

Abstract
We have used flash photolysis of caged Ca2+ and membrane capacitance measurements to probe exocytosis in chromaffin cells at low concentrations of intracellular Ca2+ ([Ca2+]i) (<10 μM). We observed a small pool of granules that is more sensitive to [Ca2+]i than the previously described “readily releasable pool.” Upon activation of PKC, this “highly Ca2+-sensitive pool” is enhanced in size to a greater extent than the readily releasable pool but is eliminated upon expression of a C-terminal deletion mutant (Δ9) of synaptosome-associated protein of 25 kDa (SNAP-25). Thus, in chromaffin cells, PKC enhances exocytosis both by increasing the number of readily releasable vesicles and by shifting vesicles to a highly Ca2+-sensitive state, enabling exocytosis at sites relatively distant from Ca2+ channels.
Neurons and neuroendocrine cells release signaling molecules by means of exocytosis in response to stimuli that cause membrane depolarization and Ca2+ influx. Understanding the spatial and temporal relationship between Ca2+ influx and exocytosis and the modulation of this relationship by second messengers is necessary to having a fundamental understanding of how cells process and transmit information.
The kinetics of exocytosis in response to a constant stimulus are often complex in that an initially high rate of exocytosis decays with multiple exponential components. In response to membrane depolarization, the fastest component of release occurs from what has been called the “immediately releasable pool” (IRP) of vesicles (1–6). A number of experiments suggest that the IRP consists of fusion-competent vesicles located near Ca2+ channels; however, another possibility is that these vesicles are particularly sensitive to intracellular Ca2+ concentration ([Ca2+]i).
Photorelease of caged Ca2+ elevates [Ca2+]i uniformly throughout the cell; therefore, the proximity of vesicles to Ca2+ channels is irrelevant, and the relationship between [Ca2+]i and exocytosis can be directly probed. The fastest kinetic component of exocytosis in response to photoelevation of [Ca2+]i has been ascribed to fusion of the “readily releasable pool” (RRP) of vesicles. In chromaffin cells, the size of the RRP is 4- to 5-fold greater than the IRP. No distinct kinetic component of exocytosis corresponding to the IRP has been identified in caged Ca2+ experiments, which supports the view that it is the proximity of vesicles to Ca2+ channels, rather than heterogeneity among vesicles in Ca2+ sensitivity, that determines which vesicles are released first upon membrane depolarization (6). However, exocytosis evoked by [Ca2+]i levels less than several μM has not been extensively studied with caged Ca2+ experiments in chromaffin cells. Understanding the kinetics of exocytosis evoked by low [Ca2+]i levels is particularly relevant for understanding the release of vesicles located relatively distant from Ca2+ channels.
We have probed the [Ca2+]i dependence of exocytosis at low μM levels in chromaffin cells and have found a small “highly Ca2+-sensitive pool” (HCSP) that had previously been overlooked. Release from the HCSP, like the RRP, involves soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins because it is eliminated upon expression of a mutant of synaptosome-associated protein of 25 kDa (SNAP-25) lacking the nine C-terminal amino acids (Δ9SNAP-25). Depleting the HCSP with a modest elevation of [Ca2+]i does not reduce the subsequent response to brief depolarization, supporting the hypothesis that short depolarizations release vesicles located near Ca2+ channels.
Neurons and neuroendocrine cells can modulate the rate of exocytosis in response to a given Ca2+ stimulus through either a change in the number of vesicles in releasable pools or a shift in the Ca2+ sensitivity of release. Both the IRP and the RRP are increased in size upon activation of PKC in chromaffin cells, whereas the [Ca2+]i sensitivity of release from the RRP is not altered (7). PKC is also reported to increase the size of the RRP in hippocampal (8, 9) and retinal bipolar terminals (10). In contrast, in calyx-type synapses, PKC is reported to increase the sensitivity of release to depolarization without affecting the size of the RRP (11–13). Here, we show that the HCSP is increased in size to a greater extent than the RRP upon activation of PKC. Thus, in chromaffin cells, PKC both increases the number of releasable vesicles and increases the fraction of vesicle that is in a highly Ca2+-sensitive state.
Materials and Methods
Cell Preparation and Solutions.
