

Abstract
Polycystin-1 plays an essential role in renal tubular morphogenesis, and disruption of its function causes cystogenesis in human autosomal-dominant polycystic kidney disease (ADPKD). We demonstrated that polycystin-1 undergoes cleavage at G protein coupled receptor proteolytic site in a process that requires the receptor for egg jelly domain. Most of the N-terminal fragment remains tethered at the cell surface, although a small amount is secreted. PKD1-associated mutations in the receptor for egg jelly domain disrupt cleavage, abolish the ability of polycystin-1 to activate signal transducer and activator of transcription-1, and induce tubulogenesis in vitro. We conclude that the cleavage of polycystin-1 is likely essential for its biologic activity.
Autosomal-dominant polycystic kidney disease (ADPKD) is a common Mendelian disorder affecting 1 of every 1,000 people (1). It can be caused by mutations of at least two genes, PKD1 and PKD2 (2). The hallmark of the disease is the development of multiple bilateral cysts in kidney, resulting in progressive renal failure in 50% of patients by their sixth decade of age. ADPKD is a systemic disease with many extra-renal manifestations including liver cysts, pancreatic cysts, and intracranial aneurysms (1).
Gene-targeting experiments suggest that PKD1 may function to regulate terminal differentiation of tubular structures in kidney and liver (3) and maintain the structural integrity of the vasculature (4). Our in vitro studies have shown that the PKD1 gene product polycystin-1 can slow growth, promote resistance to apoptosis, and induce spontaneous tubulogenesis in Madin–Darby canine kidney (MDCK) cells (5). Recently, we showed that polycystin-1 regulates the cell cycle by means of direct activation of the Janus kinase-signal transducer and activator of transcription (JAK-STAT)-signaling pathway (6).
Polycystin-1 contains a large N-terminal extracellular region with a combination of functional motifs, an odd number of transmembrane segments (TMs), and an ≈200-amino acid intracellular carboxyl terminus (7). A large proportion of the extracellular amino terminus is composed of 15 tandemly repeated polycystic kidney disease (PKD) repeats, which can form homophilic interactions (8). The cytoplasmic carboxyl terminus of polycystin-1 contains a coiled coil domain that is a site of interaction with polycystin-2 (9). We recently showed (10) that this interaction targets polycystin-2 to the cell surface where the complex functions as a cation channel. The C terminus also binds to a number of other intracellular proteins, including Gα-subunits of heterotrimeric G proteins (11), which is consistent with a function for polycystin-1 as a G protein-coupled receptor (12).
Situated between the last PKD repeat and the first TM is the receptor for egg jelly (REJ) domain, a structure of unknown function, which was originally described in sea urchin receptor for egg jelly (suREJ1; ref. 13). Immediately following this domain in both polycystin-1 and suREJ1 is a G protein-coupled receptor proteolytic site (GPS) domain (13, 14). The GPS domain was first demonstrated to be the internal cleavage site for the neuronal G protein-coupled receptor (GPCR) latrophilin/CL-1, with the actual cleavage site at HL^T (where “^” identifies the position of cleavage) ≈20 residues N-terminal from the membrane (15). Latrophilin/CL-1 belongs to the long-N-terminal family B GPCR-related seven-transmembrane receptor (LNB-TM7) family of proteins, a group related to family B GPCRs that is extraordinary for having unusually large and complex N-terminal extracellular regions (16). The relative position of the GPS domain is similar in polycystin-1 and members of the LNB-TM7 family, and cleavage at the site is predicted for both. Recently, suREJ3, a distant member of polycystin family in sea urchin, was shown to undergo cleavage, presumably at the GPS domain (17). The conserved nature of the GPS domain in LNB-TM7s suggests that cleavage at this site is important for their function.
In this paper, we demonstrate that polycystin-1 undergoes cleavage at the GPS domain and provide compelling evidence that the cleavage is likely essential for the normal function of polycystin-1.
Materials and Methods
Generation of Polycystin-1 Cleavage Mutant Constructs.
The construct WT was the base plasmid for the generation of the cleavage mutants. WT was derived from pCI-PKD1-Flag (10) by eliminating the BglII site of the vector. The cleavage mutants were generated in a two-step PCR procedure by using pfu DNA polymerase and WT as template. 5′ and 3′ PCR products with compatible ends were generated with the desired mutant nucleotide included in one of the primers, digested with the appropriate enzymes, and then cloned into the same sites of WT in a tri-molecular ligation reaction. Details for specific constructs are available upon request. The FL cDNAs encoding the WT and cleavage mutant polycystin-1 proteins were subcloned into a modified episomal expression vector pREP10 (Invitrogen) to generate episomal expression constructs for each protein.
