

Abstract
A novel enzyme, l-sorbosone dehydrogenase 1 (SNDH1), which directly converts l-sorbosone to l-ascorbic acid (l-AA), was isolated from Ketogulonicigenium vulgare DSM 4025 and characterized. This enzyme was a homooligomer of 75-kDa subunits containing pyrroloquinoline quinone (PQQ) and heme c as the prosthetic groups. Two isozymes of SNDH, SNDH2 consisting of 75-kDa and 55-kDa subunits and SNDH3 consisting of 55-kDa subunits, were also purified from the bacterium. All of the SNDHs produced l-AA, as well as 2-keto-l-gulonic acid (2KGA), from l-sorbosone, suggesting that tautomerization of l-sorbosone causes the dual conversion by SNDHs. The sndH gene coding for SNDH1 was isolated and analyzed. The N-terminal four-fifths of the SNDH amino acid sequence exhibited 40% identity to the sequence of a soluble quinoprotein glucose dehydrogenase from Acinetobacter calcoaceticus. The C-terminal one-fifth of the sequence exhibited similarity to a c-type cytochrome with a heme-binding motif. A lysate of Escherichia coli cells expressing sndH exhibited SNDH activity in the presence of PQQ and CaCl2. Gene disruption analysis of K. vulgare indicated that all of the SNDH proteins are encoded by the sndH gene. The 55-kDa subunit was derived from the 75-kDa subunit, as indicated by cleavage of the C-terminal domain in the bacterial cells.
l-Ascorbic acid (l-AA) is an essential nutrient for humans. It is known that l-AA is directly converted from aldonolactones, such as l-gulono-γ-lactone and l-galactono-γ-lactone, by aldonolactone dehydrogenase/oxidases in plants and mammals (except some primates, including humans). The l-gulono-γ-lactone oxidase that catalyzes the conversion of l-gulono-γ-lactone to l-AA was isolated from rat and goat livers and characterized by Nishikimi et al. (19). An l-galactono-γ-lactone oxidase of yeast origin was described by Bleeg (4). In addition, l-sorbosone is also known to be a potential precursor of l-AA in plants. An NADP-dependent l-sorbosone dehydrogenase (SNDH) that converts l-sorbosone to l-AA in spinach leaves has been reported (17), but the enzyme has not been purified or characterized in detail.
In current industrial l-AA production processes, as summarized by Hancock and Viola (13), 2-keto-l-gulonic acid (2KGA) is a key intermediate that is chemically converted to l-AA. All of the processes require a large amount of energy and organic solvent, and thus a cheaper and environmentally conscious substitute process, such as enzymatic conversion, is desirable. It has also been reported that l-sorbosone is converted to 2KGA in bacteria. Many microbial l-sorbosone dehydrogenases, including enzymes from Acetobacter liquefaciens (27), Gluconobacter melanogenus UV10 (14), and Gluconobacter oxydans T-100 (23), have been isolated and characterized. Moreover, Ketogulonicigenium vulgare DSM 4025 has been reported to produce aldehyde dehydrogenase (ALDH) (15) and l-sorbose/l-sorbosone dehydrogenases (SSDHs) (3), which are responsible for sequentially converting l-sorbose to 2KGA via l-sorbosone.
The metabolic pathways from primary sugars in K. vulgare DSM 4025 have been studied in our laboratory. Recently, it was reported that K. vulgare DSM 4025 has the ability to produce l-AA from sugars such as d-sorbitol, l-sorbose, l-gulose, and l-sorbosone (30). One l-AA-producing enzyme that catalyzes oxidation of l-gulono-γ-lactone was isolated previously (29), but the detailed pathway from primary sugars to l-AA was unclear. In this study, we report identification of novel SNDHs that directly convert l-sorbosone to l-AA and their genes in K. vulgare DSM 4025.
MATERIALS AND METHODS
Chemicals.
l-Sorbose, l-sorbosone, d-glucosone, l-gulono-γ-lactone, and 2KGA were supplied by F. Hoffmann-La Roche, Switzerland. Pyrroloquinoline quinone (PQQ) was purchased from Mitsubishi Gas Chemical, Japan. Other chemicals used in this study were commercially available.
Bacterial strains and plasmids.
The bacterial strains and plasmids used in this study are listed in Table 1. Strain DSM 4025, formerly classified in the genus Gluconobacter (35) and recently proposed to be reclassified in the new genus Ketogulonicigenium (34), was used for preparation of the SNDHs and chromosomal DNA.
TABLE 1.
Bacterial strains and plasmids
| Bacterial strain or plasmid | Relevant characteristic(s)a | Source or reference |
|---|---|---|
| E. coli strains | ||
| VCS257 | Derivative of DP50supF [supE44 supF58 hsdS3(rB−mB−) dapD8 lacY1 glnV44 Δ(gal-uvrB)47 tyrT58 gyrA29 tonA53 Δ(thyA57)] | Stratagene |
| JM109 | recA1 endA1 gyrA96 thi hsdR17 supE44 relA1 Δ(lac-proAB)/F′ [traD36 proAB+lacIqlacZΔM15] | Takara Bio |
| DH5α | F− φ80d lacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) phoA supE44 λ−thi-1 gyrA96 relA1 | Takara Bio |
| HB101 | leuB6 proA2 recA13 thi-1 ara-14 lacY1 galK2 xyl-5 mtl-1 rpsL20 supE44 Δ(mcrC-mrr) | Takara Bio |
| M15 | Nals Strs Rifs Thi− Lac− Ara+ Gal+ Mtl− F− RecA+ Uvr+ Lon+ | QIAGEN |
| DSM 4025 derivatives | ||
| DSM 4025 | K. vulgare strain | 34 |
| GOMTR1 | Rifr, spontaneous mutant of DSM 4025 | This study |
| GOMTR1SN::Km | Rifr Kmr, sndH gene disruptant of GOMTR1 | This study |
| Plasmids | ||
| pVK100 | ATCC 37156, RK2 (IncP) derivative, 23 kb, Kmr Tcr, cos | ATCCb |
| pRK2013 | RK2 (IncP) derivative, 45 kb, Kmr, mob | ATCC |
| pSUP202 | Apr Tcr Cmr, mob | 28 |
| pSUPSN | pSUP202 carrying sndH | This study |
| pSUPSN::Km | pSUP202 carrying sndH disrupted with Kmr gene cassette | This study |
| pQE30 | Six-His tag, Apr | QIAGEN |
| pUC18/19 | Apr | Takara Bio |
| pUC4K | Apr Kmr | TOYOBO |
Apr, ampicillin resistance; Cmr, chloramphenicol resistance; Kmr, kanamycin resistance; Tcr, tetracycline resistance; Rifr, rifampin resistance.
ATCC, American Type Culture Collection.
Media and culture conditions.
K. vulgare DSM 4025 cells were grown at 27°C for 4 days on B8 agar containing 8% (wt/vol) l-sorbose, 0.25% MgSO4 · 7H2O, 1.75% corn steep liquor (Nissyoku, Japan), 5% live baker's yeast (Oriental, Japan), 0.5% urea, 0.5% CaCO3, and 2% agar. The cells were also cultivated in 2 liters of medium containing 8% l-sorbose, 0.05% glycerol, 0.25% MgSO4 · 7H2O, 3% corn steep liquor, 0.4% yeast extract, and 0.15% antifoam agent at 30°C and pH 7.0 using a 3-liter jar fermentor. After 48 h, 38.4 g (wet weight) of the cells was obtained from 6 liters of broth. For conjugal transfer of plasmids, DSM 4025 derivative recipients were cultivated in T broth consisting of 3% Trypticase soy broth (Difco, United States) and 0.3% yeast extract (Difco) at 30°C for 24 h. Transconjugants were cultivated on M agar consisting of 5% mannitol, 1.75% corn steep liquor, 5% live baker's yeast, 0.25% MgSO4 · 7H2O, 0.5% CaCO3, 0.5% urea, and 2% agar (pH 7.0) at 27°C for 24 h. Escherichia coli strains were grown in Luria-Bertani (LB) medium with vigorous shaking at 30°C. Antibiotics were used in the medium at the following concentrations: ampicillin, 100 μg/ml; kanamycin (Km), 50 μg/ml; rifampin, 100 μg/ml; and tetracycline, 2 μg/ml.