Bovine adrenal chromaffin cells were prepared as described (14) and seeded on 25-mm glass coverslips coated with poly(d)-lysine in DMEM (GIBCO) supplemented with 10% (vol/vol) FBS and 1% penicillin/streptomycin. Cells were kept in a 37°C incubator with 5% CO2 and used 1–3 days after preparation. All reagents were obtained from Sigma, unless otherwise stated. The standard bath solution for experiments that included membrane depolarization consisted of 140 mM NaCl, 5.5 mM KCl, 1 mM MgCl2, 10 mM CaCl2, and 10 mM Hepes titrated to pH 7.2 with NaOH. For experiments without depolarization, the bath solution contained 1 mM CaCl2.
The standard pipette solution contained 120 mM N-methyl-glucamine, 120 mM l-glutamic acid, 8 mM NaCl, and 40 mM Hepes titrated to pH 7.2 with extra N-methyl-glucamine. The Ca2+-indicator dyes fura-2 FF (K+ salt, Teflabs, Austin, TX) and bis-fura-2 (K+ salt, Molecular Probes) were mixed at an equimolar ratio (10 mM) and stored at −20°C. This stock solution was diluted in standard pipette solution to yield a final concentration of 100 μM for each dye. In caged Ca2+ experiments, 0.5–1 mM nitrophenyl-EGTA (15), 80% loaded with CaCl2, together with 2 mM Na2ATP and 1 mM MgCl2, were added to the pipette solution.
[Ca2+]i Calibration.
The Ca2+ indicator combination was calibrated in vivo in a manner similar to that used in ref. 16. Eight solutions with free [Ca2+]i of 0, 0.31, 1.2, 3.6, 7.2, 28, and 127 μM and 10 mM were prepared by using Ca2+ buffers EGTA (Kd = 150 nM at pH 7.2, total concentration = 10 mM), N-hydroxyethylethylenediaminetriacetic acid (HEDTA, Kd = 3.6 μM, concentration = 10 mM), or 2-ol-N,N′-tetraacetic acid (DPTA, Kd = 81 μM, concentration = 30 mM; ref. 17). Whole-cell recordings were made by using each of the calibration solutions in the pipette, and the ratio of fluorescence emission (535 ± 25 nm) for 340- and 365-nm excitation was recorded after allowing 2 min for the pipette solution to diffuse into the cell. Three to five recordings were made for each calibration solution. The average ratio values were fit to the following equation to find the five unknown parameters (16):
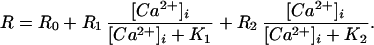 |
Inversion of Eq. 1 was used to calculate [Ca2+]i from the ratio R in caged Ca2+ experiments. We did not apply any corrections for the perturbation of the calibration upon photolysis of nitrophenyl-EGTA (18), because we used low cage concentrations and light intensities to achieve the μM [Ca2+]i used in this study.
Electrophysiology and Photometry.
Whole-cell patch-clamp measurements were performed at room temperature (≈23°C) by using an EPC-9 patch-clamp amplifier and PULSE acquisition software (HEKA Electronics, Lambrecht/Pfalz, Germany). Pipettes (2–4 MΩ) were pulled from Kimax glass capillaries, coated with wax, and fire-polished. The pipette potential was held at a dc potential of −70 mV, except during membrane depolarization. Capacitance measurements were performed by using the “sine + dc” method implemented in PULSE software (19, 20). The assumed reversal potential was 0 mV, and the sinusoid had an amplitude of 25 mV and a frequency of 1,500 Hz.
A monochromator (Polychrome II or IV, TILL Phototonics, Planegg, Germany/ASI, Eugene, OR) coupled to the epifluorescence port of an Olympus IX 50/70 microscope with a fiber-optic cable excited the Ca2+ indicators at 340 and 365 nm. A 2-port condensor (TILL/ASI) combined the monochromator excitation path with that of a flash lamp (JML-C1, Rapp OptoElectronic, Hamburg, Germany or TILL/ASI). A 40 × 1.15 numerical aperture (N.A.) water immersion lens (model U-APO, Olympus) focused the excitation light and collected fluorescent light. The fluorescent light (535 ± 25 nm) was measured by using a photomultiplier tube or photodiode mounted in a viewfinder (TILL/ASI).
Data analysis and curve fitting were performed by using IGOR software (WaveMetrics, Lake Oswego, OR). Results and histograms are expressed as means ± SEM.
Results
The HCSP Is Rapidly Released at Low [Ca2+]i.
