

Abstract
Chronic liver disorders represent a serious health problem, considering that 300 million people worldwide are hepatitis B virus carriers, and 8,000–10,000 patients per year, in the U.S. alone, die as a result of liver failure caused by hepatitis C infection. Nitric oxide synthase (NOS) regulates hepatic vasculature; however, the patterns of expression and activity of NOS proteins in healthy and diseased human livers are unknown. Sections of diseased (n = 42) and control livers (n = 14) were collected during orthotopic liver transplants and partial hepatectomy. The diseased sections included alcoholic cirrhosis, viral hepatitis, cholestasis, acute necrosis, and uncommon pathologies including α1-anti-trypsin disorder. The endothelial NOS (eNOS), inducible NOS (iNOS), and neuronal NOS (nNOS) were studied by using the citrulline assay, Western immunoblot, immunohistochemistry, and in situ hybridization. The systemic generation of plasma NO metabolites was measured by HPLC. In control livers, Ca2+-dependent and –independent NOS activities were identified by Western analysis as eNOS and iNOS, respectively. The eNOS was uniformly distributed in the hepatocytes and also detected in the endothelium of hepatic arteries, terminal hepatic venules, sinusoids, and in biliary epithelium. The iNOS was detected in hepatocytes and localized mainly in the periportal zone of the liver acinus. This pattern of distribution of eNOS and iNOS in normal liver was confirmed by in situ hybridization. In diseased livers, there was a significant increase in Ca2+-independent NOS with the corresponding strong appearance of iNOS in the cirrhotic areas. The eNOS was translocated to hepatocyte nuclei. Thus, eNOS and iNOS proteins are differentially expressed in healthy human liver, and this expression is significantly altered in cirrhotic liver disorders.
Nitric oxide (NO) is a short-living biological mediator generated from l-arginine by NO synthase (NOS). The NOS family of enzymes identified to date includes constitutively expressed endothelial NOS (eNOS or type 3 NOS) and neuronal NOS (nNOS or type 1 NOS), as well as inducible NOS (iNOS or type 2 NOS). NO exerts a broad spectrum of physiological functions, including regulation of vascular reactivity, platelet and leukocyte activation, neurotransmission, regulation of cellular proliferation, and nonspecific immunity reactions (1). Inappropriate release, metabolism, or actions of NO have been associated with diverse vascular, ischemic, thrombotic, and inflammatory pathologies (1). In the liver, NO is generated by eNOS and iNOS, and this generation can mediate a number of physiological and disease reactions involving this organ (2).
Chronic liver disorders represent a serious health concern. The scale of this problem is emphasized by the fact that currently there are 300 million people worldwide who carry hepatitis B virus and are at risk of dying from liver failure (3). Indeed, in the U.S. alone, it is estimated that 8,000–10,000 patients per year die as a result of end-stage chronic liver cirrhosis and failure caused by infection with hepatitis C virus (4). Animal models have been used to study the role of NO in the pathogenesis of inflammatory liver injury (5, 6). Whereas NO generated by eNOS is clearly beneficial for liver function regulating blood flow and blood cell interactions, the inducible NO synthesized by iNOS has been shown to have both beneficial and detrimental effects for liver homeostasis (2).
Much less is known about NOS function in human liver. Human-inducible NOS was first cloned from human hepatocytes in culture (7). These data revealed that human-inducible NOS shared an 80% sequence homology with murine macrophage-derived mac-NOS. Although it has been suggested that NO may be a causative factor in hemodynamic changes detected in patients with liver cirrhosis (8), most human studies involved the use of cultured hepatocytes, blood cells, or liver biopsies to study NOS mRNAs (9–13). As with most proteins, the presence of mRNA for NOS isoforms provides indicative but not definitive evidence for the expression of enzyme activity. A recent study (14) reported that the activity of constitutive NOS in liver biopsies from cirrhotic patients was reduced. However, NOS proteins were not identified in this study, nor were there any correlations made between the activity of NOS and the underlying liver disorders.
Therefore, we have investigated the expression of NOS proteins, their cellular localization, and enzymatic activities in surgical sections of apparently normal and diseased human livers obtained from patients with common and rare end-stage liver disorders. In addition, we measured plasma levels of NO metabolites, nitrite, and nitrate to compare liver-derived NO with systemically generated NO.
Materials and Methods
Patients and Materials.
