

Abstract
The nature of diatomic ligand recombination in heme proteins is elucidated by using a Landau–Zener model for the electronic coupling in the recombination rate constant. The model is developed by means of explicit potential energy surfaces calculated by using density functional theory (DFT). The interaction of all possible spin states of the three common diatomic ligands, CO, NO, and O2, and high-spin heme iron is compared. The electronic coupling, rebinding barrier, and Landau–Zener force terms can be obtained and used to demonstrate significant differences among the ligands. In particular the intermediate spin states of NO (S = 3/2) and O2 (S = 1) are shown to be bound states. Rapid recombination occurs from these bound states in agreement with experimental data. The slower phases of O2 recombination can be explained by the presence of two higher spin states, S = 2 and S = 3, which have a small and relatively large barrier to ligand recombination, respectively. By contrast, the intermediate spin state for CO is not a bound state, and the only recombination pathway for CO involves direct recombination from the S = 2 state. This process is significantly slower according to the Landau–Zener model. Quantitative estimates of the parameters used in the rate constants provide a complete description that explains rebinding rates that range from femtoseconds to milliseconds at ambient temperature.
The binding of diatomic ligands to heme has been studied for over 100 years (1). The dissociation of carbon monoxide (CO), nitric oxide (NO), and oxygen (O2) from the iron of heme occurs by both a thermal and photolytic process (2–4). In the case of flash photolysis, the fate of the ligand depends on the competition between the intrinsic (or geminate) recombination rate constant and protein relaxation in a docking site roughly 3.5 Å from the iron as well as ligand escape from the protein (5–8). Recombination of these ligands occurs by the reverse of the thermal process. The time dependence of recombination serves as a probe of the intrinsic properties of the diatomic ligand-iron bond formation and dynamic processes that occur in the protein that surrounds the heme cofactor (9–11). The startlingly different recombination dynamics of CO, NO, and O2 have been attributed to spin states in earlier work (3, 12). In the present study, the ground state potential energy surfaces for the potential energy surfaces of all possible spin states are calculated by using density functional theory (DFT) and compared within the context of a Landau–Zener (L–Z) model for the intrinsic recombination process of each of the ligands. The observed recombination kinetics of CO, NO, and O2 can be influenced by protein dynamics if the intrinsic recombination rate constant is slower than these dynamics (10, 13–16). When a combination of temperature and viscosity dependence is used, the general time scale for the intrinsic rate constants can be extracted, and it is these rate constants that are compared in the present work.
The rebinding of CO occurs as a single exponential process with a time constant of approximately τCO ≈ 1 μs at ambient temperature and at a solvent viscosity below the intrinsic viscosity of the globin (<4 cP; 1 P = 0.1 Pa⋅s) (17–19). The geminate recombination process is slower than both protein relaxation and CO escape. The escape rate constant is ≈25 times faster than the geminate rate constant at ambient temperature leading to small geminate yield, Φgem ≈ 0.04 (17). The CO ligands that escape from the globin enter the solvent. For CO ligands that have escaped into the solvent, recombination occurs as a bimolecular process with a yield of Φbi ≈ 0.96. At solution concentrations of [CO] ≈ 1 mM, the bimolecular recombination lifetime is τCO ≈1 ms. As the solvent viscosity is increased, ligand escape slows and the geminate yield increases (10, 18–23). Protein relaxation slows as well and both protein relaxation and geminate CO recombination become nonexponential (10, 16, 24–26). However, the average time constant for the intrinsic geminate CO recombination process remains close to 1 μs.
NO recombination is biphasic and occurs with time constants of τNO ≈ 10 ps and τNO ≈ 200 ps under ambient conditions (3, 14, 15, 27). The bimolecular yield for NO is small Φbi < ≈0.01. The time constant for the rapid phase of NO recombination (τNO ≈ 10 ps) is comparable to the most rapid phase of protein relaxation at a viscosity of 1 cP (8, 28). Two models for the origin of the biexponential kinetics can be divided into a distal effect; two different potential energy wells within the distal pocket of the protein, (13, 15, 29) and a proxmial effect; a time-dependent barrier caused by iron out-of-plane motion (14, 30). As the solvent viscosity is increased, NO recombination becomes increasingly single exponential with a time constant of τNO ≈10 ps (15). Because solvent relaxation slows with increasing viscosity, it follows that NO recombination occurs more rapidly than protein relaxation at high solvent viscosity. The intrinsic rate constant τNO ≈ 10 ps for NO recombination is ≈5 orders of magnitude greater than that for CO. Even more rapid NO recombination has been observed as a single exponential process in heme proteins that have constrained distal structures that restrict NO diffusion (31).
Diatomic oxygen recombination is highly nonexponential at ambient temperature with a rapid phase of geminate recombination, τgem1,O2 ≈ 3 ps (3), a longer picosecond phase, τgem2,O2 ≈ 20–200 ps (3, 12, 32), and a bimolecular process, τbi,O2 ≈ 1 ms, with a yield of Φbi > 0.05. Given the wide range in time scales for O2 recombination, there is still some uncertainty regarding the yields for the various phases (32, 33). However, generally speaking, the recombination can be divided into thirds, one-third ultrafast, one-third picosecond, and one-third nanosecond or slower (3, 32, 33). Bimolecular recombination appears to occur as a fraction of the slowest phase and depends on solvent conditions (34). The ultrafast process in O2 recombination has been ascribed to recombination to a singlet state (3). The slower phases have been ascribed to the higher spin states (3, 12).
The experimental data exhibit dramatic differences in the intrinsic geminate recombination rate constant. These differences have been attributed to the spin state differences of photolyzed ligands; CO (S = 0), NO (S = 1/2), and O2 (S = 1) (3, 12). Table 1 presents a mechanistic overview of a hypothesis for spin-dependent recombination rates that explains the differences between CO, NO, and O2 recombination rates and the experimental time constants for the intrinsic processes. In the present study spin-dependent channels of recombination have been verified by the combination of DFT and the L–Z model for the crossing of the potential energy surfaces.
Table 1.