PAGE.
Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) was performed with 12.5% polyacrylamide gels using the method of Laemmli (16), and proteins were stained with Coomassie brilliant blue R-250 (CBB). For activity staining, native PAGE (pH 9.4) was performed with a 10% polyacrylamide gel by using a method based on the method of Davis (7) at 4°C. The gel was immersed in a staining solution (100 ml) consisting of 100 mM potassium phosphate buffer (pH 7.0), 4 mg nitroblue tetrazolium, 14 mg phenazine methosulfate (PMS), and 200 mg l-sorbosone with gentle shaking at 25°C for 20 min. The reaction was stopped by immersing the gel in 7% (vol/vol) acetic acid.
Enzyme assays.
For the product assay, reaction mixtures consisting of 1.0 mM PMS, 25 mM potassium phosphate buffer (pH 7.0), 1.0 μM PQQ, 1.0 mM CaCl2, 50 mM l-sorbosone, and an enzyme solution were prepared. Each reaction mixture was incubated at 30°C for 30 to 60 min, and the reaction was stopped with metaphosphoric acid at a final concentration of 2%. The amount of l-AA produced was measured at a wavelength of 264 nm using a Tosoh high-performance liquid chromatography (HPLC) system-equipped YMC-Pack Polyamine-II column with a mobile phase consisting of 50 mM NH4H2PO4 and acetonitrile (30:70). For measurement of 2KGA, the reaction product was treated with 1% o-phenylenediamine at 95°C for 15 min and measured at a wavelength of 340 nm using a YMC-Pack Pro C18 column with a mobile phase consisting of H2O and methanol (82:18). One unit of enzyme activity was defined as the amount of enzyme required to produce 1 μmol of the product per min. The protein concentration was determined by using a CBB protein assay solution (Nacalai Tesque).
For a photometric assay of dehydrogenation activity, a basic reaction mixture containing 0.1 mM 2,6-dichlorophenolindophenol (DCIP), 1.0 mM PMS, 50 mM potassium phosphate buffer (pH 7.0), and 1.0 μM PQQ was prepared. For determination of the substrate specificity, 1.0 mM CaCl2 and 2 to 100 mM (final concentration) substrate were added to the basic mixture immediately before the assay. For investigation of the effects of inhibitors, 2 mM l-sorbosone and 5 mM (final concentration) inhibitor or 1.0 mM (final concentration) metal ion were added to the basic mixture. The reaction was started at 25°C, and the enzyme activity was measured by determining the initial rate of reduction of DCIP at 600 nm using a Shimadzu UV-VIS recording spectrophotometer. One unit of enzyme activity was defined as the amount of enzyme that catalyzed the reduction of 1 μmol DCIP per min.
Purification of the enzymes.
K. vulgare DSM 4025 cell paste (19.2 g) was suspended in 100 ml of 50 mM potassium phosphate (pH 7.0) and passed through a French pressure cell at 1,500 kg force/cm2. The resulting cell extract (CFE) was centrifuged at 100,000 × g for 60 min to prepare the soluble fraction. The dialyzed soluble fraction was placed on a column (3 by 50 cm) of Whatman DE-52 DEAE-cellulose. SNDHs were eluted with a linear gradient (800 ml) of 0 to 0.8 M NaCl and concentrated with an ultrafiltration device (Centriprep-10; Amicon, United States). The dialyzed enzyme fraction was placed on a column (3 by 25 cm) of CL-6B DEAE-Sepharose. SNDHs were eluted with a linear gradient (600 ml) of 0.3 to 0.6 M NaCl. The eluted SNDH fractions were dialyzed and placed on a column (1.5 by 25 cm) of Q-Sepharose (Pharmacia). A linear gradient (375 ml) of 0.3 to 0.6 M NaCl was applied. SNDH1 and SNDH2 were eluted at 0.56 M NaCl (Q1 fraction SN), and SNDH3 was eluted at 0.45 M NaCl. The enzyme fractions were desalted and concentrated, and 1.5 M (final concentration) ammonium sulfate was added. After centrifugation (15,000 × g), the supernatant was placed on a hydrophobic Resource ISO column (1.0 ml; Pharmacia) equilibrated with buffer containing 1.5 M ammonium sulfate. SNDH1 and SNDH2 were eluted with 1.12 to 1.10 M and 1.15 to 1.13 M ammonium sulfate, respectively. SNDH3 was eluted with 1.04 to 1.00 M ammonium sulfate. The enzyme fractions were dialyzed and stored at −20°C.
Alternatively, SNDH1 was purified by native PAGE from Q1 fraction SN. The SNDH1 band was excised from a slab gel and electrically eluted using Max-Yield (Atto, Japan) in a running buffer (pH 8.3) containing 5 mM Tris and 3.8 mM glycine. The eluted enzyme solution was ultrafiltered in order to replace the buffer.
Measurement of molecular mass.
The molecular masses of the native enzymes were estimated using HPLC with a size exclusion gel column (TSK gel G3000SWXL; Tosoh). Cyanocobalamin (1,350 Da), myoglobin (16,950 Da), ovalbumin (45,000 Da), bovine gamma globulin (158,000 Da), and thyroglobulin (670,000 Da) were used as molecular mass standards. The molecular mass of the 55-kDa subunit was determined by liquid chromatography-electric spray injection mass spectrometry (Finnigan LCQ; Thermo Finnigan, California).
Analysis of prosthetic groups.
The PQQ groups of the SNDHs were identified by the method of Duine et al. (10). Prosthetic groups of the enzymes were extracted with 0.5 M NaH2PO4 (pH 1.0) and 67% (wt/vol) methanol. Each extract was subjected to PQQ analysis by reverse-phase HPLC using a YMC-Pack Pro C18 AS-312 column at a wavelength of 313 nm.
Heme c was identified by the reduced-minus-oxidized difference spectrum. Reduced and oxidized enzyme solutions at a concentration of 50 μg/ml in 50 mM potassium phosphate buffer (pH 7.0) containing 10 mM sodium hydrosulfite and 10 mM ammonium persulfate, respectively, were prepared for measurement of the difference spectrum with a Shimadzu UV-2200.
Determination of the N-terminal sequences of the proteins.
Purified SNDH1 or SNDH2 (10 μg each) was subjected to SDS-PAGE, and the protein band was electroblotted on a polyvinylidene difluoride membrane (Bio-Rad, California). To remove the blocked glutaminyl residue at the N terminus, the protein blotted on the membrane was incubated with pyroglutamate aminopeptidase (Sigma) at 30°C for 24 h. The treated membrane was subjected to N-terminal amino acid sequencing with an automated amino acid sequencer (ABI model 490; Perkin-Elmer Corp., Connecticut).
Construction and screening of the gene library.