Peripheral venous blood and liver tissue specimens were collected from 42 patients who received orthotopic liver transplantation for end-stage liver disease between the months of January 1997 to May 1998. Postoperative pathology reports were reviewed, confirming a distribution of common and relatively common disease groups including viral hepatitis, alcoholic cirrhosis, cholestasis, fulminant hepatic failure, hemochromatosis, cryptogenic cirrhosis, and autoimmune hepatitis (collectively referred to herein as “major liver disorders”). In addition, uncommon forms of liver disease, including α1-anti-trypsin disorder, epithelioid hemangioendothelioma, and Budd–Chiari syndrome (collectively referred to herein as “rare liver disorders”), were also diagnosed. Each patient provided written informed consent before his/her recruitment into the study.
Control liver tissue was obtained from 14 adult patients who received a partial hepatectomy for selected intrahepatic malignancies (hepatocellular carcinoma, metastatic colon carcinoma, and metastatic renal cell carcinoma) and benign lesions (ecchinococcal cyst). Only those patients diagnosed with localized tumors were considered. Appropriate control specimens were obtained from the outermost region of the tumor-free margin from the excised lobe. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and those of the Research Ethics Board of the University of Alberta, Faculty of Medicine.
All liver tissue samples including those obtained from patients undergoing liver transplant were collected intraoperatively at the time of excision, at which time they were sectioned, snap frozen with liquid nitrogen, and then stored at −80°C until assayed. Venous blood was obtained from healthy adult donors who had not taken any drugs for at least 2 weeks before collection. Samples of venous blood from transplant patients were collected before the administration of immunosuppressive therapy. At the time of collection of donor and patient blood, 9 ml of blood was added to 1 ml of 3.15% sodium citrate, followed by blood centrifugation at 800 × g for 10 min at room temperature. Plasma was aliquoted and stored at −80°C until assayed.
NOS Activity.
NOS activities were measured by the rate of conversion of U-[14C]l-arginine to U-[14C]l-citrulline (15). Briefly, liver samples (0.5 g wet weight) were homogenized by sonication (VibraCell, Danbury, CT) in 1 ml of ice-cold homogenization buffer (pH 7.4) containing 50 mM Tris⋅HCl, 320 mM sucrose, 1 mM DTT, 10 μg/ml leupeptin, 10 μg/ml soybean trypsin inhibitor, 2 μg/ml aprotinin, followed by centrifugation at 10,000 × g for 20 min at 4°C. After centrifugation, 40 μl of supernatant was incubated at 37°C for 20 min in assay buffer (pH 7.4) containing 50 mM KH2PO4, 1 mM MgCl2, 0.2 mM CaCl2, 1 mM l-citrulline, 20 μM l-arginine, 1.5 mM DTT, 1.5 mM NADPH, 10 μM tetrahydrobiopterin, 10 μM FAD, 10 μM FMN, and U-[14C]l-arginine [0.5 μCi/ml (1 Ci = 37 GBq), Amersham Pharmacia]. The specificity of l-arginine conversion by NOS to l-citrulline was further confirmed by using 1.2 mM Nω-nitro-l-arginine methyl ester, a selective inhibitor of NOS (Alexis, San Diego, CA). Additionally, 1.5 mM EGTA, a calcium-chelating agent, was used to differentiate between Ca2+-dependent and -independent isoforms of NOS. All enzyme activities were expressed as pmol of product generated per minute per mg of protein. The limit of detection of this method was 0.05 pmol/min/mg protein.
Western Immunoblots of NOS.
The expression of NOS isoforms in liver sections was measured as described (15). Briefly, homogenized samples (80 μg of protein each) were subjected to SDS/7% PAGE under reducing conditions. Proteins were transferred onto poly(vinylidene difluoride) membranes (Schleicher & Schuell) by using a TransBlot Cell system (Bio-Rad). The eNOS and nNOS were identified by using respective polyclonal antibodies (0.2 μg/ml, Santa Cruz Biotechnology), whereas monoclonal antibodies (0.05 μg/ml, Transduction Laboratories, Lexington, KY) were used for the iNOS. All blots were developed simultaneously with an enhanced chemiluminescence kit (Amersham Pharmacia); the density of bands was quantified by using a ScanJet 3c scanner (Hewlett–Packard) and SigmaGel measurement software (Jandel, San Rafael, CA). Band densities were expressed in arbitrary units per mg of protein.
Immunohistochemistry of NOS.
