

Abstract
Dnmt3L is required for the establishment of maternal methylation imprints at imprinting centers (ICs). Dnmt3L, however, lacks the conserved catalytic domain common to DNA methyltransferases. In an attempt to define its function, we coexpressed DNMT3L with each of the two known de novo methyltransferases, Dnmt3a and DNMT3B, in human cells and monitored de novo methylation by using replicating minichromosomes carrying various ICs as targets. Coexpression of DNMT3L with DNMT3B led to little or no change in target methylation. However, coexpression of DNMT3L with Dnmt3a resulted in a striking stimulation of de novo methylation by Dnmt3a. Stimulation was observed at maternally methylated ICs such as small nuclear ribonucleoprotein polypeptide N (SNRPN), Snrpn, and Igf2r/Air, as well as at various nonimprinted sequences present on the episomes. Stimulation of Dnmt3a by DNMT3L was also observed at endogenous sequences in the genome. Therefore, DNMT3L acts as a general stimulatory factor for de novo methylation by Dnmt3a. The implications of these findings for the function of DNMT3L and Dnmt3a in DNA methylation and genomic imprinting are discussed.
DNA methylation at CpG dinucleotides in mammalian cells is a heritable, epigenetic modification involved in fundamental processes such as the control of gene expression, the silencing of transposable elements, X-inactivation, tumorigenesis, and genomic imprinting (1). Dnmt3a and Dnmt3b are the two DNA methyltransferases known to possess de novo methylation activity in mammalian cells (2, 3). During the process of genomic imprinting, specific loci acquire differential DNA methylation in a parent-of-origin manner. It is believed that this differential methylation is then used by cells to maintain the monoallelic expression patterns that characterize imprinted genes. Recently, a third member of the Dnmt3 family, the DNA methyltransferase-like protein Dnmt3L, was shown to be required for the establishment of maternal methylation imprints in mouse (4, 5). Offspring from Dnmt3L−/− female mice die early during development and show a lack of methylation specifically at maternally methylated imprinting centers (ICs). Dnmt3L, however, lacks the conserved residues known to be involved in DNA methyltransferase activity and is inactive on its own (ref. 5 and see below). This observation has led to the proposal that Dnmt3L might act as a regulator of methylation at imprinted loci rather than as a bona fide DNA methyltransferase (4). More recently, it was shown that Dnmt3L physically interacts with Dnmt3a and Dnmt3b (5), making it likely that Dnmt3L exerts its function through either or both of the de novo methyltransferases. However, the function of Dnmt3L in DNA methylation is unknown.
In an attempt to define the role of Dnmt3L, we coexpressed the human DNMT3L protein together with each of the known de novo methyltransferases, Dnmt3a and DNMT3B, in human cells. The effect of coexpression on DNA methylation was assessed by using replicating minichromosomes as targets. These episomes carried various IC regions that are known to be maternally methylated, thus allowing us to test for specific methylation at these loci. Our results show that whereas coexpression of DNMT3L with DNMT3B results in little or no change of methylation of target sequences, coexpression of DNMT3L with Dnmt3a results in a marked stimulation of the de novo methylation activity of Dnmt3a. Furthermore, stimulation was observed irrespective of DNA sequence, which suggests that DNMT3L acts as a general stimulatory factor for Dnmt3a.
Materials and Methods
Episomes and Expression Vectors.
All IC-containing episomes carry the EBNA1/OriP replication system, a hygromycin resistance marker driven by the HSVTk promoter, and a pBR322-derived replicon including the ampicillin resistance gene. The various IC regions were cloned under the Rous sarcoma virus (RSV)-LTR promoter in the indicated orientation (the RSV-LTR promoter is located between OriP and the hygromycin gene). In all cases, the target regions used for methylation analysis corresponded to the repetitive, maternally methylated regions found at each IC. Plasmids pFC19 and pFC30 carry a 940-bp DNA fragment of the human small nuclear ribonucleoprotein polypeptide N (SNRPN) gene located immediately downstream of exon 1 (GenBank accession no. U41384, coordinates 14275–15215) cloned in the physiological (pFC30) or nonphysiological (pFC19) orientation. Plasmid pFC49 contains an ≈1.1-kb fragment of the murine Snrpn gene located immediately downstream of exon 1 (GenBank accession no. AF081460, coordinates 2731–3860) cloned in the nonphysiological orientation. Plasmid pFC60 carries an ≈1.3-kb DNA fragment located immediately downstream of the murine Air promoter (GenBank accession no. L06446, coordinates 870–2242) cloned in the nonphysiological orientation. Plasmid pCLH22 was described (6) and carries the luciferase gene.