K. vulgare chromosomal DNA was digested partially with EcoRI, ligated to cosmid vector pVK100, and used for in vitro packaging to construct the gene library. For preparation of the gene probe, PCR amplification was performed with the chromosomal DNA and degenerate oligonucleotide DNA primers 5′-CARGGYAACCCSGTBGA-3′ and 5′-GTYTCGTTNGCRCCVAC-3′, which were synthesized according to the N-terminal sequence of the 75-kDa subunit using Taq polymerase (Ex Taq; Takara Bio, Japan). A 41-bp DNA fragment encoding the N-terminal amino acids of SNDH was specifically amplified and cloned into vector pCR-TOPO to obtain plasmid pMTSN2. The DNA probe for hybridization was prepared using a PCR digoxigenin (DIG) labeling kit (Roche Diagnostics GmbH, Germany), pMTSN2 DNA as the template, and primers 5′-CAGGGTAACCCGGTC-3′ and 5′-GACTCGTTTGCGCCC-3′. E. coli colonies carrying the gene library were blotted on a Biodyne A membrane (1.2 μm; Pall Biosupport Corp., New York). The membrane filter was incubated with a DIG-labeled DNA probe at 30°C for 12 h, and this was followed by washing with 2× SSC (0.3 M NaCl plus 30 mM sodium citrate, pH 7.0) containing 0.1% SDS. The dried membrane was subjected to DIG chemiluminescent detection (Roche Diagnostics).
Subcloning and DNA sequence analysis.
Plasmid DNA was prepared by the alkaline SDS method (24) or by using an automatic DNA isolation system (PI-50; Kurabo, Japan). The 8.0-kb PstI fragment, which contained the intact sndH gene, was subcloned into the pUC18 vector to obtain pUCSNP4. The 2.1-kb EcoRI-HindIII fragment containing the 1.0-kb N-terminal region of the sndH gene was cut out from pUCSNP4 and cloned into pUC19 to obtain pUCSN2001. Deletion plasmids containing the truncated sndH gene were prepared using a kilo-sequence deletion kit (Takara Bio). Dideoxy chain termination with Cy5-dATP (Amersham Pharmacia Biotech) was used to determine the DNA sequence.
Disruption of the sndH gene in K. vulgare DSM 4025.
pSUPSN was constructed by inserting the 8.0-kb PstI fragment containing the sndH gene into suicide vector pSUP202 (28). A kanamycin resistance gene cassette (Km cassette) from pUC4K was then inserted into the EcoRI site of the sndH gene in pSUPSN. The resulting plasmid, pSUPSN::Km, was introduced into GOMTR1 by conjugal transfer (26) with helper strain E. coli HB101 harboring pRK2013 and donor strain E. coli DH5α harboring pSUPSN::Km. The sndH null mutant was selected as Kmr, Rifr, and Tcs.
Preparation of anti-SNDH antiserum and immunological analysis.
The sndH coding sequence was cloned into pQE30, yielding pQESN8, which encoded a recombinant SNDH protein with a six-His tag at the N terminus. The recombinant protein was expressed in E. coli M15 with isopropyl-β-d-thiogalactopyranoside (IPTG) induction, purified with an Ni-nitrilotriacetic acid agarose column (QIAGEN), and used to immunize rabbits.
After SDS-PAGE of the cell extracts, the protein bands on the gel were electroblotted on a nitrocellulose filter by using a semidry electroblotting apparatus (Trans-blot SD; Bio-Rad, United States). The subunits of SNDHs were detected using anti-SNDH rabbit antiserum and an alkaline phosphatase conjugate substrate kit (Bio-Rad).
Nucleotide sequence accession number.
The nucleotide sequence data have been deposited in the DDBJ, EMBL, and GenBank databases under accession number AB205130.
RESULTS
Purification of three SNDHs.
Five major activity bands that reduced dyes using l-sorbosone as the substrate were obtained by native PAGE of the cell extract of K. vulgare DSM 4025 (Fig. 1A). Two of these bands, bands for SSDH and ALDH, have been characterized previously in our laboratory. SSDHs comprising four types of isozymes are common PQQ-dependent enzymes in K. vulgare cells and catalyze in three steps: d-sorbitol to l-sorbose, l-sorbose to l-sorbosone, and l-sorbosone to 2KGA (3). ALDH converts l-sorbosone to 2KGA (15). However, the other three enzyme bands (SNDH1, SNDH2, and SNDH3) have not been characterized previously. Recently, it was found that K. vulgare DSM 4025 could produce l-AA from l-sorbosone (30). Therefore, we analyzed these bands for activity that converts l-sorbosone to l-AA.
FIG. 1.

PAGE analysis of SNDHs. (A) Activity staining of the l-sorbosone-dehydrogenating enzymes of K. vulgare DSM 4025 cell extract on a native 10% polyacrylamide gel. The locations of SNDH, SSDH, and ALDH proteins are indicated on the left. (B) SDS-PAGE (12.5% polyacrylamide gel) analysis of purified SNDHs. Lane MW contained a mixture of molecular mass markers. Purified SNDH1 (1.0 μg), SNDH2 (1.3 μg), and SNDH3 (0.75 μg) were loaded into the lanes. The proteins were stained with CBB.
We purified the SNDHs by conventional chromatography. The dialyzed soluble fraction (specific activity for l-AA production, 16.3 mU/mg protein from l-sorbosone) of K. vulgare cells was used for purification. All three SNDHs eluted at almost the same NaCl concentration in DEAE anion-exchange chromatography. SNDH3 was isolated from SNDH1 and SNDH2 using Q-Sepharose anion-exchange chromatography. Finally, SNDH1 and SNDH2 were separated by hydrophobic chromatography. Thus, SNDH1, SNDH2, and SNDH3 were purified almost to homogeneity and appeared slightly reddish. The purified SNDH1, SNDH2, and SNDH3 migrated to their original positions on a native polyacrylamide gel (Fig. 1A). It was found that the purified SNDHs simultaneously converted l-sorbosone to l-AA and to 2KGA. SNDH1 had specific activities at pH 7.0 for production of l-AA and 2KGA from l-sorbosone of 3.01 and 0.60 U/mg protein, respectively; the corresponding values for SNDH2 were 3.26 and 0.74 U/mg protein, respectively, and the corresponding values for SNDH3 were 4.63 and 1.16 U/mg protein, respectively. SDS-PAGE revealed that SNDH1 and SNDH3 were composed of 75-kDa and 55-kDa subunits, respectively (Fig. 1B). However, SNDH2 was a heterooligomer of 75-kDa and 55-kDa subunits. Gel filtration chromatography analysis showed that the molecular masses of SNDH1, SNDH2, and SNDH3 were 150,000 to 230,000, 190,000 to 250,000, and 100,000 to 150,000 Da, respectively (data not shown). Gel filtration chromatography with SDS revealed that SNDH2 contained equal amounts of the two subunits. The molecular mass of the 55-kDa subunit was determined to be 49,601 Da by mass spectrum analysis.
Prosthetic groups of SNDHs.
HPLC analysis of the acidified methanol extract of SNDH1 produced a peak with detection at 313 nm at the same retention time as an authentic PQQ sample (data not shown), indicating that SNDH1 has PQQ as a prosthetic group. When SNDH1 was recovered from a native polyacrylamide gel by electroelution, apo-SNDH protein was obtained. Apo-SNDH protein did not exhibit activity and migrated as a single band on a native polyacrylamide gel (Fig. 2A, lanes 1 and 3). When apo-SNDH was treated with 1 μM PQQ and 1 mM CaCl2 at 25°C, approximately one-half of the apo-SNDH was converted to holo-SNDH within 10 min (Fig. 2A, lane 4). The reconstituted holoenzyme showed activity (lane 2). Holo-SNDH migrated slightly faster than apo-SNDH.