Generation of Antibody.
To generate a polyclonal α-leucine-rich repeat (LRR) antibody, a cDNA fragment encoding the LRR domain corresponding to residues 27–200 was cloned into pET28c (Novagen). After expression in Escherichia coli, the protein was affinity-purified by using Ni-agarose beads according to the instruction of the manufacturer (Qiagen, Chatsworth, CA) and then used to immunize rabbits. α-CT has been described (5).
Cleavage Assay.
The construct was transfected into various mammalian cells by using Lipofectamine Plus (Life Technologies, Rockville, MD). The cells were lysed in buffer [20 mM sodium phosphate, pH 7.2/150 mM NaCl/1 mM EDTA/10% (vol/vol) glycerol/0.5% Triton X-100] containing complete protease inhibitor cocktails (Roche Molecular Biochemicals) for 1 h on ice. The cleared supernatant was subjected to immunoprecipitation (IP) by using either agarose-conjugated α-Flag (Sigma) or affinity-purified α-CT or α-GFP (Roche Molecular Biochemicals). The IP product was resolved on a 4% or 3–8% SDS gel, electro-blotted to a poly(vinylidene difluoride) membrane, and probed with various antibodies.
Immunoflorescence.
Human embryonic kidney (HEK) cells were transfected with the expression constructs and were fixed 24 h later in 3% (wt/vol) paraformaldehyde and 2% sucrose in PBS (pH 7.4) for 10 min, followed by incubation in 1% (wt/vol) SDS in TBS to permeabilize membranes. After preincubation with 5% (wt/vol) BSA in TBS, cells were incubated with α-CT for 30 min at 37°C, washed, and stained with Rhodamine-conjugated anti-mouse IgG antibody for 30 min, then washed thoroughly and imaged. Florescence images were obtained by using a confocal microscope, Noran OZ (Noran Instruments, Middleton, WI).
In Vitro Tubulogenesis Assay.
MDCK cells were transfected with episomal expression construct by using Lipofectamine 2000 (Invitrogen). Two days after transfection, cells were cultured for up to 2 weeks under selection by using hygromycin (150 μg/ml) to eliminate untransfected cells. Surviving colonies were pooled and grown to confluence. Then, they were trypsinized and cultured in a collagen type I mixture for up to 2 weeks as described (5).
Miscellaneous.
Surface biotinylation experiments were performed according to the manufacturer's instructions with slight modification (Pierce). The pulse-and-chase experiment used Tran35S-Label and CELLect Met/Cys Deficient media (ICN). The luciferase assays were carried out as described (6).
Results
Polycystin-1 Cleavage at the GPS Domain and Tethering of the NTF.
The GPS domain is located immediately N-terminal to the first TM of polycystin-1 (Fig. 1 A and B). Cleavage at the consensus site HL^T was predicted to result in an ≈325 kDa N-terminal fragment (NTF) of 3,048 amino acids and an ≈150 kDa C-terminal fragment (CTF) of 1,254 amino acids (Fig. 1B). To test this hypothesis, we transiently expressed the construct WT, which encodes full-length (FL) polycystin-1 with a C-terminal Flag-epitope, in several mammalian cell lines (HEK, HeLa, COS1, CHO, and HepG2). Recombinant polycystin-1 was immunoprecipitated with α-Flag or an antibody that recognizes the C terminus of polycystin-1 (α-CT) and analyzed by Western blotting by using the same antibodies. Identical results were obtained with each cell line, and the HEK data are shown representatively in Fig. 1C. Both α-CT and α-Flag detected FL polycystin-1 of ≈520 kDa, which is consistent with our previous result (18), and a smaller fragment of ≈150 kDa of comparable signal intensity. Similar sized fragments were detected by α-CT in an MDCK cell line (C8/68; ref. 5) with stable expression of untagged FL polycystin-1 but not in the control cell line (F6). These results suggest that polycystin-1 is, in fact, cleaved at the GPS domain. The presence of the FL protein indicates that the cleavage reaction was not complete, however.
Fig 1.