Expression vectors for the murine wild-type Dnmt3a (pMT3aMyc) or its mutated counterpart (pMT3aMut) have been described (3). Note that the murine Dnmt3a and its human homologue are 90% identical. The wild-type human DNMT3B cDNA (corresponding to GenBank accession no. AF176228) was amplified from plasmid pRev-TRE/hD3B (a generous gift from T. Bestor, Columbia University, New York) and cloned into pcDNA3myc, resulting in plasmid pHSA3BMyc. The human wild-type DNMT3L cDNA was amplified from IMAGE clone 1541874 and cloned into pcDNA3myc, resulting in plasmid pD3LMyc.
Cell Lines, Transfections, and Episome Recovery.
The 293/EBNA1 cell line was used in all experiments, and transfections were performed by using the calcium phosphate method (6, 7). For cotransfections, 500 ng of each desired plasmid was added to each 35-mm well after thorough mixing. Cells were allowed to grow for 2–3 days before being replated in a 100-mm-diameter plate. On reaching confluence, cells were harvested for episomal DNA extraction according to the Hirt method (8). No selection was applied during cell culture. All transfections were done in duplicate, and all experiments were independently reproduced. Expression of Dnmt3a, DNMT3B, and DNMT3L after transient transfection into 293/EBNA1 cells was detected by Western blotting after immunoprecipitation by using the Myc antibody. No significant change in the expression of Dnmt3a or DNMT3B could be detected on cotransfection of DNMT3L (data not shown).
Analysis of DNA Methylation on Hirt Harvests by Southern Blotting.
The recovered DNA was digested overnight with an excess of HpaII enzyme (New England Biolabs). Then, the samples were fractionated through 0.8–1.2% agarose gels, depending on the size of the expected bands, and the DNA was Southern transferred and probed by using 32P-radiolabeled probes, as indicated. The membranes were exposed to a Molecular Dynamics screen. Scanning and data analysis were performed by using a PhosphorImager 445SI (Molecular Dynamics) running IMAGEQUANT V.5.0.
Analysis of DNA Methylation at Sequences in the Genome.
The firefly luciferase gene flanked by the RSV-LTR promoter was integrated into the genome of 293/EBNA1 cells at two different loci (O13 and O21; ref. 9) and was used to measure methylation after Dnmt3a or Dnmt3a + DNMT3L expression vectors were transfected in. For this, cells were seeded in 100-mm-diameter plates and subjected to transient transfection by using the calcium phosphate method (transfections were performed in duplicates). After transfection (3.5 days), the cells were harvested, and genomic DNA was extracted by using a standard SDS/proteinase K method, followed by phenol and chloroform extractions. The DNA (≈10 μg per sample) was digested with HindIII and HhaI or HindIII and HpaII for 16 h with an excess enzyme and then fractionated by agarose gel electrophoresis. The DNA was Southern transferred to a nylon membrane and hybridized to a 32P-radiolabeled probe complementary to the luciferase coding sequence. CpG methylation at HhaI or HpaII sites results in fragments of increased size after exposure of the Southern blots to a PhosphorImager screen. The membrane was then stripped and reprobed with an ≈3-kb region corresponding to most of the HOXA9 gene and upstream sequences to check for methylation at an endogenous gene (data not shown).
Bisulfite Genomic Sequencing.
Bisulfite genomic sequencing was carried out as described (10, 11). Samples were digested by PstI before sodium bisulfite treatment. For the human SNRPN IC, a 287-bp fragment of the top strand of plasmid pFC19 containing 23 CpG sites was analyzed (GenBank accession no. U41384, coordinates 14426–14713). For plasmid pCLH22, an ≈250-bp fragment of the top strand of the luciferase gene containing 28 CpG sites (corresponding to region 2 in ref. 12) was analyzed. For both plasmids, an ≈150-bp region of the neighboring EBNA1 gene containing 11 CpG sites was also analyzed on the top strand (corresponding to region 4 in ref. 12). Sequencing was performed on several independent DNA harvests, and the results were pooled. Molecules in which nonconverted cytosines were found at non-CpG sites were not taken into account.