We used the whole-cell patch-clamp configuration to load the Ca2+ cage nitrophenyl-EGTA (0.5–1 mM, 80% loaded with Ca2+) into bovine adrenal chromaffin cells. The pipette solution also contained a combination of a high-affinity Ca2+ indicator (bis-fura-2, 100 μM) and a low-affinity indicator (fura-2 FF, 100 μM) to allow fluorescence [Ca2+]i measurement over a wide dynamic range (≈50 nM–200 μM; ref. 16). [Ca2+]i was measured both before and after flash photolysis of the cage, and exocytosis was assayed with membrane capacitance measurements.
We find that photoelevation of [Ca2+]i to values below ≈10 μM evokes a small (≈20 fF) response (Fig. 1 A–C) with a rate constant of release nearly two orders of magnitude faster than what has been reported for release of the RRP in chromaffin cells (16, 21). A slower but larger phase of exocytosis is also evident (Fig. 1 B and C) that has an amplitude (≈100 fF) and kinetic rate comparable to published descriptions of the RRP. We will refer to the population of granules released most rapidly at low [Ca2+]i levels as the HCSP.
Fig 1.

A small but fast phase of exocytosis is triggered by photoelevation of [Ca2+]i to 1–10 μM. (A) Flash photolysis of caged Ca2+ (arrow) elevates [Ca2+]i to 2.0 μM and elicits a burst of exocytosis with an amplitude of 44 fF and a rate constant of 50 s−1. (B) Flash photolysis elevates [Ca2+]i to 10 μM and elicits exocytosis with two exponential components. The fast component (red) has an amplitude of 17 fF and a rate constant of 140 s−1. The slower component (blue) has an amplitude of 197 fF and a rate constant of 10 s−1. (C) The small but rapid phase of exocytosis (red oval) is also evident when [Ca2+]i is elevated relatively slowly by photolyzing the cage with illumination from a monochromator. (D) The rate constants and amplitudes of exponential fits to the data are plotted vs. the [Ca2+]i that follows flash photolysis of caged Ca2+. Unfilled circles are from single-exponential fits, whereas the triangles and asterisks represent the slow and fast components, respectively, of double exponential fits to the data. The solid line is the rate constant of release for the RRP from the model in ref. 16. The filled circles represent fits to responses elicited after exposing cells to 100 nM PMA.
Our interpretation of a kinetic phase of exocytosis as release from a vesicle “pool” follows previous models of exocytosis (see ref. 22 for a review). In these models, the rate of exocytosis declines in the face of a constant [Ca2+]i stimulus, because a functionally defined group of vesicles becomes exhausted. An alternative explanation for the rapid decline in the rate of exocytosis in the face of a constant [Ca2+]i stimulus is that the Ca2+ sensors “adapt” to a given level of [Ca2+]i (23). Another possibility is that the [Ca2+]i stimulus is, in fact, not constant if flash photolysis produces a brief “spike” in [Ca2+]i. A Ca2+ spike too brief to be resolved with fura-type Ca2+ indicators is predicted to occur if the rate of photorelease of Ca2+ exceeds the rate that [Ca2+]i binds to and equilibrates with unphotolyzed cage or endogenous Ca2+ buffers in the cell (24). One would predict that both the extent of Ca2+ sensor adaptation and the magnitude of the photolysis-induced spike in [Ca2+]i critically depend on the rate of cage photolysis and [Ca2+]i elevation. For example, a slower rate of photolysis will produce less exocytosis if the Ca2+ sensors rapidly adapt. Little or no spike in [Ca2+]i will occur if Ca2+ is released slowly enough so that it has ample time to bind to Ca2+ buffers and maintain [Ca2+]i in equilibrium. Fig. 1C presents a sample trace, typical of 12 recordings, where slow elevation of [Ca2+]i to 1 μM produces an ≈20 fF highly Ca2+-sensitive burst of exocytosis similar in magnitude to that seen with flash photolysis. In this example, [Ca2+]i is elevated to 1 μM over a period of ≈200 ms by using a monochromator rather than a flash lamp to photolyze the cage. These observations are consistent with the hypothesis that the highly Ca2+-sensitive release process corresponds to exocytosis of a discrete population of vesicles.