Overview of mechanisms of ligand recombination in Im-FeP-XO where X = C, N, or O
| Species | Recombination process | Type | Time scale |
|---|---|---|---|
| Im-FeP-O2 | 1 → 0 | Elementary | 3 ps |
| Im-FeP-O2 | 2 → 1 → 0 | Sequential | 100 ps |
| Im-FeP-O2 | 3 → 2 → 1 → 0 | Sequential | Slow |
| Im-FeP-NO | 3/2 → 5/2 | Elementary | 10 ps |
| Im-FeP-NO | 5/2 → 3/2 → 1/2 | Sequential | 100 ps |
| Im-FeP-CO | 2 → 0 | Superexchange | 1 μs |
Methods
The model system used in this study consists of an imidazole (Im) ligand bound to iron-porphine (FeP) trans to a diatomic ligand XO where X = C, N, or O. The optimized ground state geometries and potential energy surfaces of Im–FeP–XO (X = C, N, or O) were obtained by using the BLYP functional (35, 36) as implemented in DMOL3 (Molecular Simulations, Waltham, MA) (37, 38). All calculations were carried out on the SGI/Cray Origin 2000 and IBM SP supercomputers at the North Carolina Supercomputer Center (NCSC). Geometry optimizations were carried out without constraints until the energy difference was <10−6 a.u. on subsequent iterations (see supporting information, which is published on the PNAS web site, www.pnas.org). Numerically tabulated basis sets of double-ζ plus extra polarization (DNPP) quality were used as described in the supporting information. For the DNPP basis there are 4 basis functions for H (1s, 2s, 2p, 3d), 7 basis functions C, N, O [1s, 2s (2), 2p (2), 3d, 4f], and 12 basis functions for Fe [1s, 2s, 2p, 3s, 3p, 3d (2), 4s (2), 4p, 4f, 5g] (L. Bartolotti, personal communication). The potential energy surfaces were calculated for both Fermi (canonical ensemble) (39) and Thermal (grand canonical ensemble) treatments of the density functional (40). The grand canonical (Thermal) option always converged to a lower overall energy. The grand canonical calculation was carried out at a finite temperature of kBT = 0.02 Hartrees. Once a calculation was complete it was extrapolated to zero temperature by subtraction of the thermal electron occupation according to the grand canonical partition function. avs (Advanced Visual System) and insightii (Molecular Simulations) programs were used for visualization of the results.
The potential energy surfaces shown in Fig. 1 were plotted in units of kJ/mol vs. Å. When fit to the polynomial function
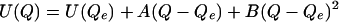 |
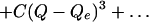 |
frequencies were calculated by using the force constants, K = 2B (units of kJ/mol/Å) and
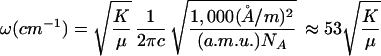 |
slopes of the potential energy surface, dH/dQ = A (units of kJ/mol/Å) as reported in Table 2. Applying a diatomic model, the reduced mass, μ = m1m2/(m1 + m2), where the porphine mass is m1 and the diatomic ligand mass is m2. The masses are in atomic mass units (a.m.u.). The conversion factors in Eq. 2 are a.m.u. = 1.672 × 10−26 kg, Å/m = 1010, NA = 6.023 × 1023 molecules per mol and c = 2.99 × 1010 cm/s. The method of fitting the potential energy surface for the extraction of parameters permits the development of the L–Z model as shown below.
Fig 1.

Potential energy surfaces (PESs) for Fe-XO recombination (X = C, N, or O). The spin states for the Fe-CO system are S = 0 (solid line), S = 1 (dashed line), and S = 2 (dotted line), which are bonding, nonbonding, and antibonding, respectively. (A) Fe-CO, QP = 0.0 Å. (B) Fe-CO, QP = 0.2 Å. (C) Fe-CO, QP = 0.4 Å. The spin states for the Fe-NO system are S = 1/2 (solid), S = 3/2 (dashed), and S = 5/2 (dotted), which are bonding, bonding for QP < = 0.2 Å, and weakly bonding, respectively. The spin states are S = 1/2, S = 3/2, and S = 5/2 for the iron and NO ligand system. (D) Fe-NO, QP = 0.0 Å. (E) Fe-NO, QP = 0.2 Å. (F) Fe-NO, QP = 0.4 Å. The spin states for the Fe-O2 ligand system are S = 0 (solid line), S = 1 (dashed line), S = 2 (dotted line), and S = 3 (wide dashed line). (G) Fe-O2, QP = 0.0 Å. (H) Fe-O2, QP = 0.2 Å. (I) Fe-O2, QP = 0.4 Å.
Table 2.
Potential energies of binding, equilibrium bond lengths, and force constants for bound states of carbonmonoxy, nitroxy, and oxy Im-FeP
| Species (spin state)
|
QP = 0.0 Å | QP = 0.2 Å | QP = 0.4 Å | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Qe | De | K | Qc | De | K | Qe | De | K | |
| CO (S = 0) | 1.72 | −189.5 | 2,420 | 1.72 | −139.5 | 2,370 | 1.73 | −83.2 | 2,205 |
| NO (S = 1/2) | 1.68 | −190.9 | 2,410 | 1.71 | −133.6 | 1,955 | 1.76 | −60.3 | 1,260 |
| NO (S = 3/2) | 1.77 | −118.4 | 1,170 | 1.84 | −71.6 | 710 | 2.47 | −30.2 | 10 |
| NO (S = 5/2) | 2.32 | −18.5 | 81 | 2.85 | −6.4 | 22 | 3.30 | −3.0 | 15 |
| O2 (S = 0) | 1.77 | −158.9 | 1,560 | 1.82 | −108.8 | 1,200 | 1.92 | −49.2 | 645 |
| O2 (S = 1) | 1.81 | −145.4 | 1,270 | 1.87 | −99.4 | 880 | 2.01 | −47.9 | 290 |
| O2 (S = 2) | 2.15 | −14.9 | 106 | 3.00 | −3.3 | 17 | 3.40 | −1.2 | 12 |
The equilibrium distance, Qc, is given in Å. The bond energy, Dc, is given in kJ/mol. The force constant for the bound state vibration, K, is given in kJ/mol/Å2. The frequency in cm−1 can be obtained from ω(cm−1) = 53 √[K/(440M/(440 + M))], where M is the mass of the diatomic in a.m.u. Appropriate unit conversions are given in Eq. 2.