Proteolytic cleavage of polycystin-1. (A) Multiple sequence alignment of GPS domains using clustal. The cleavage site is marked by an arrow. Residue numbers are shown after the name of each protein and the species from which it is derived. Bov, bovine; Ce, Caenorhabditis elegans; Fugu, Fugu rubripes; Spu, Strongylocentrotus purpuratus; LPH1, Latrophilin/CL-1. (B) A schematic diagram of the domain organization of polycystin-1. SP, signal peptide; R1 and R2–16, PKD repeats; CLD, C-type lectin domain. The predicted cleavage site with the adjacent sequence is presented at right. A black bar indicates epitopes recognized by the antibodies used in this study. The positions of Flag-epitope and the GFP-tag are indicated. WT encodes FL polycystin-1 with a Flag-epitope, whereas GFP-WT has both a GFP-tag and a Flag-tag. (C) Western blots demonstrating cleavage of exogenously expressed polycystin-1. HEK cells were either untransfected or transfected with WT or GFP-WT. F6 and C8/68 are MDCK clones with stable expression of either empty vector or human PKD1 (5). The antibodies used for IP and Western blot are indicated. (D) Western blots demonstrating cleavage and tethering of endogenous polycystin-1 in normal human kidney (NHK), human primary umbilical vein endothelial cells (HUVEC), and human primary aortic endothelial cells (HAEC). Lanes 5–8 show the reciprocal co-IP of NTF and CTF using GFP-WT. Nonspecific trapping of the products by either M2 beads or α-GFP was excluded by using an irrelevant antibody for the IP (α-myc). A myc-tagged form of PKD1 (Myc-WT) served as a positive control for the α-myc IP. (E) Autoradiograph of α-Flag IP product from WT-transfected HEK cells that were metabolically labeled for 20 min and then “chased” for various times, as indicated on top. The position of FL and NTF was confirmed on the Western blot by α-LRR (not shown). (F) Mutations in the GPS of polycystin-1. Only residues that differ from the WT polycystin-1 are shown. (G) Effect of amino acid substitutions on cleavage. Each construct carrying the mutation as indicated on top was transfected into HEK cells and assayed for cleavage as shown. E3020X (19) served as a negative control for IP because it lacks the Flag-tag.
If such a cleavage occurs, we reasoned that the corresponding NTF should be detected by α-LRR that recognizes the LRR domain contained therein. Indeed, we found this to be the case. The antibody recognized both FL and a slightly faster migrating fragment of ≈370 kDa in the protein lysates of C8/68 but not in F6 (Fig. 1C). The smaller fragment decreased to ≈325 kDa, the expected size of NTF, upon peptide N-glycosidase (PNGase) digestion (not shown). We confirmed the result by using a second FL construct with an N-terminal GFP-tag (GFP-WT). An antibody specific for GFP immunoprecipitated both the GFP-tagged version of FL polycystin-1 and its NTF cleavage product, as revealed by α-LRR.
We queried whether endogenous polycystin-1 undergoes cleavage in cells known to express PKD1 (3). We examined the cleavage of polycystin-1 in normal kidney tissue and in human primary umbilical and aortic endothelial cells by IP using α-CT. Although we were unable to detect polycystin-1 products in an unambiguous manner by using the same antibody on Western blot, we could detect FL polycystin-1 using α-LRR (Fig. 1D). Remarkably, the antibody also detected a smaller band corresponding in size to NTF. This observation suggested that NTF is tethered after cleavage, as has been previously shown for latrophilin/CL-1 (15). This possibility could be confirmed by reciprocal co-IP of NTF and CTF from cells expressing GFP-WT.
To determine whether the NTF products resulted from postcleavage tethering in vivo or from cleavage of FL polycystin-1 during or after IP, we performed a pulse-and-chase experiment and assayed the cleavage of polycystin-1 (Fig. 1E). We found that FL polycystin-1 was the only product detected immediately after the completion of pulse-labeling. Within 10–15 min of chase, however, NTF became visible and its intensity increased gradually with time, reaching a level equal to that of FL protein within 2 h. If cleavage and tethering had occurred after cell lysis during the experimental procedure, a similar cleavage pattern would have been observed for all time points. α-Flag used to precipitate polycystin-1 products bound a nonspecific band that overlapped CTF, precluding the evaluation of CTF kinetics (not shown).
Site-Specific Mutagenesis of the GPS Domain of Polycystin-1.