We fit exponentials to the time course of capacitance increases in response to flash photoelevation of [Ca2+]i between 0.7 and 10 μM. The resulting amplitudes and rate constants are plotted vs. postflash [Ca2+]i in Fig. 1D. The unfilled circles depict single-exponential fits, whereas the triangles and asterisks are fits to traces that show two exponential components. Note that the slower exponential component (triangles) has rate constants and amplitudes (100–200 fF) similar to those previously described for the RRP (solid line; ref. 16). In contrast, the fastest kinetic component (circles and asterisks) has a much smaller amplitude (≈20 fF) but faster rate constants than the RRP and, thus, represents a highly Ca2+-sensitive release process.
Photoelevation of [Ca2+]i to levels below ≈0.7 μM fails to evoke release at a measurable rate in an ≈10-s recording interval (data not shown). In contrast, [Ca2+]i elevation to ≈1.4 μM evokes exocytosis with a rate constant of ≈5 s−1. Thus, the rate of release from the HCSP is steeply dependent on [Ca2+]i for [Ca2+]i below 2 μM. However, above 2 μM, the rate of release from the HCSP is less steeply dependent on [Ca2+]i than release from the RRP.
An Intact C Terminus of SNAP-25 Is Necessary for Highly Ca2+-Sensitive Exocytosis.
Botulinum neurotoxin type A (BoNT/A) partially inhibits exocytosis from neurons and neuroendocrine cells (25). The effects of BoNT/A are mediated by selective cleavage of the SNARE protein SNAP-25 at a site 9 amino acid residues from the C terminus (26). BoNT/A eliminates fast exocytosis from the RRP in chromaffin cells, whereas a slower kinetic component of release remains (27). The effects of BoNT/A can be mimicked with expression of a mutant of SNAP-25 missing the nine C-terminal amino acid residues (Δ9SNAP-25; ref. 28). We have used the Semliki Forest virus system to express wild type (WT) and Δ9SNAP-25 in chromaffin cells (29, 30). Both forms of SNAP-25 had GFP fused to the N terminus to report gene expression. Our results (depicted in Fig. 2) demonstrate that the HCSP is not perturbed by overexpression of WT SNAP-25; however, expression of Δ9SNAP-25 potently inhibits the highly Ca2+-sensitive release process. These results support the view that the small capacitance jumps seen in response to mild elevations of [Ca2+]i reported here reflect genuine SNARE-protein-mediated exocytosis.
Fig 2.

The C terminus of SNAP-25 is essential for highly Ca2+-sensitive exocytosis. Paired experiments were performed with cells expressing either WT SNAP-25 or SNAP-25 truncated to eliminate the nine C-terminal amino acid residues (Δ9SNAP-25). The Semliki Forest virus expression system was used to express GFP-tagged SNAP-25, and only highly fluorescent cells were selected for experimentation. (A) Sample responses to flash photoelevation of [Ca2+]i from 0.34 to 4.7 μM (dashed line: Δ9SNAP-25) and from 0.40 to 2.7 μM (solid line: WT SNAP-25). (B) Amplitude of highly Ca2+-sensitive responses, defined as the fastest kinetic component of exocytosis. For WT cells, the average [Ca2+]i before and after the flash was 0.53 and 6.0 μM. For Δ9SNAP-25, average [Ca2+]i was elevated from 0.57 to 5.0 μM. Cm, membrane capacitance.
The HCSP Is Distinct from the IRP Released with Brief Depolarizing Pulses.
The fastest kinetic component of exocytosis in response to membrane depolarization in chromaffin cells has an amplitude of ≈35 fF (1, 5, 7). It has been suggested that this IRP consists of those granules located near Ca2+ channels. However, the similar amplitude of the HCSP described above raises an alternative possibility that the first granules released upon membrane depolarization are simply highly sensitive to Ca2+. The following experiment rules out this possibility.
We designed an experimental protocol to evoke exocytosis with photorelease of caged Ca2+ and with depolarization-evoked Ca2+ influx to probe the relationship between the HCSP, the IRP, and the RRP. First, we applied a pair of depolarizing pulses, 30 ms in duration, to define the size of the IRP in the cell. A typical response is depicted in Fig. 3A. Because the first pulse depletes the IRP, the change in capacitance arising from the second depolarizing pulse is typically less than one half that of the first, despite an identical amount of Ca2+ influx. Then, the IRP is allowed to refill by resting the cell for ≈60 s. Next, we used flash photolysis to elevate [Ca2+]i to 1–5 μM to release the HCSP. This step was followed by a train of brief (30 ms) depolarizing pulses to evoke further exocytosis with Ca2+ influx. A sample experiment of this type is illustrated in Fig. 3B. This experiment, typical of many dozens of this type, demonstrates that the first depolarizing pulse of the train can still evoke a robust ΔCm even after exhaustion of the HCSP. The second pulse elicits a greatly reduced ΔCm, demonstrating a normal pattern of release from the IRP. Subsequent pulses continue to evoke exocytosis that eventually slows. We interpret the total amount of depolarization-evoked exocytosis in response to 10 pulses as the magnitude of the RRP, which typically is exhausted after a few hundred milliseconds of Ca2+ influx (16, 31).