Results
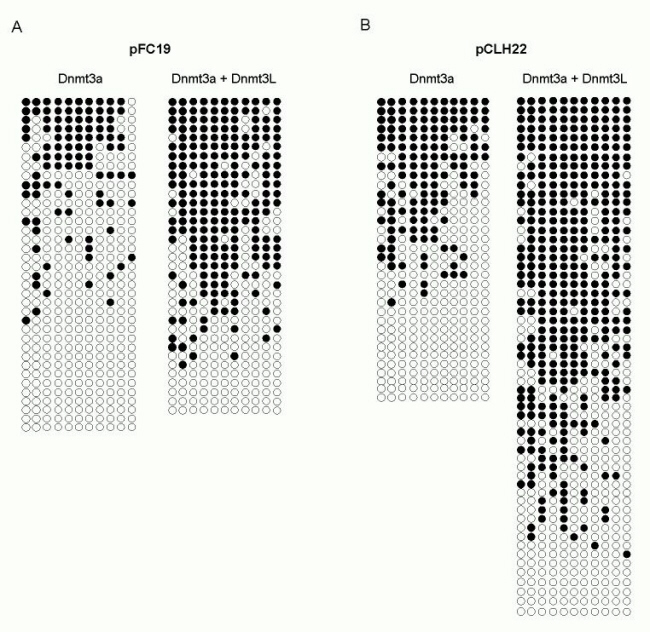
Cotransfection experiments were performed in human 293/EBNA1 cells by using, at first, a minichromosome containing an ≈1-kb portion of the human SNRPN IC as a target. The SNRPN IC is a well studied example of a maternally methylated IC (13). The region used here includes most of the differentially methylated CpG islands as well as the repetitive DNA sequences found at the SNRPN locus. This DNA fragment was cloned into a replicating Epstein–Barr virus-based episome downstream of an RSV-LTR promoter in either physiological or nonphysiological orientation (resulting in plasmids pFC30 and pFC19, respectively). After transfection, the cells were cultured for 7 days, and the DNA was recovered by Hirt harvest (8). Methylation on the transfected episomes was analyzed by restriction digests with methylation-sensitive enzymes such as HpaII and Southern blot hybridization by using the SNRPN IC as a probe. Increased-size HpaII fragments, indicative of DNA methylation, were detected irrespective of the orientation of the insert when Dnmt3a was expressed (Fig. 1A, lanes 2 and 6). This result shows that the SNRPN IC is a good target for Dnmt3a. No methylation was detected in the absence of Dnmt3a (Fig. 1A, lanes 1 and 5) or in the presence of DNMT3L alone (Fig. 1A, lanes 3 and 7). This observation suggests that human DNMT3L has no methylation activity on its own in 293/EBNA1 cells. When both Dnmt3a and DNMT3L were expressed, a larger fraction of molecules became methylated, and high molecular weight species (indicated by an arrow) corresponding to highly methylated molecules became detectable (Fig. 1A, lanes 4 and 8). The size of these large molecules is consistent with most, if not all, of the HpaII sites on the minichromosome being methylated. These observations indicate that coexpression of DNMT3L and Dnmt3a resulted in a strong stimulation of DNA methylation. This stimulatory effect clearly depends on Dnmt3a catalytic activity, because coexpression of DNMT3L and a mutant form of Dnmt3a carrying a mutation in the catalytic center showed no detectable methylation (data not shown). These data demonstrate that DNMT3L stimulates de novo methylation by Dnmt3a at the SNRPN IC in vivo.
Fig 1.

DNMT3L stimulates de novo methylation by Dnmt3a but not by DNMT3B at the SNRPN IC. DNA harvested from human 293/EBNA1 cells carrying pFC19 or pFC30 (in which a key ≈1-kb region of the SNRPN IC was cloned in either orientation) was digested with an excess of the methylation-sensitive enzyme HpaII. The resulting DNA species were separated by electrophoresis through an agarose gel, transferred to a nylon membrane, and hybridized with a probe corresponding to the SNRPN IC. Two bands (684 and 754 bp for pFC19, and 684 and 221 bp for pFC30) were expected if no methylation was present. CpG methylation prevents cleavage by HpaII and results in higher molecular weight species that can be easily separated. (A) Each target plasmid was introduced into cells alone, together with an expression vector for Dnmt3a, or together with expression vectors for both Dnmt3a and DNMT3L, as indicated. (B) Plasmid pFC19 was introduced in cells either with DNMT3B or with both DNMT3B and DNMT3L expression vectors, as indicated.
In the presence of DNMT3B alone, methylation of the SNRPN IC was readily detected (Fig. 1B, lanes 1 and 2). However, coexpression of DNMT3L with DNMT3B did not result in any appreciable change in methylation (Fig. 1B, lanes 3 and 4). Thus, it seems that expression of DNMT3L has little or no impact on the activity of DNMT3B at this target sequence.