The GPS domain is an unusual signal for cleavage in that it also includes sets of highly conserved residues distant to the predicted cleavage site HL^T (see Fig. 1A). We examined the role of these residues for cleavage by site-specific mutagenesis (Fig. 1 F and G). The analyses of the consensus HLT sequence revealed a critical role of both histidine (H-2, with -2 indicating the position 2 residues N-terminal to the cleavage site) and threonine (T + 1), as their replacement by other residues abolished the cleavage reaction. Proper proteolytic processing did not require L-1, however, as replacement by similar or larger nonpolar residues (M or F) was less detrimental. The conserved residues outside of HLT (C-6, C3015, F + 3) were also found to be important for cleavage, whereas replacement of several nonconserved residues (R–10, A + 2 not shown) had no effect. These data show that the highly conserved residues of the GPS domain common to LNB-TM7s and polycystin-1 are essential for cleavage of the latter and suggest a common proteolytic machinery and mechanism for these proteins.
Characterization of Tethering.
Because NTF remains tethered after cleavage, we examined whether it can be tethered by CTF, as has been reported for latrophilin/CL-1 (15). As shown in Fig. 2A, the soluble form of NTF, E3020X (a naturally occurring mutant with premature termination within the GPS domain; ref. 19) can be coprecipitated by CTF, when the molecules are coexpressed as separate proteins. Further confirmation was provided by colocalization studies of N-terminal GFP-tagged E3020X (GFP-E3020X) with CTF. Whereas GFP-E3020X expressed alone showed essentially intracellular GFP fluorescence, coexpression with CTF resulted in a plasma membrane pattern that overlapped that seen with α-CT. These results suggest that CTF alone was sufficient to tether NTF at or near the cell surface.
Fig 2.

Characterization of NTF tethering. (A) Tethering of NTF by CTF. (Left) E3020X was transfected either alone or cotransfected with a CTF construct that includes a C-terminal Flag sequence and assayed for cleavage as shown. Equal amounts of NTF were present in samples with and without CTF coexpression (Lysate). Nonspecific trapping was excluded by using an irrelevant antibody (α-Myc) for IP. (Right) The immunofluorescence results of HEK cells transfected with GFP-E3020X alone or cotransfected with CTF both intracellular and plasma-membrane. (B) Chemical nature of NTF tethering. Co-IP of NTF by CTF-specific α-Flag from WT-transfected HEK cells was tested under native (N), denaturing and nonreducing (D), or denaturing and reducing (D+R) conditions, as outlined (Right). (C) Concentrated culture medium of cells transfected with each of the constructs was analyzed by Western blotting by using α-LRR (Upper). WT-Flag-IP served as size control for FL and NTF. (Lower) Flag-IP for the corresponding protein lysate probed with α-CT. The intracellular level of E3020X was similar to that of WT when total protein lysates were assayed on Western blot by α-LRR (not shown).
We then examined the chemical nature of the tethering by assaying the effect of protein denaturation on the process. We found that denaturation by SDS and boiling abolished co-IP of NTF, demonstrating that the tethering of NTF is noncovalent (Fig. 2B). We obtained similar results when the experiment was performed under denaturing and reducing conditions, confirming that the coexisting 520-kDa FL product is indeed the uncleaved form of polycystin-1 and not a processed two-chain structure tethered by disulfide bonds. Given the noncovalent nature of tethering, we queried whether any NTF escapes tethering and is secreted as a soluble protein (Fig. 2C). We could detect a minute amount of NTF in the medium of cells transfected with WT, but not of cells transfected with a noncleavable form T/R(+1) (see Fig. 1) expressed at a similar level [lane T/R(+1)]. E3020X was readily secreted into the medium (lane E3020X). This result shows that a small amount of NTF is secreted but the majority remains tethered to the cell.
Polycystin-1 Cleavage Requires the REJ Domain.
The GPS domain is situated at a similar position in polycystin-1 and all LNB-TM7s, i.e., immediately N-terminal to the first TM. We sought to determine whether this domain can function as an autonomous cleavage signal in a heterologous context. When all domains N-terminal to the GPS domain in polycystin-1 were replaced with alkaline phosphatase (AP-GPS-CTF), cleavage did not occur (Table 1). This result indicates that GPS alone was not sufficient to support the cleavage reaction, and additional elements present in the N-terminal extracellular sequence of polycystin-1 are required for the process. To determine what those might be, we generated a series of constructs containing the intact GPS domain and different segments of the N-terminal extracellular region and tested for cleavage of each product (Table 1). Polycystin-1 molecules that lack either virtually the entire [Δ(2145–2963)] or part [Δ(2493–2963)] of the REJ domain failed to be cleaved. This result indicates that the intact REJ domain is required to support cleavage at the adjacent GPS domain. In contrast, polycystin-1 molecules containing the intact REJ domain but with various deletions N-terminal to the domain did undergo partial cleavage. These data show that the REJ domain is both necessary and sufficient for cleavage to occur. We confirmed that cleavage occurred at the GPS rather than at another position because of the unnatural assortment of amino acid sequences by using constructs that were known to inhibit cleavage.