FIG. 2.
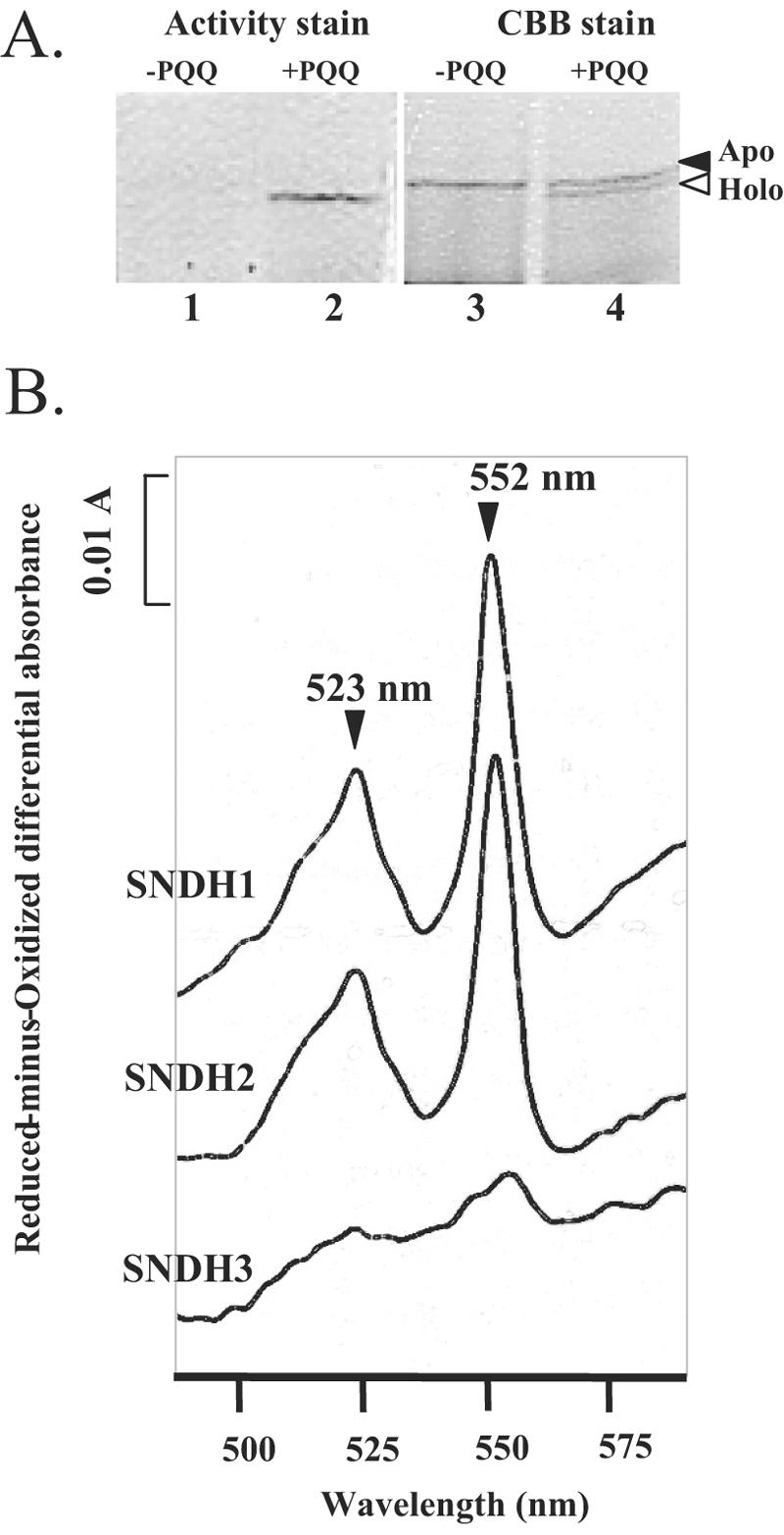
(A) Reconstruction of holo-SNDH from apo-SNDH prepared by electroelution from a native polyacrylamide gel. In each lane, 0.75 μg of the protein was used. Lanes 1 and 2, activity staining of l-sorbosone-dehydrogenating enzymes; lanes 3 and 4, protein staining with CBB. Lanes 1 and 3 contained apo-SNDH, and lanes 2 and 4 contained SNDH treated with 1 μM PQQ and 1 mM CaCl2. The solid arrowhead indicates the position of the apoenzyme. The open arrowhead indicates the position of the holoenzyme in which PQQ binds to the apoenzyme. (B) Reduced-minus-oxidized difference spectra of the purified SNDHs.
The reduced-minus-oxidized difference spectra of SNDH1 and SNDH2 showed a typical pattern for c-type cytochromes, which had two major peaks at 523 nm and 552 nm (Fig. 2B). This suggests that these enzymes contain heme c. In contrast, SNDH3 had no apparent peaks around 550 nm, suggesting a lack of heme c in this enzyme.
Characterization of the three SNDHs.
To investigate the effects of pH on the enzyme activity, 100 mM sodium citrate (pH 4.5 to 6.5), potassium phosphate (pH 6.5 to 8.5), and Tris-HCl (pH 7.5 to 9.0) were used. For conversion of l-sorbosone to l-AA, all three SNDHs had optimum reaction rates at pH 7.0 to 8.0. In contrast, for conversion to 2KGA, they exhibited less activity around neutral pHs but high activity at pHs greater than pH 8.5 (data not shown). A reaction mixture (pH 7.0) containing 50 mM l-sorbosone was incubated for 60 min at 20 to 60°C. For both production of l-AA and production of 2KGA, the optimum temperatures for the reaction were rather broad; the three enzymes exhibited similar high activities at 20 to 40°C (data not shown). The effects of metal ions [CoCl2, CuSO4, Fe(SO4)3, NiSO4, ZnCl2, MgCl2, TiCl4, and CaCl2] at a concentration of 1 mM and inhibitors (EDTA, NaN3, and sodium monoiodoacetate) at a concentration of 5 mM on the enzyme activity for l-sorbosone were examined by a photometric assay using DCIP. The effects of most metal ions and inhibitors on the activities of the three SNDHs were similar (data not shown). Monoiodoacetate commonly inactivates SNDHs. Among the metal ions tested, Cu2+ almost completely inhibited the activity of the SNDHs. No enhancement of activity by the metal ions tested was observed.
The substrate specificities of the purified SNDHs were examined using several aldehyde, alcohol, or lactone compounds at two pHs, pH 7.5 for the activity to produce l-AA and pH 9.0 for the activity to produce 2KGA (see Table S1 in the supplemental material). For this photometric assay with DCIP, a concentration of the unstable osones l-sorbosone and d-glucosone of 2 mM was used; in contrast, a concentration of the stable sugars d-glucose, l-sorbose, d-sorbitol, d-xylose, and l-gulono-γ-lactone of 100 mM was used. The three SNDHs exhibited almost no activity with l-sorbose, d-sorbitol, or l-gulono-γ-lactone, although they exhibited relatively high activities with aldoses, especially d-glucose and d-xylose.
Analysis of the sndH gene.
The N-terminal amino acid sequences of the 75-kDa and 55-kDa SNDH subunits were analyzed with a sequencer. Since the N-terminal sequences of the proteins could not be read due to probable blocked residues, the proteins were treated with pyroglutamate aminopeptidase. It was found that both the 75-kDa and 55-kDa subunits had the same N-terminal sequence from pyroglutamate (pyroQ), which was pyroQXNPVEVPVGANET. To clone the sndH gene of K. vulgare DSM 4025, forward and reverse degenerate primers corresponding to the N-terminal amino acid sequence of SNDH were synthesized and used to obtain a DNA probe. pVSN5 carrying the sndH gene was identified from the K. vulgare DSM 4025 gene library using the DNA probe. Southern blot analysis of the genome DNA digested with EcoRI, EcoRV, PstI, SmaI, BamHI, and HindIII using the sndH probe showed hybridization to a single fragment (data not shown), indicating the presence of only one copy of sndH in the K. vulgare genome.