To assess whether stimulation of Dnmt3a by DNMT3L also occurred at other imprinted loci, we repeated the cotransfection experiments by using two distinct maternally methylated ICs as targets: the mouse Snrpn IC and the Igf2r/Air IC. Detection of DNA methylation was done as described above except that the Southern blots were probed with each respective target sequence. An ≈1.1-kb fragment corresponding to the repetitive, differentially methylated region of the mouse Snrpn IC was used. Although the mouse Snrpn IC possesses a structure similar to the human SNRPN IC, their sequences are not homologous (14, 15), making this region a distinct target for methylation. The Snrpn IC showed methylation on exposure to Dnmt3a but not to a mutated version of the protein (Fig. 2A, lanes 1 and 2). In the presence of DNMT3L alone, no methylation of the Snrpn IC was observed (Fig. 2A, lane 5). However, when DNMT3L and Dnmt3a were coexpressed, highly methylated molecules were consistently observed (arrow), indicating a large stimulation of DNA methylation (Fig. 2A, lane 3). The enhanced methylation activity seen in the presence of Dnmt3a and DNMT3L was not detected when an inactive catalytic mutant of Dnmt3a was cotransfected with DNMT3L (Fig. 2A, lane 4). The mouse Igf2r/Air IC is also maternally methylated and coordinates the imprinted expression of several imprinted genes, including Igf2r (16). The region used here corresponds to most of the differentially methylated CpG islands found downstream of the Air promoter and is also very repetitive (17). As documented for the Snrpn IC, no methylation was observed at the Igf2r/Air IC in the absence of any methyltransferase or in the presence of a mutated form of Dnmt3a (Fig. 2B, lanes 1 and 4). Unlike SNRPN and Snrpn, expression of Dnmt3a alone only resulted in a modest methylation activity at the Igf2r/Air IC (Fig. 2B, lanes 2 and 3). However, coexpression of DNMT3L with Dnmt3a resulted in a pronounced increase of methylation activity as evidenced by the appearance of highly methylated molecules (Fig. 2B, lanes 5 and 6, arrow). Stimulation similarly depended on the presence of an intact Dnmt3a catalytic center, as judged by the lack of DNA methylation when a mutant Dnmt3a was cotransfected (Fig. 2B, lane 7). Thus, coexpression of DNMT3L with Dnmt3a resulted in a marked increase of de novo methylation activity by Dnmt3a at all three maternally methylated ICs tested. Note that, contrary to what was observed for Dnmt3a, no appreciable change in methylation was observed at either Snrpn or Igf2r/Air when DNMT3B and DNMT3L were coexpressed (data not shown).
Fig 2.

DNMT3L stimulates de novo methylation by Dnmt3a at maternally methylated ICs and nonimprinted sequences. Key regions of the murine Snrpn IC (A) and murine Igf2r/Air IC (B) carried by pFC49 and pFC60, respectively, were introduced into cells together with expression vectors for Dnmt3a and/or DNMT3L, as indicated. (C) pCLH22, which carries the luciferase gene, was used. The probes used to detect methylation correspond to the respective ICs or to the luciferase gene.
The appearance of highly methylated molecules in the presence of DNMT3L suggests that methylation was not constrained to the ICs but was also present on the backbone sequences of the minichromosomes. To investigate the possibility that DNMT3L stimulates methylation by Dnmt3a at nonimprinted sequences, we used pCLH22, an episome carrying the firefly luciferase gene, as a target in cotransfection experiments. As observed (3), evidence of DNA methylation was readily seen when Dnmt3a was expressed alone (Fig. 2C, lanes 1 and 2), whereas no activity was detected when a mutated version of the protein was expressed. Coexpression of DNMT3L and Dnmt3a also consistently resulted in a large stimulation of methylation at the luciferase locus (Fig. 2C, lanes 5 and 6). Stimulation also depended on the catalytic activity of Dnmt3a, because no detectable methylation was observed in cotransfections with a mutant Dnmt3a (Fig. 2C, lanes 7 and 8). Additionally, the EBNA1 locus, located ≈2.5 kb upstream of luciferase, as well as sequences derived from the pBR322 vector, located ≈2 kb downstream of luciferase on the backbone of the episomes used here, also showed a strong increase in methylation when DNMT3L and Dnmt3a were both expressed (see below; data not shown). These findings clearly demonstrate that DNMT3L stimulates de novo methylation by Dnmt3a irrespective of the target sequence.