Immunohistochemistry was carried out as described (16) by using an antigen-retrieval technique. Briefly, samples were prepared by mounting 4-μm thick slices of formalin-fixed/paraffin-embedded liver sections obtained from patients onto Aptex coated slides and then drying them overnight at 37°C. After 10 min of heating at 70°C, slides were deparaffinized with xylene, rehydrated with decreasing grades of ethanol and finally with water. Next, slides were soaked in H2O2/methanol solution for 6 min and later rinsed with water. Antigen retrieval was accomplished by microwaving slides in citrate buffer for 20 min. Counterstaining was done with Harris' hematoxylin. Blocking reagent (20% normal goat serum) was applied to each slide followed by 15 min incubation at room temp. Primary antisera consisted of mouse anti-human monoclonal antibodies for eNOS and iNOS (5 μg/ml, Transduction Laboratories). Normal mouse serum was used for negative controls. After overnight incubation at 4°C with the primary antibody, slides were rinsed with PBS for 5 min and then incubated an additional 20 min with the Link reagent (2–10 μg/ml biotinylated anti-mouse IgG, Vector Laboratories). After a 5-min PBS rinse, the streptavidin peroxidase label reagent (BioGenex Laboratories, San Ramon, CA) was applied for 20 min at room temp. A final PBS rinse was followed by a 5-min incubation with the chromagen solution 3,3′-diaminobenzidine (DAB) + H2O2. Lastly, the slides were dehydrated, cleared, and mounted for viewing.
In Situ Hybridization of NOS.
In situ hybridization was performed as described (17). Briefly, cryostat sections from biopsy specimens were permeabilized with Triton X-100 and proteinase K solution (1 μg/ml) in 0.1 Tris containing 50 mmol EDTA for 20 min at 37°C. The samples were subsequently incubated with 0.1 mol/liter triethanolamine and 0.5% acetic anhydride for 20 min at 37°C to prevent the nonspecific binding of the 35S-labeled cRNA probes. Prehybridization of the samples was carried out in 50% formamide in 2× standard saline citrate for 15 min at 37°C. Hybridization was carried out with the use of a hybridization mixture containing the appropriate sense or antisense probes (0.75 × 106 counts per min per slide) for both eNOS and iNOS. Posthybridization involved high-stringency washing of the samples in decreasing concentrations of standard saline citrate at 42°C. To remove any unbound RNA probes, the samples were washed with ribonuclease solution for 20 min at 42°C. The samples were dehydrated with increasing concentrations of ethanol and then air-dried. After this, the samples were dripped in Amersham Pharmacia LM-2 emulsion and exposed for a period of 14 days. The autoradiographs were then developed in Kodak D-19 developer, fixed, and counterstained with hematoxylin. The samples were mounted with a coverslip and examined under a graduated microscope for positive signals.
NO Metabolites: Nitrite and Nitrate (NO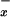 ).
).
Cell-free plasma was deproteinized by ultrafiltration by using an Ultrafree-MC micropartition system (Millipore). Samples were analyzed by using an automated HPLC system according to the method described by Green et al. (18). The method of detection was based on the nitrite reaction with Griess reagent to give a color product that could be detected at the visible wavelength of 546 nm. The nitrate content of the plasma sample was reduced to nitrite as it passed through a cadmium precolumn before being mixed with the Griess reagent; it was then analyzed on-line by using a visible light absorbance detector. The limit of detection of this method was 1.0 nmol/ml nitrite. To account for abnormalities in renal function, NO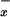 levels were also normalized to serum creatinine levels.
levels were also normalized to serum creatinine levels.
Statistical Analysis.
All data were expressed as mean ± SEM and were analyzed by INSTAT software (GraphPad, San Diego). Comparisons were performed by using ANOVA followed by a Tukey–Kramer multiple comparisons test. Differences were considered statistically significant at values of P < 0.05.
Results
Table 1, which is published as supporting information on the PNAS web site, www.pnas.org, summarizes some demographic, clinical, and laboratory parameters characterizing patients included in this study.
NOS Activity and Western Immunoblots.
The activity of Ca2+-dependent NOS in sections of control liver was 0.39 ± 0.14 pmol/min/mg protein. No significant difference in this activity was found in any of the chronic liver disorders when compared with the control levels (Fig. 1A). Western blot analysis revealed that eNOS immunoreactivity was present in the liver samples of control patients. This finding was not significantly changed in samples from patients with alcoholic cirrhosis, viral hepatitis, cholestasis, and fulminant liver failure (Fig. 1A). The activity of Ca2+-independent NOS in sections of control liver was 0.44 ± 0.16 pmol/min/mg protein (Fig. 1B). There was a significant increase in this activity in major liver disorders, except for fulminant hepatic failure. In control samples, there was a significant expression of immunoreactivity consistent with the presence of iNOS (Fig. 1B). Western blot did not reveal significant changes in the expression of iNOS immunoreactivity in any of the patient groups when compared with controls (Fig. 1B). No significant expression of nNOS immunoreactivity was detected in control or diseased specimen.