Analysis of the 3,408-bp nucleotide sequence that was determined revealed the presence of two open reading frames, which corresponded to the sndH gene encoding the SNDH protein and another gene designated deaD located downstream of the sndH gene (Fig. 3). It is likely that sndH and deaD compose an operon, since no putative promoter regions or ribosome-binding sites were found just upstream of deaD. The sndH gene consisted of 1,830 bp and encoded a 609-amino-acid polypeptide containing a putative 31-residue signal peptide. The sndH-encoded signal peptide contained (i) many hydrophobic residues, (ii) positively charged residues close to the N terminus, and (iii) an AXA motif as a signal cleavage sequence (Fig. 3). After the signal peptide, there was a QVNPVEVP sequence identical to the N-terminal amino acid sequences of the SNDHs. The putative Shine-Dalgarno sequence, AGGAGA, for the sndH gene was located 6 bp upstream of the initiation codon. There were three inverted repeat sequences, two short inverted repeat sequences (IR1 and IR2) in the N-terminal region of the deaD gene and a long inverted repeat sequence (IR3) immediately downstream of the deaD gene. Since IR3 contains AAAAA and TTT at both ends, it may function as a transcription terminator from both directions. Since SNDH has a heme-binding site in the C-terminal region (see below), the 75-kDa subunit is most likely a heme-bound mature SNDH protein. The 55-kDa subunit contained pyroQXNPVEVP as the N-terminal sequence and had a molecular mass of 49,601 Da. Since the molecular mass of a polypeptide from Gln1 to Gly453 of SNDH has been calculated to be 49,592 Da, the 55-kDa subunit is thought to be SNDH(1-453), a C-terminal truncation. The absence of a heme-binding site in SNDH(1-453) is consistent with the difference spectrum (Fig. 2B).
FIG. 3.

Nucleotide and deduced amino acid sequences for the sndH region in K. vulgare. Thin lines indicate putative promoter sequences (TAGACC and TAACCT). The putative Shine-Dalgarno (SD) sequence is indicated by a boldface line. Inverted repeat sequences are indicated by pairs of inverted arrows (IR 1, IR 2, and IR 3). Stop codons are indicated by asterisks. For sndH, the signal sequence is indicated by a dashed line, the N terminus of the mature SNDH polypeptide is indicated by an arrowhead, and the heme-binding motif is enclosed in a box. For deaD, the DEAD motif is enclosed in a box.
A homology search of the deduced amino acid sequence encoded by the sndH gene was performed with the protein database. The SNDH amino acid sequence from residue Gln1 to residue Glu446 exhibited 40% identity with the sequence of soluble quinoprotein glucose dehydrogenase (s-GDH) from Acinetobacter calcoaceticus (6, 9) (see Fig. S1 in the supplemental material). In the aligned region, 75% of the residues involved in PQQ, calcium, and glucose binding in the s-GDH reported by Oubrie (21) were conserved in SNDH. On the other hand, the remaining C-terminal region of the SNDH protein exhibited similarity (28 to 32% identity) to heme-containing proteins, such as c-type cytochrome, and cytochrome f from the cyanobacterium Plectonema boryanum (1) and the cd1-type nitrite reductase NirS from a Paracoccus denitrificans strain (8). In the same region, a heme c binding motif (CAACH) was found at positions 530 to 534 of SNDH.
An additional open reading frame (1,101 bp) designated deaD was found downstream of sndH (Fig. 3). The deaD gene encodes a 367-amino-acid protein. The deduced DeaD protein exhibited 39 to 42% identity to ATP-dependent RNA helicases (5, 11, 22, 31) from bacteria.
Expression of the sndH gene.
To express the sndH gene, we grew E. coli JM109 cells harboring pUCSNP4. The cell extract exhibited activity which converted l-sorbosone to l-AA and 2KGA when PQQ and CaCl2 were added to the reaction mixture (Table 2). Without PQQ and CaCl2, no activity was observed. This finding is consistent with the previous observation that E. coli is unable to synthesize PQQ in LB medium (18). Cell extract of E. coli JM109 harboring pUCSNP4 showed an enzyme activity of 29.3 mU/mg protein for l-AA production with the cofactors, while cell extract of JM109 cells harboring pUC18 did not show this enzyme activity. In contrast, cell extract of JM109 cells harboring pUC18 produced 2KGA (35.5 mU/mg protein), most likely due to an unidentified dehydrogenase(s) in E. coli (Table 2). The specific activity for 2KGA production in cell extract of JM109 cells harboring pUCSNP4 was 57.5 mU/mg protein.
TABLE 2.
Expression of intact and truncated sndH genes in E. coli and K. vulgare cells harboring various sndH segments
| Plasmid |
l-Sorbosone-converting activity (mU/mg CFE protein)a
|
||
|---|---|---|---|
| E. coli hostb | K. vulgare GOMTR1SN::Kmc | GOMTR1c | |
| pVSN109 | 9.80 ± 0.18 (3) | NDd | 395.6 ± 22.3 (2) |
| pVSN114 | 7.23 ± 0.18 (3) | 176.4 ± 57.7 (2) | 251.8 ± 51.4 (2) |
| pVSN106 | 3.50 ± 0.28 (3) | 135.6 ± 0.1 (2) | 114.3 (1) |
| pVSN117 | 1.18 ± 0.08 (3) | 29.8 ± 6.4 (2) | 52.1 (1) |
| pVK100 | 0.00 ± 0.00 (3) | 0.0 ± 0.0 (2) | 75.0 (1) |
| pUCSN2004 | 137.2 (1) | ||
| pUCSN2003 | 63.9 (1) | ||
| pUCSN2002 | 0.0 (1) | ||
| pUCSN2001 | 0.0 (1) | ||
| pUCSNP4 | 29.3 (1) | ||
| pUC18 | 0.0 (1) | ||
The values indicate the activities for conversion to l-AA and are means ± standard deviations. The numbers in parentheses are the numbers of experiments. For E. coli harboring pUCSNP4 and E. coli harboring pUC18 the activities for conversion to 2KGA were 57.5 and 35.5 mU/mg CFE protein, as determined by one experiment in each case.
E. coli DH5α and JM109 cells were the hosts for pVSN and pUC plasmids, respectively. Each reaction mixture (100 μl) contained 100 to 275 μg CFE protein, 50 mM l-sorbosone, 1.0 mM PMS, 1 μM PQQ, 1 mM CaCl2, and 25 mM potassium phosphate (pH 7.0) in a microtube (2 ml; Eppendorf, Germany).
Each reaction mixture (30 μl) contained 5 to 20 μg CFE protein, 50 mM l-sorbosone, 0.05 mM PMS, and 25 mM potassium phosphate (pH 7.0) in a 96-well microplate (round-bottom type; Nalge Nunc International K.K., Japan).
ND, no data.