Fig 3.

The IRP released by membrane depolarization is not highly Ca2+-sensitive. (A) Cm response to a pair of depolarizing pulses from −70 mV to +10 mV, 30 ms in duration. The IRP was allowed to recover for ≈60 s before applying the flash plus depolarization stimuli. (B) Flash photolysis of caged Ca2+ at the time indicated by the arrow elevates [Ca2+]i to 1.8 μM and releases the HCSP. This step was followed 300 ms later by a train of depolarizing pulses to +20 mV. (C) Comparison of the ΔCm resulting from a pair of depolarizing pulses before and after depletion of the HCSP according to the protocol from parts A and B. The filled bars depict the responses to the first pulse, whereas the open bars stacked on top depict the much smaller increase in Cm from the second pulse (n = 16).
Fig. 3C summarizes measurements of the size of the IRP from 16 cells using the protocol of Fig. 3 A and B. The filled bars are the response to the first depolarizing pulse, whereas the unfilled bars are the response to the second pulse. Clearly, the size of the IRP is not reduced after mild elevation of [Ca2+]i and release of the HCSP. Presumably, the HCSP cannot refill in the 300-ms interval between the flash and the first depolarizing pulse because [Ca2+]i remains elevated (data not shown). Thus, the HCSP and the IRP represent distinct vesicle pools or release processes, and the bulk of vesicles released with brief membrane depolarization, presumably located close to Ca2+ channels, are not highly Ca2+ sensitive. In contrast, it has been shown (6) that photoelevation of [Ca2+]i to higher levels rapidly releases the RRP, so that depolarization-induced Ca2+ influx fails to evoke exocytosis.
The HCSP Is Preferentially Enhanced by Activation of PKC.
Activation of PKC by phorbol esters potently enhances exocytosis from neurons and neuroendocrine cells. It has been shown that PKC increases the size of the IRP and RRP in chromaffin cells (6, 7) as well as the size of the RRP in neuronal preparations (8, 10). Is the HCSP also affected by PKC? We applied 100 nM phorbol myristate acetate (PMA) in paired experiments in 24 cells. PMA increases the size of the HCSP from 22 ± 0.8 fF to 158 ± 13 fF (Fig. 4A). Fig. 1D plots the rate constants and amplitudes of individual responses in the presence of PMA (filled circles) and demonstrates that the amplitude of the responses is much greater in PMA (note the logarithmic scale), yet the rate constants of release are similar to control. A similar effect of PMA on release from the RRP has been reported (7).
Fig 4.

The size of the HCSP is increased with activation of PKC, but PKC activity is not essential for highly Ca2+-sensitive release. (A) The size of the HCSP is larger after exposure to 100 nM PMA than the control response (CN), but this enhancement is blocked by the PKC inhibitor bisindolylmaleimide I (BI). BI (1 μM) was added to both the bath and pipette solutions 10–60 min before the experiment. n = 8 cells for each condition. (B) Exposure of cells to BI (n = 18) or the nonspecific protein kinase inhibitor K252a (K, 0.5 μM, n = 12) in the absence of PMA has no effect on the size of the HCSP. n = 17 paired CN.
Phorbol esters can have effects other than activation of PKC. For example, the Munc-13 protein has been shown to regulate exocytosis (32). Munc-13 has a phorbol ester (C1)-binding domain and is translocated to the plasma membrane upon bath application of PMA (33). We used the specific PKC inhibitor bisindolylmaleimide I (1 μM) to test whether the enhancement of the HCSP by PMA is through activation of PKC. Bisindolylmaleimide inhibits binding of ATP to PKC (34); therefore, it should have little effect on Munc-13 or other proteins with C1 domains that do not bind ATP. Fig. 4A demonstrates that bisindolylmaleimide can completely block the enhancement of the HCSP by PMA. Thus, we conclude that activation of PKC potently enhances the size of the HCSP.