Fig 1.

The activity and the corresponding protein expression of eNOS (A) and iNOS (B) in normal and diseased livers. *, P < 0.05; and **, P < 0.01, groups vs. control. (Insets) Individual Western blots of sections of control livers.
Immunohistochemistry and in Situ Hybridization of NOS.
Controls.
Fig. 2 shows the distribution of eNOS immunoreactivity in normal liver. Low-power micrograph (Fig. 2A) documents that eNOS was uniformly distributed in hepatocytes throughout all zones. Fig. 2B demonstrates the expression of eNOS in the endothelium of hepatic artery and in the epithelium of adjacent bile duct. Fig. 2C highlights the presence of eNOS in the endothelium of terminal hepatic venules and in the contiguous sinusoids.
Fig 2.

The distribution of eNOS and iNOS in normal liver as shown by immunohistochemistry. (A) Homogenous distribution of eNOS immunoreactivity in hepatocytes. THV, terminal hepatic venule; PT, portal triad (×40). (B) The presence of eNOS in the epithelial cells of biliary duct (BD) and in the endothelium of hepatic artery (A) (×400). (C) The eNOS in THV and sinusoids (S) (×400). The iNOS immunoreactivity is strongly expressed in the cytoplasm of hepatocytes surrounding the portal triad (PT), but not those associated with the terminal hepatic venule (THV). (D, ×40; E, ×100, respectively.)
Fig. 2 D and E depicts the distribution of iNOS immunoreactivity in control liver. Low-power micrograph (Fig. 2D) shows that iNOS was distributed mainly in periportal regions of the acinus. There appears to be a sharp transition between iNOS-rich and iNOS-poor regions of the acinus (Fig. 2E). In all investigated liver sections, hepatocytes constituted the major source of iNOS immunoreactivity (not shown).
To confirm that eNOS and iNOS are constitutively expressed in hepatocytes, control liver sections were examined by using in situ hybridization. The examination of control liver sections treated with 35S-labeled eNOS and iNOS cRNA probes showed autoradiographic evidence of association with hepatocytes and endothelial cells (Fig. 3 A and B). Sections of liver treated with control sense probes revealed background staining only (Fig. 3C).
Fig 3.

In situ hybridization of eNOS (A) and iNOS (B) in control liver using 35S-labeled eNOS and iNOS probes. (A) The eNOS cRNA staining is associated with hepatocytes and endothelial cells. (B) Focal iNOS cRNA staining in hepatocytes. (C) Section of liver stained with sense control iNOS cRNA probe, with background staining only. Dark field illumination with hematoxylin–eosin counterstaining (×200).
Patients.
Fig. 4 A and B demonstrates the distribution of eNOS in sections from patients with hepatitis C and alcoholic cirrhosis, respectively. Low power micrographs show that the majority of eNOS was concentrated in hepatocytes as dense granules in areas of cirrhosis (Fig. 4 A and B). Examination of the corresponding high-power micrographs (Fig. 4 A and B, Insets) showed that eNOS was translocated to hepatocyte nuclei. Similar expression of eNOS in the nuclear regions was detected in other major liver disorders (not shown).
Fig 4.

The distribution of eNOS and iNOS in diseased livers as revealed by immunohistochemistry. Dense areas of eNOS immunoreactivity (A, hepatitis; B, alcoholic cirrhosis) are present in nuclei of hepatocytes (Insets). (A and B, ×40; Insets, ×250.) Shown is the distribution of iNOS in alcoholic cirrhosis (C), α1-antitrypsin disorder (D), and biliary atresia (E). (C, ×100; D and E, ×40.) (C and D Insets) Intense staining of iNOS (×250). (E Inset, ×10.)