Plasmids pUCSN2001, pUCSN2002, pUCSN2003, and pUCSN2004 (Fig. 4), which contained intact sndH genes or sndH genes with C-terminal deletions, were constructed and introduced into E. coli JM109 cells to analyze sndH expression. As shown in lane 2 of the upper gel in Fig. 5A, two activity bands were observed in JM109 harboring pUCSN2004; the lower band corresponded to SNDH3, and the upper band did not correspond to any of the three SNDHs. A fourth activity band was also found in cell extract of K. vulgare DSM 4025 (Fig. 5A, lane 1). Although the fourth SNDH is expected to contain the 75-kDa subunit (see below), its exact nature is not known at this time. JM109 harboring pUCSN2003, which carried the truncated sndH gene to the position corresponding to Gly535, expressed only SNDH3 (lane 3). When more of the sndH coding sequence was deleted, no activity band was observed (lanes 4 and 5). In addition, JM109 harboring pUCSN2001 or pUCSN2002 showed no l-AA production from l-sorbosone (Table 2). The cell extracts were subjected to Western blot analysis with polyclonal anti-SNDH antiserum (Fig. 5A, lower gel). JM109 cells harboring pUCSN2004 produced an immunoreactive 75-kDa subunit as well as a 55-kDa subunit, while JM109 cells harboring pUCSN2003 produced only the 55-kDa subunit.
FIG. 4.

Gene organization of sndH region in K. vulgare. Km, insertion site of the kanamycin resistance gene in GOMTR1SN::Km; Heme, heme c binding motif; solid arrows, coding regions of 55-kDa and 75-kDa subunits. Phe308, Thr429, and Gly535 indicate the C termini of the truncated SNDH molecules encoded in deletion plasmids.
FIG. 5.
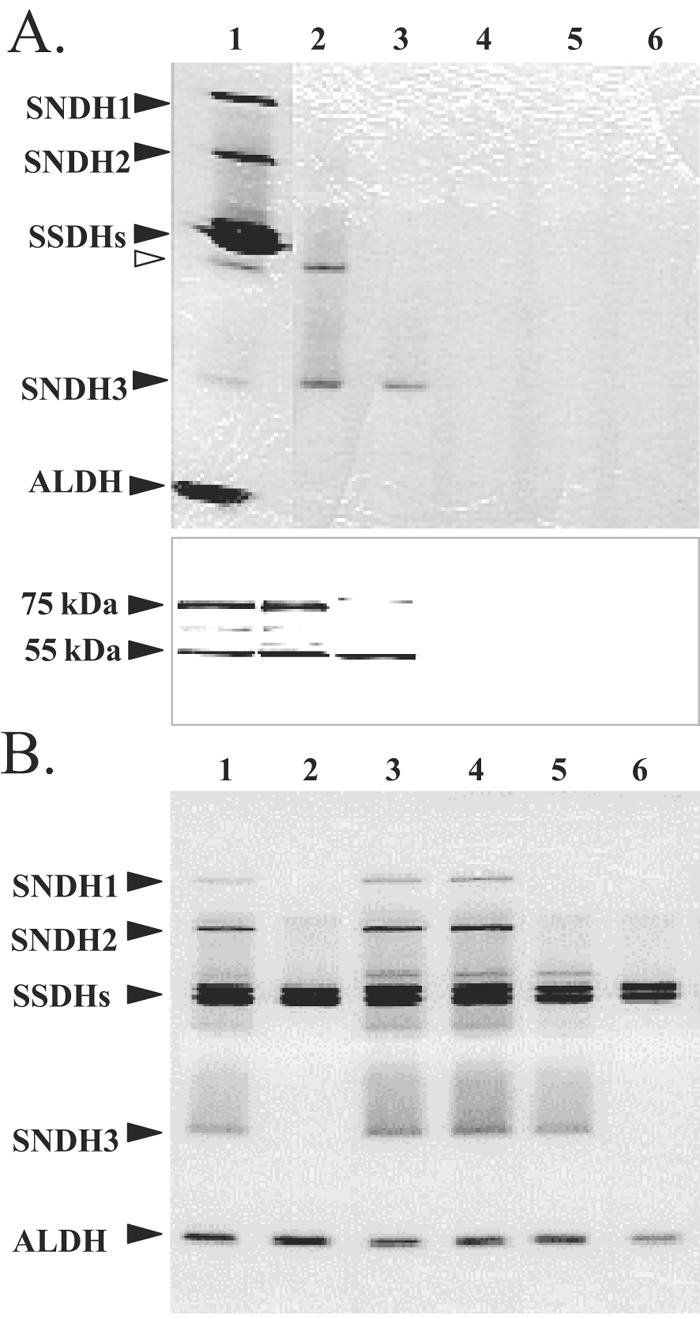
Expression of intact and truncated sndH genes in E. coli (A) and K. vulgare (B). (A) E. coli cells harboring pUCSN series plasmids: activity staining (upper gel) and Western blot analysis (lower gel) of CFE proteins. For the upper gel, CFE proteins (15 μg) were incubated with PQQ and CaCl2 for 10 min at 25°C and subjected to native PAGE (10% polyacrylamide gel), followed by activity staining using l-sorbosone. The locations of SNDH, SSDH, and ALDH proteins are indicated on the left. The open arrowhead indicates the position of the fourth SNDH band. For the lower gel, CFE proteins (5 μg) were subjected to SDS-PAGE (12.5% polyacrylamide gel) and blotted onto a polyvinylidene difluoride membrane. The protein bands were detected using rabbit polyclonal anti-SNDH antiserum. The locations of 75-kDa and 55-kDa subunits are indicated on the left. Lane 1, K. vulgare DSM 4025; lanes 2 to 6, E. coli JM109 harboring pUCSN2004 (lane 2), pUCSN2003 (lane 3), pUCSN2002 (lane 4), pUCSN2001 (lane 5), and pUC18 (lane 6). (B) K. vulgare sndH::Km cells harboring pVSN series plasmids. CFE proteins (16 μg) were subjected to native PAGE (10% polyacrylamide gel), followed by activity staining using l-sorbosone. Lane 1, K. vulgare GOMTR1; lane 2, GOMTR1SN::Km; lanes 3 to 6, GOMTR1SN::Km harboring pVSN117 (lane 3), pVSN106 (lane 4), pVSN114 (lane 5), and pVK100 (lane 6).
The sndH null mutation was introduced into K. vulgare GOMTR1, a spontaneous rifampin-resistant mutant of DSM 4025. pSUPSN::Km was constructed by insertion of a Km cassette into the EcoRI site in sndH in suicide vector pSUP202. sndH::Km was introduced into the GOMTR1 genome by conjugal transfer of pSUPSN::Km, followed by homologous recombination with wild-type sndH. Replacement of the sndH allele by sndH::Km in GOMTR1SN::Km was confirmed by PCR using sndH- and Km-specific primers. Analysis by native PAGE and activity staining of GOMTR1SN::Km cell extract showed that the sndH disruptant simultaneously lost the three SNDH bands (Fig. 5B, lane 2). The SNDH deficiency was also confirmed by an in vitro assay for activity converting l-sorbosone to l-AA (GOMTR1SN::Km harboring pVK100) (Table 2). pVSN series plasmids (Fig. 4) that had either an intact sndH gene or an sndH gene with a C-terminal deletion were constructed. They were introduced into sndH disruptant GOMTR1SN::Km cells by conjugal transfer. The cell extracts of the recombinants were analyzed by native PAGE to clarify complementation of the deficiency of the SNDHs. The disruptants complemented with the intact sndH gene had the phenotype of the parent strain (Fig. 5B, lanes 3 and 4). The recombinant with pVSN117 encoding the truncated sndH gene to the position corresponding to Gly535 expressed only SNDH3 (Fig. 5B, lane 5). These results confirmed that the three SNDH proteins are products of the sndH gene and that the 55-kDa subunit has a C-terminal truncation.