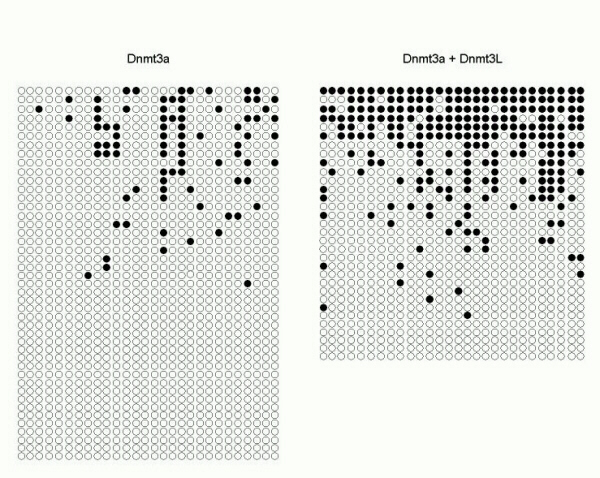
We used the bisulfite genomic sequencing method (10) to examine the stimulation of Dnmt3a methylation activity by DNMT3L at individual regions. For this, we first analyzed an ≈290-bp fragment on the G-rich, top strand of the SNRPN locus containing 23 CpG sites (carried by plasmid pFC19). Methylation by Dnmt3a alone was significant, corresponding to an overall efficiency of CpG methylation of ≈8%. In the presence of both DNMT3L and Dnmt3a, the efficiency increased on average by 4.1-fold at SNRPN (Fig. 3, Table 1). Analysis of individual CpG sites methylated by Dnmt3a alone revealed that some sites were methylated more frequently than others (i.e., the maximal ratio from the highest to the lowest methylated site was 10, Fig. 3). This finding could be attributed to sequence preference by Dnmt3a, as described in vitro (12). Note, however, that the hierarchy of sites found here does not exactly correspond to the one found in vitro, possibly because the DNA is now in chromatin and bound by proteins. Alternatively, this observation could simply reflect differences in accessibility of the various CpG sites in DNA. Stimulation of Dnmt3a by DNMT3L was seen at each site but resulted in a more uniform pattern of methylation (Fig. 3). We then performed bisulfite sequencing on episomes carrying the nonimprinted luciferase gene instead of the SNRPN IC (plasmid pCLH22) and found that CpG methylation at luciferase increased 4.1-fold in the presence of DNMT3L and Dnmt3a with an increase observed at each CpG site (Table 1; Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). For both plasmids, we also sequenced a region of the neighboring EBNA1 gene. CpG methylation in this region increased only 2-fold, which probably reflects the fact that this region is already a preferred target for methylation by Dnmt3a alone, as judged by the much higher basal methylation efficiency (Table 1; Fig. 7, which is published as supporting information on the PNAS web site; ref. 3). These results confirm that DNMT3L acts as a general stimulatory factor for de novo methylation by Dnmt3a and, further, show that the increase of methylation is not limited to the restriction sites analyzed by Southern blots. Analysis of the distribution of methylated sites among individual molecules also revealed that stimulation by DNMT3L favored the production of highly methylated molecules (Fig. 4). Both at the SNRPN IC and at the luciferase gene, the majority of molecules recovered from cells expressing Dnmt3a alone did not carry any CpG methylation, whereas most of the molecules that did carry methylation showed a mosaic profile with <25% of the potential CpG sites being methylated (Fig. 4). In the presence of Dnmt3a and DNMT3L, the proportion of unmethylated molecules was reduced, and a strong increase in the density of methylation on methylated molecules was observed. In the case of the luciferase gene, extensively methylated molecules appeared only in the presence of Dnmt3a and DNMT3L (Fig. 4B). These data are consistent with our observation that highly methylated episomes are observed only in the additional presence of DNMT3L based on Southern blots (Figs. 1 and 2).
Fig 3.

Coexpression of Dnmt3a and DNMT3L results in stimulation of methylation at all CpG sites. Methylation at the SNRPN locus was analyzed by using bisulfite genomic sequencing on samples recovered from cells carrying pFC19 and expressing Dnmt3a alone (Left) or Dnmt3a and DNMT3L together (Right). The distribution of methylated sites (•) at each of the 23 CpG sites along the top strand of the SNRPN IC is depicted. A histogram representing the total number of methylation events observed at a given site for the whole sample is shown at the bottom. Sample size was identical for both categories (53 independent molecules analyzed per group).
Table 1.
Methylation analysis
|
|
pFC19 | pCLH22 | ||||||
|---|---|---|---|---|---|---|---|---|
| SNRPN | EBNA1 | Luciferase | EBNA1 | |||||
| 3a | 3a + 3L | 3a | 3a + 3L | 3a | 3a + 3L | 3a | 3a + 3L | |
| me-CpG | 101 | 418 | 103 | 211 | 83 | 246 | 142 | 387 |
| n | 53 | 53 | 37 | 35 | 44 | 32 | 34 | 60 |
| Methylation, % | 8.3 | 34.3 | 27.5 | 54.8 | 6.7 | 27.5 | 38 | 58.6 |
| Fold stimulation | 4.1 | 2.0 | 4.1 | 1.5 | ||||
Regions of the SNRPN IC (carried by pFC19) or the luciferase gene (carried by pCLH22) were analyzed for methylation by the bisulfite sequencing method. For both episomes, a region of the neighboring EBNA1 gene was also analyzed. The number of methylated CpG sites observed for each region (me-CpG) and the sample size (n) are included in each column. The efficiency of CpG methylation (% methylation) is calculated by dividing the number of methylated sites observed by the total number of CpG sites in each region. The fold stimulation corresponds to the ratio of the efficiency observed when Dnmt3a and DNMT3L were expressed together relative to the efficiency observed when Dnmt3a was expressed alone. Note that molecules carrying nonconverted cytosines at non-CpG sites were excluded from the analysis. Such molecules were observed at low frequencies (3/106) only for the SNRPN region.