Table 1.
Mapping of N-terminal extracellular region mediating polycystin-1 cleavage
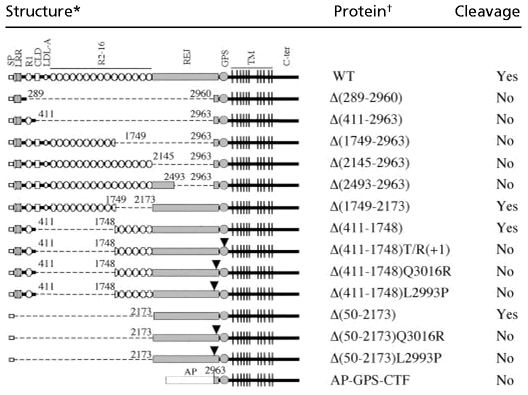 |
Schematic structures of wild-type polycystin-1 and mutant proteins. The broken line indicates deleted protein segment. AP, human-secreted placental alkaline phosphatase. The numbers indicate the amino acid positions of deletion boundaries.
Q3016R and L2993P are naturally occurring mutations that disrupt cleavage of polycystin-1 (see Fig. 3). The respective position of each substitution is indicated by an arrowhead.
Cleavage of Polycystin-1 Is Disrupted by PKD1 Mutations.
If cleavage of polycystin-1 is important for its function, then one might predict that pathologic mutations affecting the sequence at or near GPS would disrupt this process. To date, no missense mutations have been reported that alter the GPS sequence. A few mutations have been identified, however, that map within the REJ domain (Fig. 3A). Included in this set are three germline mutations (Q3016R, L2993P, and E2771K), a de novo mutation (H2921P), and an acquired somatic mutation (F2853S; refs. 19–21). Each of the residues is conserved at its relative position among many species. Therefore, we generated a series of expression constructs bearing the mutations and tested for the effect of each substitution on cleavage. We found that each of the mutations almost completely inhibited cleavage (Fig. 3B). As a corollary to the previous hypothesis, three polymorphisms (F3064L, E2996D, R2791Q; refs. 19, 22) in the same region that were suitable for testing did not affect the cleavage pattern.
Fig 3.

Effect of PKD1-associated mutations on cleavage. (A) Schematic structures of five disease-associated mutant forms of polycystin-1 and three normal variants. An arrowhead marks the relative position of the substitution in each protein. “MUT” identifies disease-associated mutations and “POLY” identifies polymorphic variants. (B) The cleavage property of each mutation was analyzed by using either α-CT (Upper) or α-LRR (Lower). The asterisk indicates an additional band of uncertain nature. (C) Cleavage-deficient forms of polycystin-1 traffic normally. GFP-WT, GFP-L2993P, and GFP-Q3016R were expressed in HEK cells and evaluated for the subcellular localization of each. (D) A cell surface biotinylation study was performed on HEK cells transfected with various constructs as indicated. Biotinylated surface proteins were captured by streptavidin beads and analyzed on Western blot using α-CT. WT/Flag-IP without biotin treatment (lane 1) served as control for FL and CTF. E3020X served as negative control for α-CT. Asterisks indicate nonspecific bands of unknown identity.
We excluded abnormal trafficking of the mutant proteins as a trivial explanation for the impaired cleavage reaction. Similar cell surface pattern of staining was seen for the N-terminally GFP-tagged mutant (GFP-L2993P and GFP-Q3016R) and normal (GFP-WT) proteins (Fig. 3C). This result was confirmed by surface biotinylation study (Fig. 3D). We found that WT FL polycystin-1, its cleavage products (CTF, lanes 2 and 3; NTF, data not shown) as well as the uncleavable Q3016R (lane 4) were biotinylated. As a control, CM130, a Golgi resident protein, was not detected in the streptavidin eluates, although it was present in the total lysates (data not shown). Taken together, the results suggest that the cleavage reaction may be essential for the normal function of polycystin-1.