We measured the production of l-AA from l-sorbosone in cell extracts from E. coli and K. vulgare DSM 4025 sndH null and wild-type strains carrying intact or truncated sndH genes on the pVSN plasmids (Fig. 4). Extracts from the recombinant bacteria showed increasing activity for conversion of l-sorbosone to l-AA in parallel with longer sndH DNA of the plasmid (Table 2). The presence of three inverted repeat sequences (IR1, IR2, and IR3) may have affected the stability of sndH mRNA in both E. coli and K. vulgare. In K. vulgare sndH null or wild-type strains, introduction of sndH genes on pVK100 resulted in high SNDH activity.
DISCUSSION
Novel enzymes, SNDHs, were isolated from K. vulgare DSM 4025 and characterized. We found that the SNDHs could convert l-sorbosone to both l-AA and 2KGA using artificial electron acceptors, such as PMS and DCIP, in vitro. Since 2KGA cannot be a substrate for these enzymes, l-AA is directly produced from l-sorbosone enzymatically. This is the first report of a purified enzyme that forms l-AA from l-sorbosone. In addition, the sndH gene encoding SNDH was cloned and characterized.
Three SNDH isozymes (SNDH1, SNDH2, and SNDH3) were purified. SNDH1 was proven to be quinohemoprotein and a homooligomer of 75-kDa subunits, SNDH2 was found to be a heterooligomeric complex of 75-kDa and 55-kDa subunits, and SNDH3 was found to be a homooligomer of 55-kDa subunits. The 75-kDa subunit was shown to be the product of sndH, which lacked the signal peptide and had a bound heme. The heme was thought to be a noncovalent prosthetic group because the 75-kDa subunit was not stained with 3,3′,5,5′-tetramethylbenzidine when the protein was treated with SDS (data not shown). The 55-kDa subunit, lacking the C-terminal domain, was found to be derived from the 75-kDa subunit. All three SNDHs were detected on a native polyacrylamide gel when K. vulgare DSM 4025 cells were pretreated with a protease inhibitor cocktail (P1860; Sigma). The 55-kDa subunit was also produced in E. coli cells. These results suggest that production of the 55-kDa subunit has biological significance.
SNDHs catalyze the conversion of l-sorbosone to l-AA at neutral pH and to 2KGA at alkaline pH. How is l-sorbosone converted to two different products? These reactions may be explained by the tautomerization of l-sorbosone. Seok et al. (25) described the tautomerization of l-xylosone, a five-carbon analogue of l-sorbosone. l-Xylosone was shown to have several tautomeric interconversions by gas-liquid chromatography-mass spectrometry. Seok et al. proposed a furanose form of 1,4-lactone for l-xylosone in their scheme. It is thought that l-sorbosone also forms a furanose structure, as well as a pyranose structure from intramolecular cyclization. We propose the hypothetical metabolic pathways for l-sorbosone shown in Fig. 6. In this scheme, l-sorbosone is converted to l-AA by SNDHs with dehydrogenation at the C-1 position (reaction 3a) when the substrate is the furanose form (1,4-lactone form). On the other hand, when l-sorbosone is the pyranose form (2,6-lactone form), it is converted to 2KGA by SNDHs (reaction 3b) with dehydrogenation at the C-1position after hydration of the aldehyde group in water.
FIG. 6.

Proposed metabolic pathways from d-sorbitol to l-AA and 2KGA in K. vulgare DSM 4025. Reaction 1 is catalyzed by SSDH (3); reaction 2 is catalyzed by ALDH (15); and reactions 3a and 3b are catalyzed by SNDHs.
The tertiary structure of s-GDH of A. calcoaceticus was solved previously (20), and the monomer was found to have a super barrel structure consisting of six four-strand antiparallel β-sheets (see Fig. S1 in the supplemental material). It is possible that the N-terminal domain of SNDH has a structure similar to that of s-GDH, as indicated by the high sequence identity. For the most part, the residues involved in cofactor and substrate binding proposed for s-GDH (21) are conserved in SNDH. In particular, the PQQ ligands Arg228, Asn229, Arg406, and Arg408 in s-GDH are invariant in SNDH (see Fig. S1 in the supplemental material). Since SNDH has an extended cytochrome domain, this enzyme has a composition similar to that of the quinohemoprotein type II alcohol dehydrogenases (type II ADH) from Comamonas testosteroni (12) and Pseudomonas putida (32). However, both the dehydrogenase domain (Gln1 to Glu446) and the cytochrome domain (Asp447 to Arg578) of SNDH have lower levels of similarity (<13% identity) with type II ADH from P. putida. A putative PQQ binding motif proposed for membrane-bound quinoproteins and soluble methanol dehydrogenases (2) was not found in SNDH. We concluded that SNDH consists of an N-terminal dehydrogenase domain similar to s-GDH and the C-terminal cytochrome c domain, which is a novel quinohemoprotein.
The presence of a signal peptide in SNDHs suggests that the proteins are located in the periplasmic space. Several related enzymes, including s-GDH and type II ADH, are also known to have a signal peptide. In bacterial cells expressing high potential sugar-oxidizing activities, location of the enzymes responsible for oxidation in the periplasmic space would be advantageous. These systems may provide effective hydride transfer from the substrates to the respiratory system deployed on membranes, like ADH and GDH; the SNDH proteins may also be involved in this electron transfer. Although the natural electron acceptor for SNDH in the cell was not identified, it is expected that the enzymes transfer electrons from the substrate via PQQ and heme c eventually to the membrane-bound cytochrome c oxidase, as reported previously for type II ADHs (33).
In this study, valuable enzymes, SNDHs that connect the pathway from primary sugars to l-AA in K. vulgare, were identified. We expect that further analysis of the SNDHs will contribute to the development of new economical l-AA production processes.