Fig. 4 C and D illustrates the expression of iNOS immunoreactivity in alcoholic cirrhosis and α1-antitrypsin disorder, respectively. In contrast to normal sections (Fig. 2 D and E), there was a uniform distribution of iNOS in the hepatocytes in cirrhotic regions. In these areas, the intensity of iNOS staining was high (Fig. 4 C and D, Insets). Relative to the strong staining of iNOS in hepatocytes, the expression of this enzyme in Kupffer cells was weak (not shown). Fig. 4E shows the expression of iNOS in the liver of a patient with biliary atresia. In the cirrhotic areas, there was a uniform distribution of iNOS in the surviving hepatocytes, whereas in noncirrhotic areas of the same liver, the zonal distribution of iNOS was preserved (Fig. 4E Inset). The eNOS and iNOS immunoreactivity in liver from patients with massive liver necrosis showed very weak and patchy staining within the areas of diffuse necrosis of hepatic tissue. The iNOS appeared to be expressed in the endothelial cells of terminal hepatic venule under these conditions (not shown).
NO Metabolites: Nitrite and Nitrate (NO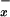 ).
).
In plasma of control subjects the levels of NO metabolites normalized to serum creatinine levels were 0.82 ± 0.03 μmol NO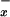 /μmol creatinine. These levels were not significantly modified in plasma of patients with viral hepatitis, alcoholic cirrhosis, and cholestasis (P > 0.05). In contrast, there was a significant decrease in NO
/μmol creatinine. These levels were not significantly modified in plasma of patients with viral hepatitis, alcoholic cirrhosis, and cholestasis (P > 0.05). In contrast, there was a significant decrease in NO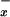 levels in plasma of patients with fulminant hepatic failure (0.32 ± 0.05 μmol NO
levels in plasma of patients with fulminant hepatic failure (0.32 ± 0.05 μmol NO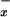 /μmol creatinine, P < 0.01).
/μmol creatinine, P < 0.01).
Discussion
We examined the expression, localization, and activity of NOS isoenzyme proteins in apparently healthy and cirrhotic surgical sections of human livers.
NOS in Normal Liver.
The citrulline assay showed that both Ca2+-dependent and Ca2+-independent NOS activities were present in normal liver. Western immunoblots determined that Ca2+-dependent and -independent NOS could be identified as eNOS and iNOS, whereas nNOS was not detectable. We then examined the source and distribution of eNOS and iNOS in normal liver. We performed immunohistochemistry on the corresponding formalin-fixed/paraffin-embedded liver slices (16), because the cellular architecture is well preserved in paraffin blocks.
The eNOS immunohistochemistry revealed that this isoform was uniformly distributed in hepatocytes. The eNOS was also present in the endothelium of hepatic arteries, terminal hepatic venules, and sinusoids. Interestingly, the epithelium of biliary ducts showed strong expression of eNOS. Thus, in addition to endothelial cells, both hepatocytes and biliary epithelium express eNOS. The functional significance of eNOS expression in the endothelium as a regulator of blood flow and cell–cell interactions has been proposed (1, 2, 8). However, to the best of our knowledge, this study provides the first evidence for the presence of eNOS in human hepatocytes. To confirm the immunohistochemistry data, we performed in situ hybridization experiments that showed the presence of staining associated with cRNA for eNOS in hepatocytes. The significance of these findings remains to be investigated; however, it is tempting to speculate that NO generation in hepatocytes may be involved in the regulation of metabolic functions of these cells. There are very few studies that have examined the role of NO in biliary epithelial cell function. It has been suggested that NO may regulate bile canalicular motility in rats (19). The present study shows the expression of eNOS in human biliary duct epithelial cells.
We found that iNOS was constitutively expressed in a zonal pattern in the normal hepatic acinus. The distribution of iNOS showed the strongest expression in the periportal region, with diminution of intensity toward the perivenous regions of the hepatic acinus. Again, by using in situ hybridization, we confirmed this constitutive expression of iNOS in control liver. The constitutive expression of iNOS protein in healthy human cells and tissues is a relatively new concept in the biology of NO. Early investigations using cell culture systems or isolated animal organs did not detect the constitutive expression of functional iNOS protein under normal conditions (1). However, the immunohistochemical examination of human airways indicates that iNOS may be expressed under physiological conditions (20).