Results
The calculated potential energy surfaces (PESs) for Im-FeP-diatomic models of the three ligands, CO, NO, and O2 are shown in Fig. 1. The PESs were calculated along the coordinate QD, which is the Fe… XO distance normal to the plane of porphine ring (X = C, N, or O). Fig. 1 shows PESs calculated at three values of the coordinate QP, which is the iron out-of-plane distance with an in-plane heme iron as the origin, QP = 0.0 Å. The PESs were calculated for all possible spin states. The binding energies for the diatomic ligands are −189.5, −190.9, and −158.9 kJ/mol for in-plane geometry (QP = 0.0 Å) of Im-FeP-CO, Im-FeP-NO, and Im-FeP-O2, respectively, as shown in Fig. 1 A, D, and G. The equilibrium Fe–XO bond lengths obtained from plots of the potential energy surfaces for bound states are given in Table 2. The triplet state of Im-FeP-CO is essentially nonbonding, and the quintet state of Im-FeP-CO is dissociative for all values of QP. Intermediate spin states for the NO and O2 adducts are bound states (Table 2). For QP < = 0.2 Å, the hextet state of Im-FeP-NO is somewhat bonding, as are the quintet and heptet states of Im-FeP-O2. Table 3 summarizes the location of the crossing points and the barrier heights estimated by using the lowest energy geometry along the QD coordinate as a reference point. For dissociative potential energy surfaces, the QD = 10 Å point was used as the reference energy.
Table 3.
Location of curve crossings and barrier heights for the thermal dissociation and association process
| Species
|
QP = 0.0 Å | QP = 0.2 Å | QP = 0.4 Å | |||
|---|---|---|---|---|---|---|
| QC | ΔU* | QC | ΔU* | QC | ΔU* | |
| CO | ||||||
| 0–1 | 3.00 | NB | 2.63 | 3.3 | 2.25 | 10 |
| 0–2 | NC | NB | 3.52 | 1.2 | 2.38 | 40 |
| 1–2 | NC | NB | NC | NB | 2.58 | 9 |
| NO | ||||||
| 1/2–3/2 | 2.26 | NB | 2.26 | NB | 2.16 | 3 |
| 1/2–5/2 | 2.6 | NB | 2.60 | NB | 2.37 | 18 |
| 3/2–5/2 | NC | NB | NC | NB | 2.77 | 8 |
| O2 | ||||||
| 0–1 | 2.10 | NB | 2.07 | NB | 1.99 | NB |
| 1–2 | 2.30 | NB | 2.24 | 8.5 | 2.12 | 41 |
| 0–2 | 2.25 | NB | 2.20 | 10 | 2.08 | 45 |
| 1–3 | 2.65 | 1 | 2.48 | 9.3 | 2.23 | 52 |
| 0–3 | 2.49 | 2 | 2.39 | 14 | 2.17 | 63 |
The quantity ΔU* is the height of the energy barrier. It is the energy of the high-spin state at the crossing point (given as QC) relative to the value of the system in the high-spin state at QD = 10.0 Å. QC given in Å; ΔU* given in kJ/mol. NC, no crossing; NB, no barrier.
The determination of the spin gap in the deoxy form of the iron porphine model is the most difficult quantitative aspect of these calculations (41, 42). Previous work has shown that the spin gap obtained is more accurately calculated by using a DNPP basis set than the DNP basis set (43). The DNPP basis set has qualitatively correct ordering with the possible exception of the Im-FeP-CO calculation for QP = 0.0 Å. For QP > = 0.2 Å, the quintet state is lower than the singlet state in energy, whereas for the QP = 0.0 Å calculation the quintet state is ≈7 kJ/mol higher in energy. Larger basis sets tend to depress the energy of the higher spin states, and thus it is possible that the current basis set does not yield the lowest possible energy. Moreover, there are details such as geometry distortions of the heme (porphine) macrocycle that cause a splitting of the dπ orbitals that are not included in the present DFT calculation (44, 45). Although vibronic coupling and vibronic distortions are important, particularly for excited state dynamics, they do not change the basic conclusions of the present study. These factors could give rise to a barrier at QP = 0.0 Å (Fig. 1A), i.e., the quintet–singlet (2–0) transition would appear as a curve crossing if the deoxy quintet state were 10 kJ/mol or more in energy (43). The relationship between the potential energy surfaces for Im-FeP-CO is quite different from that for Im-FeP-NO and Im-FeP-O2, where the S = 5/2 and S = 2 configurations, respectively, would have no barrier because of curve crossing unless they were lowered in energy by more than 50 kJ/mol. The S = 3/2 and S = 1 states of NO and O2, respectively, are bound states for QP = 0.0 Å (Fig. 1 D and G, respectively). By contrast, the neither the intermediate spin state (S = 1) nor the high spin state (S = 2) are bound states for Im-FeP-CO.
Discussion
The remarkable range of recombination lifetimes for CO, NO, and O2 can be explained by considering the differences in the spin states of the respective diatomic ligands. In the six-coordinate bound state with heme iron the spin states are S = 0, S = 1/2, and S = 0, for Im-FeP-CO, Im-FeP-NO, and Im-FeP-O2, respectively. After photolysis, the diatomic ligands move away from the iron along the coordinate QD and the spin state of the iron changes from S = 0 to S = 2 as the d-electrons populate the dz2 and dx2-y2 orbitals of the five-coordinate adduct (2). Experimental evidence suggests that iron out-of-plane motion is extremely rapid along coordinate QP (46, 47). We therefore assume that the heme iron reaches its equilibrium position in the deoxy state on a time scale that is short compared with ligand recombination. The equilibrium position is calculated to be at QP = 0.25 Å. X-ray crystal structures give QP ≈ 0.4 Å (48, 49). The calculations of Im-FeP-XO (X = C, N, and O) models consider geometries at QP = 0.2 and 0.4 Å to include two possible out-of-plane geometries. Given the rapidity of photolysis, it is reasonable to assume that the spin state of the iron is randomized relative to that of the photolyzed ligand (2, 50). The assumption that the spin states of the free ligands and the iron are uncorrelated implies that possible spin states are SFe + Sligand, SFe + Sligand − 1, … |SFe − Sligand|. In the model calculations for dissociated states the spin states are S = 1 or 2, S = 3/2 or 5/2, and S = 1, 2, or 3 for the unbound Im-FeP:CO, Im-FeP:NO, and Im-FeP:O2 adducts, respectively.