Supplementary Material
Acknowledgments
We thank Yu-Chin Pan and Michel Hanspeter (Roche, Nutley, NJ) for analyzing the molecular masses of the subunits of SNDHs. We are grateful to Teruya Komano (Tokyo Metropolitan University) for valuable counsel during this work.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Aitken, A. 1977. Purification and primary structure of cytochrome f from cyanobacterium, Plectonema boryanum. Eur. J. Biochem. 78:273-279. [DOI] [PubMed] [Google Scholar]
- 2.Anthony, C. 1992. The structures of bacterial quinoprotein dehydrogenases. Int. J. Biochem. 24:29-39. [DOI] [PubMed] [Google Scholar]
- 3.Asakura, A., and T. Hoshino. 1999. Isolation and characterization of a new quinoprotein dehydrogenase, l-sorbose/l-sorbosone dehydrogenase. Biosci. Biotechnol. Biochem. 63:46-53. [DOI] [PubMed] [Google Scholar]
- 4.Bleeg, H. S. 1966. l-Ascorbic acid in yeast and isolation of l-galactono-gamma-lactone oxidase from the mitochondria. Enzymologia 31:105-112. [PubMed] [Google Scholar]
- 5.Böddeker, N., K. Stade, and F. Franceschi. 1997. Characterization of DbpA, an Escherichia coli DEAD box protein with ATP independent RNA unwinding activity. Nucleic Acids Res. 25:537-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cleton-Jansen, A.-M., N. Goosen, K. Vink, and P. van de Putte. 1989. Cloning, characterization and DNA sequencing of the gene encoding for the Mr 50,000 quinoprotein glucose dehydrogenase from Acinetobacter calcoaceticus. Mol. Gen. Genet. 217:430-436. [DOI] [PubMed] [Google Scholar]
- 7.Davis, B. J. 1964. Disc electrophoresis. II. Method and application to human serum proteins. Ann. N. Y. Acad. Sci. 121:404. [DOI] [PubMed] [Google Scholar]
- 8.de Boer, A. P., W. N. Reijnders, J. G. Kuenen, A. H. Stouthamer, and R. J. van Spanning. 1994. Isolation, sequencing and mutational analysis of a gene cluster involved in nitrite reduction in Paracoccus denitrificans. Antonie Leeuwenhoek 66:111-127. [DOI] [PubMed] [Google Scholar]
- 9.Dokter, P., J. Frank, and J. A. Duine. 1986. Purification and characterization of quinoprotein glucose dehydrogenase from Acinetobacter calcoaceticus L.M.D. 79.41. Biochem. J. 239:163-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duine, J. A., J. Frank, and P. E. Verwiel. 1981. Characterization of the second prosthetic group in methanol dehydrogenase from Hyphomicrobium X. Eur. J. Biochem. 118:395-399. [DOI] [PubMed] [Google Scholar]
- 11.Fleischmann, R. D., M. D. Adams, O. White, R. A. Clayton, E. F. Kirkness, A. R. Kerlavage, C. J. Bult, J. F. Tomb, B. A. Dougherty, J. M. Merrick, et al. 1995. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269:496-512. [DOI] [PubMed] [Google Scholar]
- 12.Groen, B. W., M. A. van Kleef, and J. A. Duine. 1986. Quinohaemoprotein alcohol dehydrogenase apoenzyme from Pseudomonas testosteroni. Biochem. J. 234:611-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hancock, R. D., and R. Viola. 2002. Biotechnological approaches for l-ascorbic acid production. Trends Biotechnol. 20:299-305. [DOI] [PubMed] [Google Scholar]
- 14.Hoshino, T., T. Sugisawa, and A. Fujiwara. 1991. Isolation and characterization of NAD(P)-dependent l-sorbosone dehydrogenase from Gluconobacter melanogenus UV10. Agric. Biol. Chem. 55:665-670. [Google Scholar]
- 15.Hoshino, T., and T. Sugisawa. February 1997. Aldehyde dehydrogenase enzyme. U.S. patent 5,776,742.
- 16.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 227:680-685. [DOI] [PubMed] [Google Scholar]
- 17.Loewus, M. W., D. L. Bedgar, A. Saito, and F. A. Loewus. 1990. Conversion of l-sorbosone to l-ascorbic acid by a NADP-dependent dehydrogenase in bean and spinach leaf. Plant Physiol. 94:1492-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsushita, K., J. C. Arents, R. Bader, M. Yamada, O. Adachi, and P. W. Postma. 1997. Escherichia coli is unable to produce pyrroloquinoline quinone (PQQ). Microbiology 143:3149-3156. [DOI] [PubMed] [Google Scholar]
- 19.Nishikimi, M., B. M. Tolbert, and S. Udenfriend. 1976. Purification and characterization of l-gulono-gamma-lactone oxidase from rat and liver. Arch. Biochem. Biophys. 175:427-435. [DOI] [PubMed] [Google Scholar]
- 20.Oubrie, A., H. J. Rozeboom, K. H. Kalk, J. A. Duine, and B. W. Dijkstra. 1999. The 1.7Å crystal structure of the apo form of the soluble quinoprotein glucose dehydrogenase from Acinetobacter calcoaceticus reveals a novel internal conserved sequence repeat. J. Mol. Biol. 289:319-333. [DOI] [PubMed] [Google Scholar]
- 21.Oubrie, A. 2003. Structure and mechanism of soluble glucose dehydrogenase and other PQQ-dependent enzymes. Biochim. Biophys. Acta 1647:143-151. [DOI] [PubMed] [Google Scholar]
- 22.Peng, H. L., M. J. Hsieh, C. L. Zao, and H. Y. Chang. 1994. Nucleotide sequence and expression in Escherichia coli of the Klebsiella pneumoniae deaD gene. J. Biochem. 115:409-414. [DOI] [PubMed] [Google Scholar]
- 23.Saito. Y., Y. Ishii, H. Hayashi, Y. Imao, T. Akashi, K. Yoshimasa, Y. Noguchi, S. Soeda, M. Yoshida, M. Niwa, J. Hosoda, and K. Shimomura. 1997. Cloning of the genes coding for l-sorbose and l-sorbosone dehydrogenases from Gluconobacter oxydans and microbial production of 2-keto-l-gulonate, a precursor of l-ascorbic acid, in a recombinant G. oxydans strain. Appl. Environ. Microbiol. 63:454-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory. Cold Spring Harbor, N.Y.
- 25.Seok, Y.-J., S.-T. Kim, K.-S. Yang, W.-K. Huh, and S.-O. Kang. 1996. Characterization of enediol-containing tautomers of l-xylosone. J. Carbohydr. Chem. 15:1073-1083. [Google Scholar]
- 26.Shinjoh, M., and T. Hoshino. 1995. Development of a shuttle vector and a conjugative transfer system for Gluconobacter oxydans. J. Ferment. Bioeng. 79:95-99. [Google Scholar]
- 27.Shinjoh, M., N. Tomiyama, A. Asakura, and T. Hoshino. 1995. Cloning and nucleotide sequencing of the membrane-bound l-sorbosone dehydrogenase gene of Acetobacter liquefaciens IFO 12258 and its expression in Gluconobacter oxydans. Appl. Environ. Microbiol. 61:413-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon, R., U. Priefer, and A. Pühler. 1983. A broad host range mobilization system for in vitro genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology 1:784-791. [Google Scholar]
- 29.Sugisawa, T., S. Ojima, P. K. Matzinger, and T. Hoshino. 1995. Isolation and characterization of a new vitamin C producing enzyme (l-gulono-γ-lactone dehydrogenase) of bacterial origin. Biosci. Biotechnol. Biochem. 59:190-196. [Google Scholar]
- 30.Sugisawa, T., T. Miyazaki, and T. Hoshino. 2005. Microbial production of l-ascorbic acid from d-sorbitol, l-sorbose, l-gulose, and l-sorbosone by Ketogulonicigenium vulgare DSM 4025. Biosci. Biotechnol. Biochem. 69:659-662. [DOI] [PubMed] [Google Scholar]
- 31.Toone, W. M., K. E. Rudd, and J. D. Friesen. 1991. deaD, a new Escherichia coli gene encoding a presumed ATP-dependent RNA helicase, can suppress a mutation in rpsB, the gene encoding ribosomal protein S2. J. Bacteriol. 173:3291-3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toyama, H., A. Fujii, K. Matsushita, E. Shinagwa, M. Ameyama, and O. Adachi. 1995. Three distinct quinoprotein alcohol dehydrogenases are expressed when Pseudomonas putida is grown on different alcohols. J. Bacteriol. 177:2442-2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toyama, H., F. S. Mathews, O. Adachi, and K. Matsushita. 2004. Quinohemoprotein alcohol dehydrogenases: structure, function, and physiology. Arch. Biochem. Biophys. 428:10-21. [DOI] [PubMed] [Google Scholar]
- 34.Urbance, J. W., B. J. Bratina, S. F. Stoddard, and T. M. Schmidt. 2001. Taxonomic characterization of Ketogulonigenium vulgare gen. nov., sp. nov. and Ketogulonigenium robustrum sp. nov., which oxidize l-sorbose to 2-keto-l-gulonic acid. Int. J. Syst. Evol. Microbiol. 51:1059-1070. [DOI] [PubMed] [Google Scholar]
- 35.Yin, G., Z. Tao, Z. Yan, W. Ning, C. Wang, and S. Wang. June 1990. Fermentation process. U.S. patent 4,935,359.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.