Cleavage Mutants Have Impaired Function.
We tested this hypothesis by using an in vitro tubulogenesis model that recapitulates some cardinal features of polycystin-1 function in vivo (5). We had previously demonstrated that polycystin-1 can induce formation of tubule-like structures in MDCK cells, which otherwise form spherical cysts when cultured in 3-dimensional collagen gels (5). Therefore, we expressed a series of cleavage-deficient polycystin-1 proteins in MDCK cells by using a population-based episomal expression system and tested for the ability of each mutant protein to induce tubulogenesis (Fig. 4A). As expected, we found that cells of the parental MDCK line and those transfected with empty vector formed cyst-like structures, whereas cells transfected with WT consistently developed tubules (Fig. 4 B and C). In contrast, 88–99% of the structures formed by cells expressing either R4227X or the cleavage mutants were cysts. R4227X is a mutant form of polycystin-1 previously shown to be incapable of associating with polycystin-2 (10), unable to function as a GPCR (12), or unable to activate the JAK-STAT pathway (6). These results suggest that cleavage of polycystin-1 is required for its tubulogenic properties as measured by this assay.
Fig 4.

Effect of cleavage-disrupting mutations on functional properties of polycystin-1. (A) Various forms of polycystin-1 were expressed in MDCK cells by using an episomal expression system and detected by α-LRR antibody on Western blot. E3020X was expressed and loaded in parallel to identify the position of NTF. (B) Quantification of the types of structures formed by MDCK cells expressing various PKD1 constructs when grown in collagen. The parental cell line and cells transfected with empty vector served as negative controls. The total number of structures examined for each experiment is shown at the bottom. (C) Representative examples of the types of structures formed by MDCK cells described in B. (Bars = 100 μm.) (D) Transcriptional activity of STAT1 was measured in HEK cells by using a luciferase reporter assay. The various constructs were cotransfected with pTK-IRF1-Luc and pCMV β-gal. Luciferase activity was measured 24 h after transfection, and activity was normalized to the level of β-gal expression. The results of a single experiment performed in triplicate are presented.
We have recently shown that polycystin-1 signals, at least in part, through direct activation the JAK-STAT pathway (6). We used a luciferase reporter assay because we had previously shown that its results were predictive of those obtained by using a comprehensive approach. We found that WT induced four- to fivefold higher relative luciferase activity than either mutant construct (Fig. 4D). The results obtained for F2853S and R4227X were indistinguishable.
Discussion
In this study, we have shown that polycystin-1 is internally cleaved very rapidly after its synthesis in vivo. Although the cleavage site is not formally determined, both the predicted size of the N- and C-terminal cleavage products and the results of the site-specific mutagenesis assays strongly suggest that cleavage occurs at the consensus HL^T sequence of the GPS domain. We have provided multiple independent lines of evidence that strongly suggest that this process is essential for the full biological activity of polycystin-1. We speculate that cleavage of polycystin-1 results in the creation of a high-affinity binding pocket for as-yet unidentified ligands, as has been suggested for other receptor molecules (23–25). Cleavage of polycystin-1 may also be required for efficient signal transduction after ligand binding (23).
One remarkable finding is that the GPS domain alone is not sufficient to support the cleavage and requires the adjacent REJ domain for the reaction to occur. The REJ domain also occurs in suREJ3 (17) and probably plays a similar role for cleavage of that molecule. It is not found in LNB-TM7s, however. Interestingly, all known LNB-TM7 proteins contain a structurally undefined region of various length immediately N-terminal to the GPS domain, and which corresponds to the REJ domain in polycystin-1 (16); we speculate that this region might have a similar function. The mechanism by which the REJ domain supports cleavage at the GPS domain is unknown. We hypothesize that the REJ domain and the analogous region of LNB-TM7s share a common structural fold that serves to expose the GPS domain for the proteolytic machinery. Alternatively, this structure might bind to other molecules that affect the cleavage at the GPS domain.
One unique aspect of the polycystin-1 cleavage was that it was incomplete in all cell types under the conditions tested. Given that endogenous polycystin-1 had a similar property, this observation is unlikely to be an artifact resulting from over-expression of recombinant protein, as has been described for presenilin-1 (26). This result prompts consideration as to what factors might be responsible for regulating cleavage and whether these are dynamically controlled. One possibility is that posttranslational modifications of polycystin-1 may alter its conformation in such a way that it no longer is capable of undergoing cleavage. An alternative explanation is that there is another molecule that binds to FL polycystin-1 and thereby inhibits its cleavage.