Fig 4.

Coexpression of Dnmt3a and DNMT3L results in extensive DNA methylation patterns. The percentage of CpG methylation observed at the SNRPN IC (A) or at the luciferase gene (B) was determined in individual molecules analyzed by bisulfite sequencing. Molecules were grouped into five classes depending on the percentage of methylation observed, as shown on the x axis. The fraction of each class within the total sample size is indicated on the y axis. Methylation of the target region resulted from Dnmt3a alone (gray bars) or Dnmt3a and DNMT3L together (black bars).
To verify that stimulation of Dnmt3a by DNMT3L was not restricted to sequences present on episome-based vectors, we used two cell lines derived from 293/EBNA1 cells in which the luciferase gene was integrated into the genome (9) and in which Dnmt3a or Dnmt3a + DNMT3L expression vectors were then introduced by transfection, as described above. Methylation at luciferase was then assessed by Southern blot after digestion of the genomic DNA with HhaI, which is sensitive to CpG methylation (the DNA was also digested with HindIII). In the absence of Dnmt3a, no methylation was observed at the luciferase locus in either cell line (Fig. 5, lanes 1 and 6). When Dnmt3a was expressed, weak methylation was observed, as judged by the appearance of fragments with increased size (Fig. 5, lanes 2, 3, 7, and 8). When both Dnmt3a and DNMT3L were expressed, high molecular weight species became clearly visible, reflecting a higher density of methylated CpG residues (Fig. 5, lanes 4, 5, 9, and 10). Similar results were obtained on digestion of the DNA with HpaII instead of HhaI (data not shown). Thus, DNMT3L stimulates de novo methylation by Dnmt3a at the luciferase gene integrated in the genome. Reprobing the membrane with a probe complementary to the endogenous nonimprinted HOXA9 gene showed a similar result (data not shown). In contrast, however, we did not detect any change in methylation at the endogenous SNRPN IC on expression of Dnmt3a or Dnmt3a + DNMT3L, despite the presence of unmethylated HhaI and HpaII sites, presumably from the unmethylated paternal allele (data not shown). This observation suggests that the endogenous SNRPN IC might be protected from de novo methylation. These data show that methylation by Dnmt3a and stimulation of Dnmt3a by DNMT3L occur at sequences present on episome-based vectors as well as at sequences present in the genome, with the possible exception of some endogenous sequences such as the SNRPN IC, which might be protected from de novo methylation.
Fig 5.

Stimulation by DNMT3L is detected at the luciferase gene integrated in the genome. Two cell lines (O13 and O21) carrying the luciferase gene at distinct loci were transfected with Dnmt3a or Dnmt3a + DNMT3L expression vectors. After transfection (3.5 days), genomic DNA was harvested and digested with excess HindIII and HhaI, a methylation-sensitive enzyme. The resulting DNA species were then separated by agarose gel electrophoresis, Southern transferred onto a membrane, and hybridized to a probe complementary to the luciferase gene. The presence of Dnmt3a and/or DNMT3L expression vectors was as indicated. CpG methylation prevents cleavage by HhaI and results in higher molecular weight species that can be easily distinguished.
Discussion
In this study, we have investigated the role of the DNMT3L protein in DNA methylation, particularly in relation to the two known de novo DNA methyltransferases, Dnmt3a and DNMT3B. Our results constitute evidence for a functional role for DNMT3L in DNA methylation. We have found that DNMT3L strongly stimulates de novo methylation by Dnmt3a at all tested DNA sequences. Stimulation of Dnmt3a by DNMT3L results in an increased fraction of methylated DNA molecules and in a higher density of methylation on these molecules. Unlike Dnmt3a, however, coexpression of DNMT3L with DNMT3B did not result in any significant change in methylation activity.