The zonal distribution of iNOS in hepatocytes is a subject of considerable interest. Despite similar histological appearance, hepatocytes localized in distinct regions of the liver acinus seem functionally different from each other. The origin of the concept of hepatocyte heterogeneity can be dated back to 1846 (21). In the 1950s, Rappaport introduced a concept of the liver acinus as the structural unit of liver that can be subdivided further into three functional zones (1, 2, and 3; ref. 22). The general consensus deriving from this study was that the main factor regulating the functional differences between zones was their perfusion with blood of varying composition. Thus, in this context, functional hepatocyte heterogeneity may be seen as an adaptation to a changing sinusoidal microenvironment. The histochemical and enzymology studies performed in the 1950s and 1960s gave rise to the concept of metabolic zonation that could result from zonal gene expression early in life (21). The hepatic enzymes mediating the glucagon-sensitive glucose-forming pathways and the urea cycle exemplify the concept of metabolic zonation (21). The pathological significance of hepatocyte heterogeneity is best illustrated by the observation that there is zonal sensitivity of hepatocytes to toxic damage by hypoxia and hepatotoxins, including acetaminophen, halothane, CCl4, and bromobenzene (23, 24).
The iNOS seems to be yet another enzyme that is heterogeneously distributed in liver. The expression of iNOS is cytokine-regulated, and endotoxin plays a major role in iNOS induction. Indeed, cytokine stimulation leads to expression of hepatic iNOS in cultured human hepatocytes (7). It is possible that constitutive induction of hepatic iNOS is caused by constant stimulation of hepatocytes with bacterial and/or chemical products that are absorbed from the intestine to the portal circulation and then distributed in the liver. Periportal hepatocytes might encounter the greatest concentration of endotoxin when compared with the perivenous regions leading to zonal induction of iNOS. An alternative explanation of periportal localization of iNOS could be based on zonal iNOS gene expression early in life.
The physiological significance of iNOS heterogeneity remains to be studied; however, three possibilities deserve some consideration. First, it may be that iNOS expression under physiological conditions merely reflects the effects of differential oxygen content and intestinal toxins to which hepatocytes are exposed. Second, generation of NO by iNOS would make it readily available for local reactions of defense and nonspecific immunity (1, 2). Third, the induction of iNOS may represent a mechanism through which hepatocytes control the degree of apoptosis in the liver (2).
NOS in Cirrhotic Liver.
The measurement of Ca2+-dependent eNOS activity did not reveal any significant changes in the activity of this enzyme in patients with viral hepatitis, alcoholic cirrhosis, and cholestasis. The corresponding immunoblot showed that the expression of eNOS protein was similar to control in alcoholic cirrhosis and cholestasis.
The immunohistochemical analysis showed that major as well as rare liver disorders examined in this study were associated with profound changes in the cellular distribution of eNOS, leading to its translocation to hepatocyte nuclei. Interestingly, growth factors such as vascular-endothelial growth factor are known to cause nuclear translocation of eNOS in vascular endothelium (25). The significance of this observation is as yet unclear. Again, nuclear translocation of eNOS may merely reflect “growth factor storm” that is characteristic of chronic liver inflammation and cirrhosis (26, 27). Alternatively, the eNOS translocation may be a part of a liver defense mechanism aimed at limiting the effects of growth factors by decreasing the rate of cellular proliferation or apoptosis that is known to be regulated by NO (2).
The iNOS activity was significantly elevated in viral hepatitis, alcoholic cirrhosis, and cholestasis. Interestingly, the degree of elevation was similar in these disease states despite their very divergent etiopathogenesis. Thus, increased activity of iNOS may be a common denominator of end-stage liver disorders of various etiologies.
We observed a lack of correlation between increased activity and unaltered Western immunoreactivity of iNOS. Similar discrepancy involving the activity and expression of iNOS can be detected in other types of pathologies, such as hyperoxia-induced inflammation in rats (15).
The immunohistochemistry of iNOS in chronic common end-stage liver disorders was in close agreement with the enzyme activity data showing that the cirrhotic nodules strongly and uniformly expressed iNOS. This strong and nonzonal expression of iNOS was also present in the sections of rare liver disorders such as α1-antitrypsin deficiency. Relative to the strong staining of iNOS in hepatocytes, its expression in resident hepatic macrophages (Kupffer cells) was weak. Interestingly, in cell culture systems, iNOS is rapidly induced by cytokines in Kupffer cells (2). Therefore, the Kupffer cell iNOS is unlikely to contribute in a significant way to the pool of inducible NO generated in cirrhotic liver disorders.
The reasons for nonzonal distribution of iNOS in end-stage liver disorders are most likely related to cirrhosis. The liver section obtained from a patient with extrahepatic biliary atresia supports this hypothesis. Biliary atresia is a pediatric disorder characterized by obstruction of extrahepatic biliary ducts. The obstruction leads to the inflammation and cirrhosis of some but not all regions of the liver (28). The examination of distribution of iNOS demonstrated that cirrhotic sections of liver obtained from this subject showed a marked loss of the zonality that still persisted in noncirrhotic regions of the same liver.