The potent effect of PKC stimulation suggests the possibility that the highly Ca2+-sensitive release process requires PKC phosphorylation of one or more proteins involved in exocytosis. To test the possibility that the “basal” (non-PMA-stimulated) HCSP reflects a phosphorylated state, we evaluated the effect of bisindolylmaleimide in the absence of PMA. Fig. 4B demonstrates that both bisindolylmaleimide and the broad-spectrum serine/threonine protein kinase inhibitor K252a (500 nM) have no apparent effect on the size of the HCSP. Assuming that protein phosphatases are constitutively active in cells, it seems unlikely that phosphorylation by PKC is required for highly Ca2+-sensitive release. Rather, PKC promotes highly Ca2+-sensitive release.
We repeated the protocol of Fig. 4 before and after application of PMA to determine the relative enhancement of the HCSP and RRP. Fig. 5A presents a sample trace that demonstrates that a sizable fraction of releasable vesicles are highly sensitive to Ca2+ in the presence of PMA. Fig. 5B summarizes results from 23 cells. Whereas both vesicle pools are enhanced by PMA, the HCSP is enhanced by a much larger percentage. In control cells, the HCSP contributes 13% of the total response of 172 fF, whereas in PMA, the HCSP contributes 29% of a total response of 485 fF.
Fig 5.

The size of the HCSP is increased to a greater extent than the RRP upon activation of PKC. (A) Sample trace after the protocol of Fig. 3B. Flash photoelevation of [Ca2+]i to 4 μM (arrow) reveals a large HCSP. (B) Summary of control responses (open bars, n = 12) and responses in the presence of PMA (filled bars, n = 11). The fractional increase in the HCSP is much greater than the fractional increase in the RRP (note the log scale). The mean [Ca2+]i before photolysis was 0.68 μM for control cells and 0.74 μM for cells in PMA. After photolysis, the mean [Ca2+]i was 3.3 μM for control and 4.1 μM in PMA. Control cells had an initial mean ICa of 294 pA, whereas cells in PMA had a mean initial ICa of 207 pA.
Discussion
We have observed a rapid, small-amplitude capacitance increase in chromaffin cells in response to photoelevation of [Ca2+]i to values less than 10 μM. We believe that this increase reflects genuine SNARE protein-dependent exocytosis, because it is ablated with expression of a C-terminal mutant of SNAP-25 and is modulated in size by activation of PKC. The amplitude of the capacitance response is insensitive to the rate of [Ca2+]i elevation, supporting the hypothesis that the highly Ca2+-sensitive response results from exocytosis of a discrete pool of chromaffin granules. Heterogeneity in the Ca2+ sensitivity of exocytosis within a single cell has been observed in nonexcitable cells such as neutrophils (35) and sea urchin eggs (36). The existence of vesicle subpopulations with differing sensitivity to [Ca2+]i has also been indirectly inferred in neuronal preparations (e.g., ref. 37).
One reason why highly Ca2+-sensitive exocytosis may have been overlooked in previous reports in chromaffin cells is the relatively small amplitude of this fast response compared with the ≈10-fold larger RRP. Thus, a discrete kinetic component attributable to release from the HCSP is not easily resolved for [Ca2+]i > 10 μM because release from the RRP becomes dominant (e.g., Fig. 1B).
Are Vesicles Located Near Ca2+ Channels More or Less Sensitive to [Ca2+]i than Releasable Vesicles Located Far from Ca2+ Channels?
The fastest kinetic component of depolarization-triggered exocytosis in neuroendocrine cells has been attributed to release from an IRP of vesicles. In chromaffin cells the IRP is ≈35 fF (15–30 vesicles) in size (1, 5, 7). No kinetic component comparable in magnitude to the IRP has previously been noted in caged Ca2+ experiments (e.g., ref. 6). In addition, depolarization-evoked release from the IRP is less sensitive to the introduction of exogenous intracellular Ca2+ buffers than the slower phase of release (1, 3–5). These observations and others have led to the hypothesis that the IRP is the subset of the RRP located near Ca2+ channels. The experimental protocol of Fig. 3 demonstrates that exhaustion of the HCSP does not significantly decrease the size of the IRP. Therefore, the bulk of vesicles released with brief membrane depolarization, presumably located close to Ca2+ channels, are not highly Ca2+-sensitive.