A L–Z model, presented first in a study of CO recombination kinetics (43) provides a unified approach to the treatment of the kinetics of the three diatomic ligands. The L–Z approach is ideal for treatment of cases where the electronic coupling can be either in the adiabatic or nonadiabatic regime. For large electronic coupling, the adiabatic basis is most convenient. In this basis, the nuclei follow one potential energy surface in the transition from reactants to products. For small electronic coupling, the system must jump from one potential energy surface to the other and the nonadiabatic basis is most convenient. The L–Z model for the electronic coupling bridges both regimes by including both the nuclear velocity of moving through the transition state (TS) and the differential force felt by the nuclei as the move through the crossing region. For nonadiabatic transitions, the nuclei move rapidly through the transition region with a frequency ω and jump from one surface to the other with a frequency given by the L–Z probability. As nuclear motion slows down the nuclei spend more time in the crossing region and the process begins to resemble an adiabatic process in which the electronic state gradually takes on the character of the product state. Although the probability is larger for adiabatic transfer, the L–Z model is also valid for the probability in the nonadiabatic regime. Given the large differences in electronic coupling between the three diatomic ligands considered here, the L–Z approach is well-suited to describe the nature of coupling in the crossing regions shown in potential energy surfaces in Fig. 1. The L–Z approach begins with the definition of the TS rate constant for diatomic ligand recombination (51):
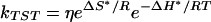 |
where ΔS* and ΔH* are the activation entropy and enthalpy, respectively. The activation enthalpy, ΔH* is related to the activation internal energy, ΔU* calculated in Table 3 by ΔH* = ΔU* + RT. The activation entropy can be obtained from the partition function and is discussed elsewhere (43). The entropy factor, eΔS*/R does not differ significantly for the three diatomic ligands, CO, NO, and O2. The prefactor η is the L–Z form of the electronic factor:
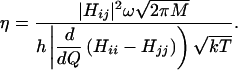 |
The velocity in the direction of the iron is 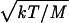 according to the kinetic theory of gases, where M is the total mass M of each diatomic. The relative velocities of the CO, NO, and O2 ligands are 0.94:0.97:1. The attempt frequency ω depends on whether the photolyzed state is a dissociative or bound state (Eq. 2). For a dissociative potential surface ω is obtained from the frequency of iron doming, because this motion moves the iron along the coordinate QP, which is required to minimize the barrier along QD. If the photolyzed state is a bound state, then ω is the frequency for oscillation of the ligand in the bound state along QD (Eq. 2). For these cases, the frequency for reaching the crossing point is much larger because Fe-CO, Fe-NO and Fe-O2 frequencies are typically ≈500 cm−1 (52, 53). If the ligand is not in a bound state, the attempt frequency for approach to the TS corresponds to heme doming with a frequency of ≈50–100 cm−1 (54–56). The frequencies in the bound states can be estimated by calculating the curvature, i.e., the second derivative of the potential energy surface at the minimum (Eq. 1). For example, using the assumption of a diatomic harmonic oscillator with a mass of the Im-FeP (440 a.m.u.) and the CO (28 a.m.u.), the calculated frequency is 508 cm−1, which is good agreement with the experimental value of 507 cm−1 for the Fe-CO stretch in myoglobin (53). The L–Z model indicates that bound states have a frequency factor ω that is ≈1 order of magnitude larger than the frequency factor for dissociative states. According to the DFT method and the L–Z model, both the electronic coupling, Hij, and the attempt frequency, ω, are significantly larger for NO and O2 ligands in bound states compared with CO, which is in a dissociative state. Thus, NO and O2 are predicted to have much more rapid intrinsic geminate recombination rates in agreement with experimental observations.
according to the kinetic theory of gases, where M is the total mass M of each diatomic. The relative velocities of the CO, NO, and O2 ligands are 0.94:0.97:1. The attempt frequency ω depends on whether the photolyzed state is a dissociative or bound state (Eq. 2). For a dissociative potential surface ω is obtained from the frequency of iron doming, because this motion moves the iron along the coordinate QP, which is required to minimize the barrier along QD. If the photolyzed state is a bound state, then ω is the frequency for oscillation of the ligand in the bound state along QD (Eq. 2). For these cases, the frequency for reaching the crossing point is much larger because Fe-CO, Fe-NO and Fe-O2 frequencies are typically ≈500 cm−1 (52, 53). If the ligand is not in a bound state, the attempt frequency for approach to the TS corresponds to heme doming with a frequency of ≈50–100 cm−1 (54–56). The frequencies in the bound states can be estimated by calculating the curvature, i.e., the second derivative of the potential energy surface at the minimum (Eq. 1). For example, using the assumption of a diatomic harmonic oscillator with a mass of the Im-FeP (440 a.m.u.) and the CO (28 a.m.u.), the calculated frequency is 508 cm−1, which is good agreement with the experimental value of 507 cm−1 for the Fe-CO stretch in myoglobin (53). The L–Z model indicates that bound states have a frequency factor ω that is ≈1 order of magnitude larger than the frequency factor for dissociative states. According to the DFT method and the L–Z model, both the electronic coupling, Hij, and the attempt frequency, ω, are significantly larger for NO and O2 ligands in bound states compared with CO, which is in a dissociative state. Thus, NO and O2 are predicted to have much more rapid intrinsic geminate recombination rates in agreement with experimental observations.
