for the subunit joining function found for mammalian eIF5B. By switching the nucleotide specificity of eIF5B from GTP to XTP, we demonstrated that there at least two GTP-dependent steps in eukaryotic translation initiation. Accordingly, GTP hydrolysis by eIF2 facilitates proper AUG codon selection, a process mediated by base-pairing interactions in prokaryotes; whereas, bacterial IF2 and eIF5B share a conserved function in facilitating subunit joining in the final step of translation initiation.
Acknowledgments
We thank members of the Dever, Pestova, and Hinnebusch laboratories for helpful discussions, Keith Remo for assistance in plasmid construction, and Alan Hinnebusch for comments on the manuscript. This work was supported in part by National Institutes of Health Grant R01GM63940 (to T.V.P.).
Abbreviations
IF, initiation factor
eIF, eukaryotic IF
EF, elongation factor
MP, methionyl-puromycin
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Hershey J. W. B. & Merrick, W. C. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J. W. B. & Mathews, M. B. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 33–88.
- 2.Merrick W. C. (1979) J. Biol. Chem. 254, 3708-3711. [PubMed] [Google Scholar]
- 3.Lorsch J. R. & Herschlag, D. (1999) EMBO J. 18, 6705-6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi S. K., Olsen, D. S., Roll-Mecak, A., Martung, A., Remo, K. L., Burley, S. K., Hinnebusch, A. G. & Dever, T. E. (2000) Mol. Cell. Biol. 20, 7183-7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee J. H., Choi, S. K., Roll-Mecak, A., Burley, S. K. & Dever, T. E. (1999) Proc. Natl. Acad. Sci. USA 96, 4342-4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi S. K., Lee, J. H., Zoll, W. L., Merrick, W. C. & Dever, T. E. (1998) Science 280, 1757-1760. [DOI] [PubMed] [Google Scholar]
- 7.Pestova T. V., Lomakin, I. B., Lee, J. H., Choi, S. K., Dever, T. E. & Hellen, C. U. T. (2000) Nature 403, 332-335. [DOI] [PubMed] [Google Scholar]
- 8.Merrick W. C., Kemper, W. M. & Anderson, W. F. (1975) J. Biol. Chem. 250, 5556-5562. [PubMed] [Google Scholar]
- 9.Sprang S. R. (1997) Annu. Rev. Biochem. 66, 639-678. [DOI] [PubMed] [Google Scholar]
- 10.Roll-Mecak A., Shin, B. S., Dever, T. E. & Burley, S. K. (2001) Trends Biochem. Sci. 26, 705-709. [DOI] [PubMed] [Google Scholar]
- 11.Hwang Y. W. & Miller, D. L. (1987) J. Biol. Chem. 262, 13081-13085. [PubMed] [Google Scholar]
- 12.Cavallius J. & Merrick, W. C. (1998) J. Biol. Chem. 273, 28752-28758. [DOI] [PubMed] [Google Scholar]
- 13.Godefroy-Colburn T., Wolfe, A. D., Dondon, J., Grunberg-Manago, M., Dessen, P. & Pantaloni, D. (1975) J. Mol. Biol. 94, 461-478. [DOI] [PubMed] [Google Scholar]
- 14.Luchin S., Putzer, H., Hershey, J. W., Cenatiempo, Y., Grunberg-Manago, M. & Laalami, S. (1999) J. Biol. Chem. 274, 6074-6079. [DOI] [PubMed] [Google Scholar]
- 15.Tomsic J., Vitali, L. A., Daviter, T., Savelsbergh, A., Spurio, R., Striebeck, P., Wintermeyer, W., Rodnina, M. V. & Gualerzi, C. O. (2000) EMBO J. 19, 2127-2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang H., Yoon, H., Hannig, E. M. & Donahue, T. F. (1997) Genes Dev. 11, 2396-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Algire M. A., Maag, D., Savio, P., Acker, M. G., Tarun, S. Z., Sachs, A. B., Asano, K., Nielsen, K. H., Olsen, D. S., Phan, L., et al. (2002) RNA 8, 382-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinnebusch A. G. (1996) in Translational Control, eds. Hershey, J. W. B., Mathews, M. B. & Sonenberg, N. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 199–244.
- 19.Kozak M. (1999) Gene 234, 187-208. [DOI] [PubMed] [Google Scholar]
- 20.Abastado J. P., Miller, P. F., Jackson, B. M. & Hinnebusch, A. G. (1991) Mol. Cell. Biol. 11, 486-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grant C. M., Miller, P. F. & Hinnebusch, A. G. (1994) Mol. Cell. Biol. 14, 2616-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hinnebusch A. G. (1985) Mol. Cell. Biol. 5, 2349-2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mueller P. P. & Hinnebusch, A. G. (1986) Cell 45, 201-207. [DOI] [PubMed] [Google Scholar]