Another remarkable feature of the polycystin-1 cleavage is the observation that most of the NTF product of polycystin-1 is tethered to the membrane-bound CTF in a noncovalent fashion, similar to that seen for LNB-TM7 family members latrophilin/CL-1 (15), CD97 (27), and for suREJ3 (17). As a corollary, a small amount of NTF can escape tethering and be secreted into the medium. These observations raise the intriguing possibility that the binding of NTF to CTF may be dynamic and subject to localized changes in the microenvironment present at the cell surface, as has been suggested for CD97 (27). It is possible that either additional molecules or other local effects may modulate release of tethered NTF and thereby affect signal transduction by polycystin-1.
Acknowledgments
We thank A. Bowers for technical assistance; W. Luo and M. Liu for helpful discussion; S. Ahkter for assistance with the biotinylation study; M. Delannoy for confocal microscopy; D. Shortle, L. Onuchic, and S. McGaughey for critical reading of the manuscript; and members of the Germino laboratory for helpful advice. This work was supported by the National Institutes of Health, the American Heart Association, the Polycystic Kidney Disease Foundation, the National Kidney Foundation, and the Arrison Foundation. The investigators are members of the Johns Hopkins University Polycystic Kidney Disease Center of Excellence (NIH P50-DK57325). G.G.G. is the Blum Scholar of the Johns Hopkins University School of Medicine.
Abbreviations
GPCR, G protein-coupled receptor
MDCK, Madin–Darby canine kidney
TM, transmembrane segment
PKD, polycystic kidney disease
GPS, GPCR proteolytic site
LNB-TM7, long-N-terminal family B GPCR-related seven-transmembrane receptor
LRR, leucine-rich repeat
REJ, receptor for egg jelly
IP, immunoprecipitation
NTF, N-terminal fragment
CTF, C-terminal fragment
FL, full-length
α-CT, antibody that recognizes the C terminus of polycystin-1
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Gabow P. A. (1993) N. Engl. J. Med. 329, 332-342. [DOI] [PubMed] [Google Scholar]
- 2.Peters D. J. & Sandkuijl, L. A. (1992) Contrib. Nephrol. 97, 128-139. [DOI] [PubMed] [Google Scholar]
- 3.Boulter C., Mulroy, S., Webb, S., Fleming, S., Brindle, K. & Sandford, R. (2001) Proc. Natl. Acad. Sci. USA 98, 12174-12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim K., Drummond, I., Ibraghimov-Beskrovnaya, O., Klinger, K. & Arnaout, M. A. (2000) Proc. Natl. Acad. Sci. USA 97, 1731-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boletta A., Qian, F., Onuchic, L. F., Bhunia, A. K., Phakdeekitcharoen, B., Hanaoka, K., Guggino, W., Monaco, L. & Germino, G. G. (2000) Mol. Cell 6, 1267-1273. [DOI] [PubMed] [Google Scholar]
- 6.Bhunia A. K., Piontek, K., Boletta, A., Liu, L., Qian, F., Xu, P. N., Germino, F. J. & Germino, G. G. (2002) Cell 109, 157-168. [DOI] [PubMed] [Google Scholar]
- 7.Hughes J., Ward, C. J., Peral, B., Aspinwall, R., Clark, K., San Millan, J. L., Gamble, V. & Harris, P. C. (1995) Nat. Genet. 10, 151-160. [DOI] [PubMed] [Google Scholar]
- 8.Ibraghimov-Beskrovnaya O., Bukanov, N. O., Donohue, L. C., Dackowski, W. R., Klinger, K. W. & Landes, G. M. (2000) Hum. Mol. Genet. 9, 1641-1649. [DOI] [PubMed] [Google Scholar]