The electronic factor Hij is also an important factor that gives rise to spin-dependent rate constants. For a transition involving the change of only a single spin (e.g., triplet - singlet) i = j ± 1. Transitions that involve two or more changes in spin may occur by a sequential or superexchange mechanism (57–61). In a sequential mechanism there are two processes both having i = j ± 1. In a superexchange mechanism, there is a single electronic coupling matrix element Hij, where i = j ± 2. For example, a quintet–singlet coupling has the following relationship to the single spin coupling for 0–1 or 1–2:
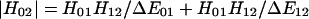 |
Because both 0 → 1 and 1 → 2 involve a single spin change, we assume that H01 ≈ H12 and ΔE01 ≈ ΔE12, then H02 ≈ 2H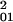 /ΔE01. This simple model can be related to the potential energy surfaces calculated for CO, NO, and O2 rebinding. To determine ΔE01 we examine the energy gap from 0–1 at the 0–2 crossing point (or the energy from 1/2–3/2 at the 1/2–5/2 crossing point for NO). The magnitude of ΔE01 is given in Table 4. A typical value is ΔE01 ≈ 500–1,000 cm−1 so that if H01 ≈ 1 kJ/mol (80 cm−1), then H02 ≈ 0.001 kJ/mol (0.08 cm−1). Given the close proximity of the crossing points for the three potential energy surfaces shown in Fig. 1C, there is likely a lower bound estimate for the superexchange coupling. Because the L–Z rate constant is proportional to the square of the coupling matrix element, the superexchange model predicts a singlet–triplet (or quartet–doublet) transition could be as large as 106 times larger than the singlet–quintet (or hextet–doublet).
/ΔE01. This simple model can be related to the potential energy surfaces calculated for CO, NO, and O2 rebinding. To determine ΔE01 we examine the energy gap from 0–1 at the 0–2 crossing point (or the energy from 1/2–3/2 at the 1/2–5/2 crossing point for NO). The magnitude of ΔE01 is given in Table 4. A typical value is ΔE01 ≈ 500–1,000 cm−1 so that if H01 ≈ 1 kJ/mol (80 cm−1), then H02 ≈ 0.001 kJ/mol (0.08 cm−1). Given the close proximity of the crossing points for the three potential energy surfaces shown in Fig. 1C, there is likely a lower bound estimate for the superexchange coupling. Because the L–Z rate constant is proportional to the square of the coupling matrix element, the superexchange model predicts a singlet–triplet (or quartet–doublet) transition could be as large as 106 times larger than the singlet–quintet (or hextet–doublet).
Table 4.
Energy of the spin gap of the intermediate-spin state relative to the low-spin state at the reactive crossing point
| Ligand | QP = 0.0 Å | QP = 0.2 Å | QP = 0.4 Å |
|---|---|---|---|
| NO ΔE1/2–3/2, kJ/mol (cm−1) | 17 (1,420) | 17 (1,420) | 14 (1,170) |
| CO ΔE01, kJ/mol (cm−1) | NA | 7.5 (630) | 16 (1,340) |
| O2 ΔE01, kJ/mol (cm−1) | 6 (500) | 4 (330) | 3 (250) |
For CO and O2, the energy of the S = 0 → S = 1 spin gap, ΔE01 is given at QC, the crossing point of the S = 0 → S = 2 potential energy surfaces. Fig. 1C shows one example of a crossing point for the high-spin (S = 2) to low-spin (S = 0) state for CO. For NO the energy of the S = 1/2 → S = 3/2 spin gap, ΔE1/2–3/2 is given at QC, the crossing point of the S = 1/2 → S = 5/2 potential energy surfaces. NA, not applicable.
The L–Z force or difference in slope of the potential surfaces, |∂/∂Q(Hii − Hjj)|, has a relatively small effect on the relative magnitude of the rate constants. The potential energy surfaces were used to estimate the change in the slope at the crossing point, |∂/∂Q(Hii − Hjj)| given by the A coefficients of the respective potential energy surfaces fit to Eq. 1. The differences in slope are relatively small when various diatomic ligands are compared (see supporting information). The 0–2 and 1/2–5/2 crossings of O2 and NO, respectively can be compared with the 0–2 crossing of CO for the geometry QP = 0.4 Å. The reason for this choice is that the spin gap of all of the ligands is comparable at this geometry. For the double spin flip transitions corresponding to Fig. 1 C, F, and I, the slopes are all comparable, |∂/∂Q(Hii − Hjj)| = 19.7, 9.9, and 16.0 kJ/mol/Å for Im-FeP-CO, Im-FeP-NO, and Im-FeP-O2, respectively. Thus, the difference in slope may give rise to, at most, a factor of 2 greater rate constant for the NO ligand relative to CO in a superexchange mediated recombination step.
The various factors that contribute to the L–Z rate constant and prefactor (Eqs. 3 and 4) are combined in the calculated rate constants given in Table 5, which were calculated by using a consistent set of assumptions for all of the ligands and spin states. The calculations were carried out for two different proximal iron out-of-plane displacements with the exception of the in-plane geometry (QP = 0.0 Å) for Im-FeP-CO, which shows no curve crossing in the DFT calculated potential energy surfaces. Because quintet state of Im-FeP-CO is dissociative (Fig. 1A) the result for QP = 0.0 Å would be quite similar to that given in Table 5 for QP = 0.4 Å, and therefore in agreement with experiment as has been shown in detailed experimental studies of CO recombination (43). The superexchange mechanism is required for CO because S = 1 state is not a bound state and cannot be a true intermediate. Both O2 and NO have bound intermediate states as shown in Fig. 1 and Table 2, consistent with the existence of a sequential recombination pathway. The L–Z approach suggests several factors that give rise to rapid phases of NO and O2 rebinding relative to CO. In the proper spin state of NO or O2, a bound state is formed that changes the dynamics of the curve crossing by increasing the effective frequency ω by an order of magnitude. The smaller L–Z force and small barriers for NO and O2 also play a major role. All of these features are predicted by the potential energy surfaces in Fig. 1 using the L–Z model.
Table 5.