Dnmt3L has been described as necessary for the establishment of maternal methylation imprints, because loss of Dnmt3L function resulted in a loss of methylation at several maternally methylated ICs (4, 5). These observations are consistent with the hypothesis that Dnmt3L can recognize maternal ICs and recruit a de novo methyltransferase to carry out methylation at these loci. Our observation that DNMT3L stimulates de novo methylation by Dnmt3a at all tested DNA sequences, however, seems incompatible with a role in recognition and targeting of ICs. The general stimulation of Dnmt3a activity by DNMT3L suggests, instead, that the role of DNMT3L in DNA methylation may not be limited to ICs but rather that it could extend on a genome-wide level. To date, the only sequence for which DNA methylation levels were found to decrease in the absence of Dnmt3L directly in oocytes corresponds to the maternally methylated Snrpn IC (4). The inference that Dnmt3L is specifically required for the establishment of methylation imprints at maternally methylated sequences stems from the fact that inspection of several types of highly repetitive DNA sequences such as intracisternal A particle retroposons, pericentric satellite DNA repeats, and endogenous provirus sequences, as well as paternally methylated imprinted sequences, did not reveal any change in DNA methylation in the absence of Dnmt3L (4, 5). Some of these repetitive sequences, however, are known to be methylated by DNMT3B (18, 19), which, in our hands, is not affected by the presence of DNMT3L, at least for the sequences we examined. Furthermore, it seems apparent that methylation at maternally and paternally imprinted sequences is established according to two different pathways (20). Finally, most of the analyses of methylation changes were not performed directly in oocytes but in embryos derived from Dnmt3L−/− females, which could have reacquired DNA methylation later during postimplantation development. Therefore, it is possible that a closer inspection of methylation changes in the absence of Dnmt3L in oocytes will reveal the existence of DNA sequences other than ICs that are methylated by Dnmt3a and Dnmt3L. In this regard, we note that Dnmt3L−/− males are sterile and show severe hypogonadism, which suggests that Dnmt3L is necessary for the differentiation of spermatogonial cells (4, 5). Whether Dnmt3L exerts its effect through the control of DNA methylation or through another pathway remains to be established. Alternatively, it is possible to reconcile all existing data by proposing that Dnmt3a and Dnmt3L are targeted to maternally methylated ICs by a third, as yet unknown factor. Interestingly, a hitherto unknown trans-acting factor required for the establishment of correct maternal imprints in oocytes was recently reported (21). The identity of this factor and whether it actually takes part in IC recognition await further study. If such a targeting factor exists, our observation that DNMT3L stimulates Dnmt3a irrespective of the sequence context could be caused by its absence from the cell line used here.
As shown here, the stimulation conferred by DNMT3L on the de novo methylation activity of Dnmt3a allows for extensive methylation patterns to be imposed. On maturation, growing oocytes express Dnmt3L and Dnmt3a (4, 5), and methylation profiles at maternally methylated ICs progress from mosaic to dense patterns (22). This progression corresponds well to what we observe when methylation is carried out solely by Dnmt3a (mosaic patterns) compared with when DNMT3L is also expressed (dense patterns). The biological requirement for the establishment of a dense methylation pattern at ICs can be understood in the light of the various genome-wide methylation reprogramming events that characterize early development. Both paternal and maternal genomes undergo programmed demethylation during the first cell divisions after formation of the zygote (23). In this context, it was established that the regions that are most likely to “survive” this global zygotic demethylation phase correspond to the most densely methylated regions (24). It is possible that the function of DNMT3L in the context of genomic imprinting is to stimulate Dnmt3a activity at ICs to achieve the high levels of methylation required to ensure the survival of these imprints through the following programmed demethylation phase. This proposal agrees with the facts that both Dnmt3a and DNMT3L are expressed in developing oocytes and that Dnmt3a and Dnmt3b alone are not sufficient for establishing methylation imprints (2, 4, 5). Furthermore, it was recently shown that Dnmt3L and Dnmt3a physically interact and that loss of Dnmt3a function in oocytes (in an otherwise Dnmt3b+/− background) resulted in loss of maternal imprints (5). Our data, together with these observations, suggest that Dnmt3a, through its functional interaction with DNMT3L, is the primary methyltransferase responsible for methylation at maternally methylated ICs.
In the male germ line, the paternal alleles of maternally methylated ICs are resistant to methylation and remain unmethylated during development, therefore establishing the asymmetry that sustains imprinted gene expression in the soma. Interestingly, Dnmt3L and Dnmt3a are also expressed in male germ cells and in the early embryo (2, 5). This observation suggests that maternally methylated ICs are protected from methylation in the male germ line and possibly in somatic cells by as yet unknown factors. Here, we showed that although methylation by Dnmt3a and its stimulation by DNMT3L clearly occur at two distinct genomic targets (the integrated luciferase gene and the endogenous HOXA9 gene), we did not observe increased methylation at the endogenous SNRPN IC. These results are consistent with data obtained for embryonic stem (ES) cells, in which overexpression of DNMT1 led to methylation of numerous sequences with the exception of certain ICs, including the SNRPN IC (20, 25). The mechanism by which ICs are protected from methylation remains to be elucidated.