Conversely, can one conclude that the IRP is less sensitive to [Ca2+]i than the bulk of releasable vesicles? If the IRP has the same proportion of highly Ca2+-sensitive vesicles as the bulk population, then ≈10% would be lost by photoelevation of [Ca2+]i to the low μM range following the protocol of Fig. 3. This magnitude of reduction in the size of the IRP could not be easily resolved following the protocol of Fig. 3 because of pulse-to-pulse and cell-to-cell variability of responses. Further experiments are needed to determine the fraction of the IRP that is highly Ca2+-sensitive.
Effects of Activation of PKC on Exocytosis.
The most direct quantitative demonstration of the [Ca2+]i sensitivity of release is plots of the rate constant vs. [Ca2+]i obtained from caged Ca2+ experiments (e.g., Fig. 1D). From these data, one can conclude that PKC activation does not enhance the Ca2+-sensitivity of release from either the RRP (7) or HCSP (present work) per se. Nevertheless, PKC activation enhances the size of the HCSP to a greater extent than the RRP; therefore, there is a net increase in the fraction of vesicles that are in the highly Ca2+-sensitive state. Thus, consideration of the vesicle population as a whole reveals a net increase in the Ca2+-sensitivity of release upon PKC activation. A similar shift in vesicle release probability from a “reluctant” to “highly sensitive” state by cAMP has been reported in the calyx of Held (37). In contrast, a direct shift in the Ca2+-sensitivity of exocytosis by PKC is observed in gonadotropes (38). Either mechanism is consistent with experiments at calyx synapses that indicate that PKC increases the sensitivity of vesicles to depolarization-evoked exocytosis (11–13).
The potent modulation of the HCSP by PKC could have a large impact on exocytosis elicited by physiological stimuli. The release probability per action potential will increase in proportion to the increase in the number of highly Ca2+-sensitive vesicles located near Ca2+ channels. In addition, the net increase in the Ca2+ sensitivity of vesicles upon PKC activation will tend to recruit vesicles located further from Ca2+ channels to be released. Thus, the spatiotemporal pattern of exocytosis may be altered by PKC activation. In addition, exocytosis from the HCSP is likely to be the dominant mechanism at low or basal levels of [Ca2+]i.
Acknowledgments
We thank Xiuzhi Tang for preparing chromaffin cells and Bertil Hille, Tao Xu, Tzyh-Chang Hwang, and Erwin Neher for critically reading the manuscript. In addition, we thank the laboratory of Erwin Neher for the generous gift of Semliki Forest virus for expressing wild-type and Δ9SNAP-25. This work was supported by National Institutes of Health Grant NS40453 (to K.D.G.).
Abbreviations
IRP, immediately releasable pool
[Ca2+]i, intracellular Ca2+ concentration
RRP, readily releasable pool
HCSP, highly Ca2+-sensitive pool
SNAP-25, synaptosome-associated protein of 25 kDa
PMA, phorbol myristate acetate
See commentary on page 16522.
References
- 1.Horrigan F. & Bookman, R. (1994) Neuron 13, 1119-1129. [DOI] [PubMed] [Google Scholar]
- 2.Hsu S. F. & Jackson, M. B. (1996) J. Physiol. (Paris) 494, Part 2, 539-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mennerick S. & Matthews, G. (1996) Neuron 17, 1241-1249. [DOI] [PubMed] [Google Scholar]
- 4.Giovannucci D. R. & Stuenkel, E. L. (1997) J. Physiol. (Paris) 498, Part 3, 735-751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moser T. & Neher, E. (1997) J. Neurosci. 17, 2314-2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voets T., Neher, E. & Moser, T. (1999) Neuron 23, 607-615. [DOI] [PubMed] [Google Scholar]
- 7.Gillis K. D., Mößner, R. & Neher, E. (1996) Neuron 16, 1209-1220. [DOI] [PubMed] [Google Scholar]
- 8.Stevens C. F. & Sullivan, J. M. (1998) Neuron 21, 885-893. [DOI] [PubMed] [Google Scholar]
- 9.Waters J. & Smith, S. J. (2000) J. Neurosci. 20, 7863-7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berglund K., Midorikawa, M. & Tachibana, M. (2002) J. Neurosci. 22, 4776-4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yawo H. (1999) J. Physiol. 515, Part 1, 169-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu X. S. & Wu, L. G. (2001) J. Neurosci. 21, 7928-7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oleskevich S. & Walmsley, B. (2000) J. Physiol. 526, Part 2, 349-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Z. & Neher, E. (1993) J. Physiol. 469, 245-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellis-Davies G. C. & Kaplan, J. H. (1994) Proc. Natl. Acad. Sci. USA 91, 187-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voets T. (2000) Neuron 28, 537-545. [DOI] [PubMed] [Google Scholar]
- 17.Martell A. E. & Smith, R. H., (1974) Critical Stability Constants (Plenum, New York).