Comparison of calculated and experimental rate constants
| Transition | QP, Å | Hij, kJ/mol | ω, kJ/mol (cm−1) | ∂ΔH/∂Q, kJ/mol/Å | η, s−1 | ΔH*, kJ/mol | k, s−1 | kobs, s−1 |
|---|---|---|---|---|---|---|---|---|
| CO (2 → 0) | 0.4 | 0.05 | 0.6 (50) | 19.7 | 2.2 × 108 | 20 | 7.3 × 104 | 1 × 106 |
| NO (3/2 → 1/2) | 0.4 | 1 | 6.7 (354) | 9.9 | 1.3 × 1012 | 5.5 | 1.4 × 1011 | 1–2 × 1011 |
| NO (3/2 → 1/2) | 0.0 | 1 | 9.3 (492) | 2.3 | 7.5 × 1012 | 2.5 | 2.8 × 1012 | 1–2 × 1011 |
| NO (5/2 → 3/2) | 0.4 | 1 | 0.7 (39) | 2.0 | 6.8 × 1011 | 5.5 | 7.5 × 1010 | 5 × 109 |
| NO (5/2 → 1/2) | 0.4 | 0.05 | 0.7 (39) | 9.9 | 3.4 × 108 | 20.5 | 8.6 × 104 | 5 × 109 |
| NO (5/2 → 1/2) | 0.0 | 0.05 | 1.72 (91) | 3.0 | 2.5 × 109 | 2.5 | 9.6 × 108 | 5 × 109 |
| O2 (1 → 0) | 0.4 | 1 | 6.5 (345) | 3.8 | 3.2 × 1012 | 2.5 | 1.2 × 1012 | 3 × 1011 |
| O2 (1 → 0) | 0.0 | 1 | 4.6 (246) | 4.2 | 2.1 × 1012 | 2.5 | 7.6 × 1011 | 3 × 1011 |
| O2 (2 → 1) | 0.4 | 1 | 0.6 (50) | 11.1 | 1.0 × 1011 | 20 | 3.3 × 107 | 1 × 1010 |
| O2 (2 → 1) | 0.0 | 1 | 1.9 (100) | 10.4 | 3.2 × 1011 | 2.5 | 1.1 × 1010 | 1 × 1010 |
| O2 (2 → 0) | 0.4 | 0.2 | 0.6 (50) | 16.0 | 2.8 × 109 | 20 | 9.1 × 105 | 1 × 1010 |
| O2 (2 → 0) | 0.0 | 0.2 | 1.9 (100) | 14.7 | 9.0 × 109 | 2.5 | 3.2 × 109 | 1 × 1010 |
| O2 (3 → 0) | 0.4 | 0.01 | 0.6 (50) | 23.8 | 6.9 × 106 | 20 | 2.3 × 103 | Small |
| O2 (3 → 0) | 0.0 | 0.01 | 0.6 (50) | 12.7 | 1.3 × 107 | 20 | 4.3 × 103 | Small |
The calculated rate constants use the L–Z model with parameters obtained from the DFT calculations following Eqs. 1 and 2.
The superexchange coupling in the text is a lower bound. The estimate used for CO and NO calculations is H02 = 0.05H01 or H1/2,5/2 = 0.05H1/2,3/2, respectively. The estimate for oxygen is four times larger relative to CO or NO due to the fact the energy denominators in Table 3 are approximately four times smaller for O2, i.e. H02 = 0.2H01.
The binding on the distal side is sufficiently weak that the proximal side dynamics govern the approach to the transition state. Therefore, the relevant frequency in the L–Z expression is the doming mode (50 cm−1).
The barrier height is estimated based on a comparison of QP and QD potential surfaces (44).
When ΔU* = 0.0 corresponding to no barrier, then ΔH* = RT, where R is the universal gas constant and T is temperature.
The sequential mechanism is not possible for NO when QP = 0.0 Å because of the order of the curve crossings.
Conclusion
The L–Z model explains the differences in recombination kinetics among the diatomic ligands CO, NO, and O2. The model is based on quantitative estimates of the spin gaps and curve crossings shown in Fig. 1 and reported in Tables 3. There are two possible approaches to the TS in systems of the type Im-FeP-XO. In the distal approach to the TS, the diatomic ligand XO fluctuates in a potential well near the iron along coordinate QD with frequency ωFe-XO. In the proximal approach to the TS, the iron fluctuates about its equilibrium position in the deoxy state and samples various proximal geometries along the out-of-plane coordinate QP with a frequency ωFe doming. There is a competition between the barrier to the iron moving into the plane along QP and the barrier along the ligand trajectory QD. The intermediate and high spin states of NO and O2 are all bound states (except the S = 3 state of O2) as shown in Table 1. Recombination from these bound states implies that the distal approach to the barrier occurs for NO and O2 (except when the barrier is higher than the proximal Fe barrier). The presence of bound intermediate states for NO and O2 means that a sequential mechanism is applicable. As shown in Table 5, the sequential mechanism gives the correct order of magnitude for the rate constant for NO and O2. By contrast, the sequential mechanism does not apply to CO recombination, and the superexchange model applies, giving a much smaller rate constant consistent with experimental observations.
The rebinding rate for the triplet state (S = 1) of Im-FeP-O2 is the greatest of all of the ligands because it is barrierless and it is in a bound state. The calculations show agreement with experiment, both in time scale and in that recombination can occur to an out-of-plane geometry of the heme iron. The same considerations apply to intermediate spin state (S = 3/2) of NO, which is bound for QP < 0.2 Å. NO can also recombine with an out-of-plane iron (QP = 0.4 Å). It has been suggested that the TS for CO is nearly planar. Unfortunately, further assumptions must be applied to interpret the QP = 0.0 Å potential energy surfaces for Im-FeP-CO shown in Fig. 1A because there is no 2 → 0 curve crossing and the L–Z model cannot be applied. Nonetheless, the activation barrier for CO rebinding, ΔU* = 20 kJ/mol, which arises from the approach of high spin iron to the heme plane can be applied to the QP = 0.4 Å potential energy surfaces in Fig. 1C, and the rate constant calculated in Table 5 gives reasonable agreement with experiment.
The statistics of the spin states are most evident in O2, where there are three vastly different time scales for recombination. The statistical distribution of O2 spins is reflected in the distribution of kinetic processes. Roughly one-third of photolyzed O2 molecules recombine on the picosecond time scale (τO2 ≈ 3 ps) and one third recombine on a nanosecond and slower time scale that includes the bimolecular process. The bimolecular phase implies a statistical distribution of O2 ligands that escape into the solvent. The rate-limiting step for the bimolecular process is entry into the globin and the spin states (1 → 0 and 2 → 0) that recombine the most rapidly will dominate the observed recombination. Thus, the 3 → 0 direct process for O2 may not contribute at all to the observed bimolecular kinetics. Statistically, one-half of the NO ligands have the appropriate spin to recombine by the 3/2 → 1/2 pathway. The rapid recombination of the quartet (S = 3/2) state suggests that a sequential mechanism can lead to rapid NO recombination from the hextet state as well. Table 5 shows that good agreement is obtained if it assumed that the 5/2 → 3/2 transition is rate limiting for the sequential mechanism. These considerations explain the rapid (τNO ≈ 10 ps) time constant for NO recombination and the slower phase (τNO ≈ 200 ps).