- 9.Qian F., Germino, F. J., Cai, Y., Zhang, X., Somlo, S. & Germino, G. G. (1997) Nat. Genet. 16, 179-183. [DOI] [PubMed] [Google Scholar]
- 10.Hanaoka K., Qian, F., Boletta, A., Bhunia, A. K., Piontek, K., Tsiokas, L., Sukhatme, V. P., Guggino, W. B. & Germino, G. G. (2000) Nature 408, 990-994. [DOI] [PubMed] [Google Scholar]
- 11.Parnell S. C., Magenheimer, B. S., Maser, R. L., Rankin, C. A., Smine, A., Okamoto, T. & Calvet, J. P. (1998) Biochem. Biophys. Res. Commun. 251, 625-631. [DOI] [PubMed] [Google Scholar]
- 12.Delmas P., Nomura, H., Li, X., Lakkis, M., Luo, Y., Segal, Y., Fernandez-Fernandez, J. M., Harris, P., Frischauf, A. M., Brown, D. A., et al. (2002) J. Biol. Chem. 277, 11276-11283. [DOI] [PubMed] [Google Scholar]
- 13.Moy G. W., Mendoza, L. M., Schulz, J. R., Swanson, W. J., Glabe, C. G. & Vacquier, V. D. (1996) J. Cell Biol. 133, 809-817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ponting C. P., Hofmann, K. & Bork, P. (1999) Curr. Biol. 9, R585-R588. [DOI] [PubMed] [Google Scholar]
- 15.Krasnoperov V. G., Bittner, M. A., Beavis, R., Kuang, Y., Salnikow, K. V., Chepurny, O. G., Little, A. R., Plotnikov, A. N., Wu, D., Holz, R. W., et al. (1997) Neuron 18, 925-937. [DOI] [PubMed] [Google Scholar]
- 16.Stacey M., Lin, H. H., Gordon, S. & McKnight, A. J. (2000) Trends Biochem. Sci. 25, 284-289. [DOI] [PubMed] [Google Scholar]
- 17.Mengerink K. J., Moy, G. W. & Vacquier, V. D. (2002) J. Biol. Chem. 277, 943-948. [DOI] [PubMed] [Google Scholar]
- 18.Boletta A., Qian, F., Onuchic, L. F., Bragonzi, A., Cortese, M., Deen, P. M., Courtoy, P. J., Soria, M. R., Devuyst, O., Monaco, L., et al. (2001) Am. J. Kidney Dis. 38, 1421-1429. [DOI] [PubMed] [Google Scholar]
- 19.Rossetti S., Strmecki, L., Gamble, V., Burton, S., Sneddon, V., Peral, B., Roy, S., Bakkaloglu, A., Komel, R., Winearls, C. G., et al. (2001) Am. J. Hum. Genet. 68, 46-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watnick T. J., Torres, V. E., Gandolph, M. A., Qian, F., Onuchic, L. F., Klinger, K. W., Landes, G. & Germino, G. G. (1998) Mol. Cell 2, 247-251. [DOI] [PubMed] [Google Scholar]
- 21.Koptides M., Mean, R., Demetriou, K., Constantinides, R., Pierides, A., Harris, P. C. & Deltas, C. C. (2001) Hum. Mutat. 16, 176. [DOI] [PubMed] [Google Scholar]
- 22.Watnick T. J., Piontek, K. B., Cordal, T. M., Weber, H., Gandolph, M. A., Qian, F., Lens, X. M., Neumann, H. P. & Germino, G. G. (1997) Hum. Mol. Genet. 6, 1473-1481. [DOI] [PubMed] [Google Scholar]
- 23.Williams J. F., McClain, D. A., Dull, T. J., Ullrich, A. & Olefsky, J. M. (1990) J. Biol. Chem. 265, 8463-8469. [PubMed] [Google Scholar]
- 24.Krasnoperov V., Bittner, M. A., Holz, R. W., Chepurny, O. & Petrenko, A. G. (1999) J. Biol. Chem. 274, 3590-3596. [DOI] [PubMed] [Google Scholar]
- 25.Yoshimasa Y., Seino, S., Whittaker, J., Kakehi, T., Kosaki, A., Kuzuya, H., Imura, H., Bell, G. I. & Steiner, D. F. (1988) Science 240, 784-787. [DOI] [PubMed] [Google Scholar]
- 26.Thinakaran G., Borchelt, D. R., Lee, M. K., Slunt, H. H., Spitzer, L., Kim, G., Ratovitsky, T., Davenport, F., Nordstedt, C., Seeger, M., et al. (1996) Neuron 17, 181-190. [DOI] [PubMed] [Google Scholar]
- 27.Gray J. X., Haino, M., Roth, M. J., Maguire, J. E., Jensen, P. N., Yarme, A., Stetler-Stevenson, M. A., Siebenlist, U. & Kelly, K. (1996) J. Immunol. 157, 5438-5447. [PubMed] [Google Scholar]