- 18.Zucker R. S. (1992) Cell Calcium 13, 29-40. [DOI] [PubMed] [Google Scholar]
- 19.Gillis K. D. (2000) Pflügers Arch. 439, 655-664. [DOI] [PubMed] [Google Scholar]
- 20.Pusch M. & Neher, E. (1988) Pflügers Arch. 411, 204-211. [DOI] [PubMed] [Google Scholar]
- 21.Heinemann C., Chow, R. H., Neher, E. & Zucker, R. S. (1994) Biophys. J. 67, 2546-2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gillis K. D. & Chow, R. H. (1997) Semin. Cell Dev. Biol. 8, 133-140. [DOI] [PubMed] [Google Scholar]
- 23.Hsu S. F., Augustine, G. J. & Jackson, M. B. (1996) Neuron 17, 501-512. [DOI] [PubMed] [Google Scholar]
- 24.Zucker R. S. (1993) Cell Calcium 14, 87-100. [DOI] [PubMed] [Google Scholar]
- 25.Jahn R. & Niemann, H. (1994) Ann. N.Y. Acad. Sci. 733, 245-255. [DOI] [PubMed] [Google Scholar]
- 26.Binz T., Blasi, J., Yamasaki, S., Baumeister, A., Link, E., Sudhof, T. C., Jahn, R. & Niemann, H. (1994) J. Biol. Chem. 269, 1617-1620. [PubMed] [Google Scholar]
- 27.Xu T., Binz, T., Niemann, H. & Neher, E. (1998) Nat. Neurosci. 1, 192-200. [DOI] [PubMed] [Google Scholar]
- 28.Wei S., Xu, T., Ashery, U., Kollewe, A., Matti, U., Antonin, W., Rettig, J. & Neher, E. (2000) EMBO J. 19, 1279-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duncan R. R., Don-Wauchope, A. C., Tapechum, S., Shipston, M. J., Chow, R. H. & Estibeiro, P. (1999) Biochem. J. 342, Part 3, 497-501. [PMC free article] [PubMed] [Google Scholar]
- 30.Ashery U., Betz, A., Xu, T., Brose, N. & Rettig, J. (1999) Eur. J. Cell Biol. 78, 525-532. [DOI] [PubMed] [Google Scholar]
- 31.von Ruden L. & Neher, E. (1993) Science 262, 1061-1065. [DOI] [PubMed] [Google Scholar]
- 32.Martin T. F. (2002) Neuron 34, 9-12. [DOI] [PubMed] [Google Scholar]
- 33.Betz A., Ashery, U., Rickmann, M., Augustin, I., Neher, E., Sudhof, T. C., Rettig, J. & Brose, N. (1998) Neuron 21, 123-136. [DOI] [PubMed] [Google Scholar]
- 34.Toullec D., Pianetti, P., Coste, H., Bellevergue, P., Grand-Perret, T., Ajakane, M., Baudet, V., Boissin, P., Boursier, E., Loriolle, F., et al. (1991) J. Biol. Chem. 266, 15771-15781. [PubMed] [Google Scholar]
- 35.Nusse O., Serrander, L., Lew, D. P. & Krause, K. H. (1998) EMBO J. 17, 1279-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blank P. S., Cho, M. S., Vogel, S. S., Kaplan, D., Kang, A., Malley, J. & Zimmerberg, J. (1998) J. Gen. Physiol. 112, 559-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakaba T. & Neher, E. (2001) Proc. Natl. Acad. Sci. USA 98, 331-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu H., Hille, B. & Xu, T. (2002) Proc. Natl. Acad. Sci. USA 99, 17055-17059. [DOI] [PMC free article] [PubMed] [Google Scholar]