Geometric constraints by the protein can influence the observed recombination rate. Fig. 1 shows that confinement of the diatomic ligand close to the heme iron, e.g., when QD < 3.0 Å, significantly increases the energy of the highest spin states for each respective ligand. Although this particular aspect involves further assumptions, it is clear that this confinement will increase the recombination rate constant. This explains the viscosity dependence of NO recombination (15), fast NO recombination in FixL (31), and the increase in CO recombination rate constant in some constrained enviroments as well. In conclusion, spin-dependent dynamics explain the profound difference between NO and O2 recombination relative to CO recombination. The intermediate spin state (quartet or triplet) is a bound state for open shell diatomics, whereas for CO the triplet state is not a bound state and provides no direct channel that would permit a sequential mechanism. The DFT calculations of potential energy surfaces provide energetic estimates for a consistent interpretation in terms of the various spin states giving a detailed mechanistic picture for thermal recombination in heme proteins.
Supplementary Material
Acknowledgments
This work was supported by National Science Foundation Grant MCB-9874895 and the North Carolina Supercomputer Center.
Abbreviations
L–Z, Landau–Zener
TS, transition state
DFT, density function theory
Im, imidazole
FeP, iron-porphine
DNPP, double-ζ plus extra polarization
PESs, potential energy surfaces
a.m.u., atomic mass units
References
- 1.Antonini E. & Brunori, M., (1971) Hemoglobin and Myoglobin in Their Reactions with Ligands (North Holland, Amsterdam).
- 2.Franzen S., Kiger, L., Poyart, C. & Martin, J. L. (2001) Biophys. J. 80, 2372-2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrich J. W., Poyart, C. & Martin, J. L. (1988) Biochemistry 27, 4049-4060. [DOI] [PubMed] [Google Scholar]
- 4.Kholodenko Y., Volk, M., Gooding, E. & Hochstrasser, R. M. (2000) Chem. Phys. 259, 71-87. [Google Scholar]
- 5.Schlichting I., Berendzen, J., Phillips, G. N., Jr. & Sweet, R. M. (1994) Nature 371, 808-812. [DOI] [PubMed] [Google Scholar]
- 6.Srajer V., Teng, T. Y., Ursby, T., Pradervand, C., Ren, Z., Adachi, S., Schildkamp, W., Bourgeois, D., Wulff, M. & Moffat, K. (1996) Science 274, 1726-1729. [DOI] [PubMed] [Google Scholar]
- 7.Teng T.-Y., Srajer, V. & Moffat, K. (1994) Nat. Struct. Biol. 1, 701-705. [DOI] [PubMed] [Google Scholar]
- 8.Lim M., Jackson, T. A. & Anfinrud, P. A. (1995) J. Chem. Phys. 102, 4355-4366. [Google Scholar]
- 9.Tian W. D., Sage, J. T., Champion, P. M., Chien, E. & Sligar, S. G. (1996) Biochemistry 35, 3487-3502. [DOI] [PubMed] [Google Scholar]
- 10.Steinbach P. J., Ansari, A., Berendzen, J., Braunstein, D., Chu, K., Cowen, B. R., Ehrenstein, D., Frauenfelder, H., Johnson, J. B., Lamb, D. C., et al. (1991) Biochemistry 30, 3988-4001. [DOI] [PubMed] [Google Scholar]
- 11.Friedman J. & Campbell, B., (1987) Structural Dynamics and Reactivity in Hemoglobin (Springer, New York).
- 12.Greene B. I., Hochstrasser, R. M., Weisman, R. B. & Eaton, W. A. (1978) Proc. Natl. Acad. Sci. USA 75, 5255-5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaad O., Zhou, H.-X., Szabo, A., Eaton, W. A. & Henry, E. R. (1993) Proc. Natl. Acad. Sci. USA 90, 9547-9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrich J. W., Lambry, J.-C., Kuczera, K., Karplus, M., Poyart, C. & Martin, J.-L. (1991) Biochemistry 30, 3975-3987. [DOI] [PubMed] [Google Scholar]
- 15.Shreve A. P., Franzen, S., Simpson, M. C. & Dyer, R. B. (1999) J. Phys. Chem. B 103, 7969-7975. [Google Scholar]
- 16.Franzen S. & Boxer, S. G. (1997) J. Biol. Chem. 272, 9655-9660. [DOI] [PubMed] [Google Scholar]
- 17.Henry E. R., Sommer, J. H., Hofrichter, J. & Eaton, W. A. (1983) J. Mol. Biol. 166, 443-451. [DOI] [PubMed] [Google Scholar]
- 18.Ansari A., Jones, C. M., Henry, E. R., Hofrichter, J. & Eaton, W. A. (1992) Science 256, 1796-1798. [DOI] [PubMed] [Google Scholar]
- 19.Hagen S. J., Hofrichter, J. & Eaton, W. A. (1995) Science 269, 959-962. [DOI] [PubMed] [Google Scholar]
- 20.Balasubramanian S., Lambright, D. G., Marden, M. C. & Boxer, S. G. (1993) Biochemistry 32, 2202-2212. [DOI] [PubMed] [Google Scholar]
- 21.Lambright D. G., Varadarajan, R. & Boxer, S. G. (1989) J. Mol. Biol. 207, 289-299. [DOI] [PubMed] [Google Scholar]
- 22.Lambright D. G., Balasubramanian, S. & Boxer, S. G. (1991) Chem. Phys. 158, 249-260. [Google Scholar]
- 23.Austin R. H., Beeson, K. W., Eisenstein, L., Frauenfelder, H. & Gunsalus, I. C. (1975) Biochemistry 14, 5355-5373. [DOI] [PubMed] [Google Scholar]