The significance of increased activity of iNOS in cirrhosis remains to be investigated. Interestingly, overproduction of NO has been linked to the loss of cytochrome P450 content and injury in cultured rat hepatocytes (29). In contrast, pretreatment with a NO donor or preinduction of iNOS stimulated the expression of the inducible heat-shock protein HSP-70 in hepatocytes and inhibited TNFα + actinomycin-D-induced hepatocyte apoptosis (30). Moreover, increased activity of iNOS in cirrhosis could increase hepatic blood flow, thus maintaining the viability of the remaining liver cells.
The measurement of NO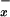 levels in plasma is thought to be a convenient way to get an insight into the systemic release of NO in humans. However, one has to be aware that the basal NO
levels in plasma is thought to be a convenient way to get an insight into the systemic release of NO in humans. However, one has to be aware that the basal NO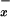 levels in human plasma are high, reflecting the dietary origin of nitrite and nitrate. The hyperdynamic circulation state that complicates liver failure may be associated with increased release of NO (8, 31). In our population of patients with diverse end-stage liver disorders, we were unable to detect significant changes in NO
levels in human plasma are high, reflecting the dietary origin of nitrite and nitrate. The hyperdynamic circulation state that complicates liver failure may be associated with increased release of NO (8, 31). In our population of patients with diverse end-stage liver disorders, we were unable to detect significant changes in NO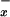 in systemic venous blood when compared with controls. Thus, plasma NO
in systemic venous blood when compared with controls. Thus, plasma NO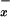 levels in the systemic circulation do not reflect well on the profound alterations in NO metabolism detected in liver parenchyma of patients with viral hepatitis, alcoholic cirrhosis, and cholestasis. Indeed, blood sampling from portal venous circulation may be necessary to get an insight into the changes in NO generation in the liver (14). In contrast to cirrhosis, a reduction of NO
levels in the systemic circulation do not reflect well on the profound alterations in NO metabolism detected in liver parenchyma of patients with viral hepatitis, alcoholic cirrhosis, and cholestasis. Indeed, blood sampling from portal venous circulation may be necessary to get an insight into the changes in NO generation in the liver (14). In contrast to cirrhosis, a reduction of NO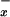 was observed in patients with acute liver necrosis, emphasizing the devastating effects of this syndrome on the generation of NO by NOS.
was observed in patients with acute liver necrosis, emphasizing the devastating effects of this syndrome on the generation of NO by NOS.
In conclusion, we have shown that both eNOS and iNOS proteins are differentially expressed in healthy human liver, and that this expression is significantly altered in chronic end-stage liver disorders of various origins.
Acknowledgments
We thank Dr. L. Jewell for helpful advice and Ms. A. Bolivar for technical assistance. This work was supported by Canadian Institutes of Health Research (CIHR) Grant 14074. M.W.R. is a CIHR Scientist, and R.M. is an Alberta Heritage Senior Medical Scholar.
Abbreviations
NOS, NO synthase
eNOS, endothelial NOS
nNOS, neuronal NOS
iNOS, inducible NOS
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.de Belder A. J. & Radomski, M. W. (1994) J. Hypertens. 12, 617-624. [PubMed] [Google Scholar]
- 2.Li J. & Billiar, T. R. (1999) Am. J. Physiol. 276, G1069-G1073. [DOI] [PubMed] [Google Scholar]
- 3.Lok A. S. (2000) J. Hepatol. 32, Suppl. 1, 89-97. [DOI] [PubMed] [Google Scholar]