Supplementary Material
Acknowledgments
We thank Drs. Ryan Irvine and Kefei Yu for critical reading of the manuscript. This work was supported in part by National Institutes of Health Grant RO1 GM60237 (to C.-L.H.). M.R.L. acknowledges support from the National Institutes of Health and the Beckman Macular Research Center.
Abbreviations
IC, imprinting center
SNRPN, small nuclear ribonucleoprotein polypeptide N
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Bestor T. H. (2000) Hum. Mol. Genet. 9, 2395-2402. [DOI] [PubMed] [Google Scholar]
- 2.Okano M., Bell, D. W., Haber, D. A. & Li, E. (1999) Cell 99, 247-257. [DOI] [PubMed] [Google Scholar]
- 3.Hsieh C. L. (1999) Mol. Cell. Biol. 19, 8211-8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bourc'his D., Xu, G. L., Lin, C. S., Bollman, B. & Bestor, T. H. (2001) Science 294, 2536-2539. [DOI] [PubMed] [Google Scholar]
- 5.Hata K., Okano, M., Lei, H. & Li, E. (2002) Development (Cambridge, U.K.) 129, 1983-1993. [DOI] [PubMed] [Google Scholar]
- 6.Hsieh C. L. (1994) Mol. Cell. Biol. 14, 5487-5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wigler M., Sweet, R., Sim, G. K., Wold, B., Pellicer, A., Lacy, E., Maniatis, T., Silverstein, S. & Axel, R. (1979) Cell 16, 777-785. [DOI] [PubMed] [Google Scholar]
- 8.Hirt B. (1967) J. Mol. Biol. 26, 365-369. [DOI] [PubMed] [Google Scholar]
- 9.Lin I. G. & Hsieh, C. L. (2001) EMBO Rep. 2, 108-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark S. J., Harrison, J., Paul, C. L. & Frommer, M. (1994) Nucleic Acids Res. 22, 2990-2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsieh C. L. (1999) Mol. Cell. Biol. 19, 46-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin I. G., Han, L., Taghva, A., O'Brien, L. E. & Hsieh, C. L. (2002) Mol. Cell. Biol. 22, 704-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicholls R. D., Saitoh, S. & Horsthemke, B. (1998) Trends Genet. 14, 194-200. [DOI] [PubMed] [Google Scholar]
- 14.Shemer R., Birger, Y., Riggs, A. D. & Razin, A. (1997) Proc. Natl. Acad. Sci. USA 94, 10267-10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gabriel J. M., Gray, T. A., Stubbs, L., Saitoh, S., Ohta, T. & Nicholls, R. D. (1998) Mamm. Genome 9, 788-793. [DOI] [PubMed] [Google Scholar]
- 16.Zwart R., Sleutels, F., Wutz, A., Schinkel, A. H. & Barlow, D. P. (2001) Genes Dev. 15, 2361-2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wutz A., Smrzka, O. W., Schweifer, N., Schellander, K., Wagner, E. F. & Barlow, D. P. (1997) Nature 389, 745-749. [DOI] [PubMed] [Google Scholar]
- 18.Xu G. L., Bestor, T. H., Bourc'his, D., Hsieh, C. L., Tommerup, N., Bugge, M., Hulten, M., Qu, X., Russo, J. J. & Viegas-Pequignot, E. (1999) Nature 402, 187-191. [DOI] [PubMed] [Google Scholar]
- 19.Hansen R. S., Wijmenga, C., Luo, P., Stanek, A. M., Canfield, T. K., Weemaes, C. M. & Gartler, S. M. (1999) Proc. Natl. Acad. Sci. USA 96, 14412-14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biniszkiewicz D., Gribnau, J., Ramsahoye, B., Gaudet, F., Eggan, K., Humpherys, D., Mastrangelo, M. A., Jun, Z., Walter, J. & Jaenisch, R. (2002) Mol. Cell. Biol. 22, 2124-2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Judson H., Hayward, B. E., Sheridan, E. & Bonthron, D. T. (2002) Nature 416, 539-542. [DOI] [PubMed] [Google Scholar]
- 22.Lucifero D., Mertineit, C., Clarke, H. J., Bestor, T. H. & Trasler, J. M. (2002) Genomics 79, 530-538. [DOI] [PubMed] [Google Scholar]
- 23.Reik W., Dean, W. & Walter, J. (2001) Science 293, 1089-1093. [DOI] [PubMed] [Google Scholar]
- 24.Howell C. Y., Steptoe, A. L., Miller, M. W. & Chaillet, J. R. (1998) Mol. Cell. Biol. 18, 4149-4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tucker K. L., Beard, C., Dausmann, J., Jackson-Grusby, L., Laird, P. W., Lei, H., Li, E. & Jaenisch, R. (1996) Genes Dev. 10, 1008-1020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}