- 24.Ansari A., Berendzen, J., Braunstein, D., Cowen, B. R., Frauenfelder, H., Hong, M. K., Iben, I. E. T., Johnson, J. B., Ormos, P., Sauke, T. B., et al. (1987) Biophys. Chem. 26, 337-355. [DOI] [PubMed] [Google Scholar]
- 25.Ansari A., Jones, C. M., Henry, E. R., Hofrichter, J. & Eaton, W. A. (1994) Biochemistry 33, 5128-5145. [DOI] [PubMed] [Google Scholar]
- 26.Lambright D. G., Balasubramanian, S. & Boxer, S. G. (1993) Biochemistry 32, 10116-10124. [DOI] [PubMed] [Google Scholar]
- 27.Petrich J. W., Lambry, J. C., Balasubramanian, S., Lambright, D. G., Boxer, S. G. & Martin, J. L. (1994) J. Mol. Biol. 238, 437-444. [DOI] [PubMed] [Google Scholar]
- 28.Lim M., Jackson, T. A. & Anfinrud, P. A. (1993) Proc. Natl. Acad. Sci. USA 90, 5801-5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlson M. L., Regan, R., Elber, R., Li, H., Phillips, G. N., Olson, J. S. & Gibson, Q. H. (1994) Biochemistry 33, 10597-10606. [DOI] [PubMed] [Google Scholar]
- 30.Kuczera K., Lambry, J. C., Martin, J. L. & Karplus, M. (1993) Proc. Natl. Acad. Sci. USA 90, 5805-5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liebl U., Bouzhir-Shima, L., Negrerie, M., Vos, M. & Martin, J.-L. (2002) Proc. Natl. Acad. Sci. USA 99, 12771-12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walda K. N., Liu, X. Y., Sharma, V. S. & Magde, D. (1994) Biochemistry 33, 2198-2209. [DOI] [PubMed] [Google Scholar]
- 33.Chatfield M. D., Walda, K. N. & Magde, D. (1990) J. Am. Chem. Soc. 112, 4680-4687. [Google Scholar]
- 34.Sato F., Sakaguchi, Y., Hayashi, H., Iizuka, T. & Shiro, Y. (1992) Biochim. Biophys. Acta 1122, 299-304. [DOI] [PubMed] [Google Scholar]
- 35.Becke A. D. (1997) J. Chem. Phys. 107, 8554-8560. [Google Scholar]
- 36.Lee C. L., Yang, W. & Parr, R. G. (1988) Phys. Rev. B 37, 785-789. [DOI] [PubMed] [Google Scholar]
- 37.Delley B. (1990) J. Chem. Phys. 92, 508-517. [Google Scholar]
- 38.Delley B. (2000) J. Chem. Phys. 113, 7756-7764. [Google Scholar]
- 39.Kohn W. & Sham, L. J. (1965) Phys. Rev. A 137, 1697-1699. [Google Scholar]
- 40.Mermin D. (1965) Phys. Rev. A 137, 1441-1443. [Google Scholar]
- 41.McMahon B. H., Stojkovic, B. P., Hay, P. J., Martin, R. L. & Garcia, A. E. (2000) J. Chem. Phys. 113, 6831-6850. [Google Scholar]
- 42.Harvey J. N. (2000) J. Am. Chem. Soc. 122, 12401-12402. [Google Scholar]
- 43.Franzen S. (2002) J. Phys. Chem. 106, 4533-4542. [Google Scholar]
- 44.Eaton W. A., Hanson, L. K., Stephens, P. J., Sutherland, J. C. & Dunn, J. B. R. (1978) J. Am. Chem. Soc. 100, 4991-5003. [Google Scholar]
- 45.Franzen S., Wallace-Williams, S. E. & Shreve, A. P. (2002) J. Am. Chem. Soc. 124, 7146-7155. [DOI] [PubMed] [Google Scholar]
- 46.Franzen S., Lambry, J. C., Bohn, B., Poyart, C. & Martin, J. L. (1994) Nat. Struct. Biol. 1, 230-233. [DOI] [PubMed] [Google Scholar]
- 47.Franzen S., Bohn, B., Poyart, C. & Martin, J. L. (1995) Biochemistry 34, 1224-1237. [DOI] [PubMed] [Google Scholar]
- 48.Vojtechovsky J., Chu, K., Berendzen, J., Sweet, R. M. & Schlichting, I. (1999) Biophys. J. 77, 2153-2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kachlova G. S., Popov, A. N. & Bartunik, H. D. (1999) Science 284, 473-476. [DOI] [PubMed] [Google Scholar]
- 50.Martin J. L., Migus, A., Poyart, C., Lecarpentier, Y., Astier, R. & Antonetti, A. (1983) Proc. Natl. Acad. Sci. USA 80, 173-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Devault D. (1980) Q. Rev. Biophys. 13, 387-564. [DOI] [PubMed] [Google Scholar]
- 52.Tsubaki M. & Yu, N.-T. (1982) Biochemistry 21, 1140-1144. [DOI] [PubMed] [Google Scholar]
- 53.Spiro T. G., Smulevich, G. & Su, C. (1990) Biochemistry 29, 4497-4508. [DOI] [PubMed] [Google Scholar]
- 54.Kozlowski P. M., Spiro, T. G. & Zgierski, M. Z. (2000) J. Phys. Chem. B 104, 10659-10666. [Google Scholar]
- 55.Li X.-Y. & Zgierski, M. Z. (1992) Chem. Phys. Lett. 188, 16-20. [Google Scholar]
- 56.Franzen S., Fritsch, K. & Brewer, S. H. (2002) J. Phys. Chem. B 106, 11641-11646. [Google Scholar]
- 57.Christensen H. E. M., Conrad, L. S., Mikkelsen, K. V., Nielsen, M. K. & Ulstrup, J. (1990) Inorg. Chem. 29, 2808-2816. [Google Scholar]
- 58.Skourtis S. S. & Mukamel, S. (1995) Chem. Phys. 197, 367-388. [Google Scholar]
- 59.Marcus R. A. (1987) Chem. Phys. Lett. 133, 471-477. [Google Scholar]
- 60.Franzen S., Goldstein, R. F. & Boxer, S. G. (1993) J. Phys. Chem. 97, 3040-3053. [Google Scholar]
- 61.Bixon M., Jortner, J. & MichelBeyerle, M. E. (1995) Chem. Phys. 197, 389-404. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}