- 4.Williams I. (1999) Am. J. Med. 107, 2S-29S. [DOI] [PubMed] [Google Scholar]
- 5.Billiar T. R., Curran, R. D., Harbrecht, B. G., Stuehr, D. J., Demetris, A. J. & Simmons, R. L. (1990) J. Leukocyte Biol. 48, 565-569. [DOI] [PubMed] [Google Scholar]
- 6.Diehl A. M. (1999) Clin. Biochem. 32, 571-578. [DOI] [PubMed] [Google Scholar]
- 7.Geller D. A., Lowenstein, C. J., Shapiro, R. A., Nussler, A. K., Di Silvio, M., Wang, S. C., Nakayama, D. K., Simmons, R. L., Snyder, S. H. & Billiar, T. R. (1993) Proc. Natl. Acad. Sci. USA 90, 3491-3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vallance P. & Moncada, S. (1991) Lancet 337, 776-778. [DOI] [PubMed] [Google Scholar]
- 9.Mihm S., Fayazi, A. & Ramadori, G. (1997) Hepatology 26, 451-458. [DOI] [PubMed] [Google Scholar]
- 10.Sanchez-Rodriguez A., Criado, M., Rodriguez-Lopez, A. M., Esteller, A., Martin de Arriba, A. & Lopez-Novoa, J. M. (1998) Clin. Sci. 94, 637-643. [DOI] [PubMed] [Google Scholar]
- 11.Galley H. F., Coomansingh, D., Webster, N. R. & Brunt, P. W. (1998) Clin. Sci. 95, 355-359. [PubMed] [Google Scholar]
- 12.Amaro M. J., Bartolome, J. & Carreno, V. (1999) Hepatology 29, 915-923. [DOI] [PubMed] [Google Scholar]
- 13.Schweyer S., Mihm, S., Radzun, H. J., Hartmann, H. & Fayyazi, A. (2000) Gut 46, 255-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarela A. I., Mihaimeed, F. M. A., Batten, J. J., Davidson, R. T. & Mathie, R. T. (1999) Gut 44, 749-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radomski A., Sawicki, G., Olson, D. M. & Radomski, M. W. (1998) Br. J. Pharmacol. 125, 1455-1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Archer S. L., Reeve, H. L., Michelakis, E., Puttagunta, L., Waite, R., Nelson, D. P., Dinauer, M. C. & Weir, E. K. (1999) Proc. Natl. Acad. Sci. USA 96, 7944-7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamid Q., Azzawi, M., Ying, S., Moqbel, R., Wardlaw, A. J., Corrigan, C. J., Bradley, B., Durham, S. R., Collins, J. V., Jeffery, P. K., et al. (1991) J. Clin. Invest. 87, 1541-1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green L. C., Wagner, D. A., Glogowski, J., Skipper, P. L., Wishnok, J. S. & Tannenbaum, S. R. (1982) Anal. Biochem. 126, 131-138. [DOI] [PubMed] [Google Scholar]
- 19.Dufour J. F. J., Turner, T. J. & Arias, I. M. (1995) Gastroenterology 108, 841-849. [DOI] [PubMed] [Google Scholar]
- 20.Watkins D. N., Peroni, D. J., Basclain, K. A. & Garlepp, M. J. (1997) Am. J. Respir. Cell Mol. Biol. 16, 629-639. [DOI] [PubMed] [Google Scholar]
- 21.Gumuccio J. J. (1989) Hepatology 9, 154-160. [DOI] [PubMed] [Google Scholar]
- 22.Rappaport A. M. (1958) Anat. Rec. 130, 637-686. [DOI] [PubMed] [Google Scholar]
- 23.Sies H. (1978) Adv. Exp. Med. Biol. 94, 561-566. [DOI] [PubMed] [Google Scholar]
- 24.Ji S., Lemasters, J. J., Christenson, V. & Thurman, R. G. (1982) Proc. Natl. Acad. Sci. USA 79, 5415-5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng Y., Venema, V. J., Venema, R. C., Tsai, N. & Caldwell, R. B. (1999) Biochem. Biophys. Res. Commun. 256, 192-197. [DOI] [PubMed] [Google Scholar]
- 26.Harada K., Shiota, G. & Kawasaki, H. (1999) Liver 19, 318-325. [DOI] [PubMed] [Google Scholar]
- 27.Okano J. I., Shiota, G. & Kawasaki, H. (1999) Liver 19, 151-159. [DOI] [PubMed] [Google Scholar]
- 28.Cotran R. S., Kumar, V. & Robbins, S. L. (1989) in Robbins Pathologic Basis of Disease, ed. Saunders Staff, W. B. (Saunders, Philadelphia), pp. 911–980.
- 29.Lopez-Garcia M. P. & Sanz-Gonzalez, S. M. (2000) FEBS Lett. 466, 187-191. [DOI] [PubMed] [Google Scholar]
- 30.Li J. & Billiar, T. R. (1999) Cell Death Differ. 6, 952-955. [DOI] [PubMed] [Google Scholar]
- 31.Guarner C., Soriano, G., Tomas, A., Bulbena, O., Novella, M. T., Balanzo, J., Vilardell, F., Mourelle, M. & Moncada, S. (1993) Hepatology 18, 1139-1143. [PubMed] [Google Scholar]