

Abstract
The tfdA gene is known to be involved in the first step of the degradation of the phenoxy acid herbicide 4-chloro-2-methylphenoxyacetic acid (MCPA) in several soil bacteria, but bacteria containing other tfdA-like genes have been isolated as well. A quantitative real-time PCR method was used to monitor the increase in the concentration of tfdA genes during degradation of MCPA in sandy topsoil and subsoil over a period of 115 days. Quantitative PCR revealed growth in the tfdA-containing bacterial community, from 500 genes g−1 soil to approximately 3 × 104 genes g−1 soil and to 7 × 105 genes g−1 soil for topsoil initially added to 2.3 mg MCPA kg−1 (dry weight) soil and 20 mg MCPA kg−1 (dry weight) soil, respectively. We analyzed the diversity of the tfdA gene during the degradation experiment. Analyses of melting curves of real-time PCR amplification products showed that a shift in the dominant tfdA population structure occurred during the degradation period. Further denaturing gradient gel electrophoresis and sequence analysis revealed that the tfdA genes responsible for the degradation of MCPA belonged to the class III tfdA genes, while the tfdA genes present in the soil before the occurrence of degradation belonged to the class I tfdA genes. The implications of these results is that the initial assessment of functional genes in soils does not necessarily reflect the organisms or genes that would carry out the degradation of the compounds in question.
Phenoxy acid herbicides have been widely used for agricultural purposes throughout the world since their development in the 1940s. In Denmark, 4-chloro-2-methylphenoxyacetic acid (MCPA) has been among the five top-selling herbicides during the last five decades. The relatively large amounts applied (1,500 to 2,000 g/ha), the weak retention of the compound in soils, and the unrestricted use during cold autumns, when degradation is slow, have resulted in frequent occurrences of MCPA as a groundwater contaminant (50). Groundwater is an important drinking water resource, especially in Denmark, where 99% of the drinking water is provided by groundwater. Hence, studies that explore the fate of potential groundwater-polluting compounds are important.
The degradation of phenoxy herbicides such as 2,4-dichlorophenoxyacetic acid (2,4-D), MCPA, and related compounds has been studied intensively during the last 40 years, with the majority of the work being done on 2,4-D. This, among other studies, has revealed detailed knowledge about the degradation and mineralization kinetics (27, 38), the catabolic pathways (6, 21, 46), the genes and enzymatic systems involved in the degradation (6, 20, 24, 35), and the phylogenetic composition of isolated microbial degraders (25, 53).
Numerous kinetic models have been developed to describe the mineralization of xenobiotic compounds in the environment (for a review, see reference 17). Typically, the models are divided into different forms of degradation kinetics that either take the growth of microorganisms into account or not. Furthermore, different models that take different forms of growth into account, depending on initial cell and substrate concentrations, have been developed (7, 52).
The soil bacterium Ralstonia eutropha JMP134, originally isolated from an Australian agricultural soil sample (44), contains genes for the complete pathway of 2,4-D and MCPA degradation. These genes are the ones most extensively studied and have become a model for the study of phenoxy acid degradation (11, 55). This strain harbors plasmid pJP4, which contains all of the structural and regulatory genes needed to convert these compounds to 2-chloromaleylacetic acid, which is further metabolized by chromosomally encoded gene products to finally yield CO2 and chloride (14, 15, 22, 31, 32, 34). The first step in the degradation pathway is initiated by an α-ketoglutarate-dependent dioxygenase, encoded by the tfdA gene, which cleaves the acetate side chain to produce the corresponding phenol 4-chloro-2-methylphenol (MCP) (Fig. 1) (18, 19, 54). Further degradation steps are initiated and regulated by gene products of the genes tfdB (45), tfdCDEF (35, 45), tfdR, and tfdS (37, 39).
FIG. 1.
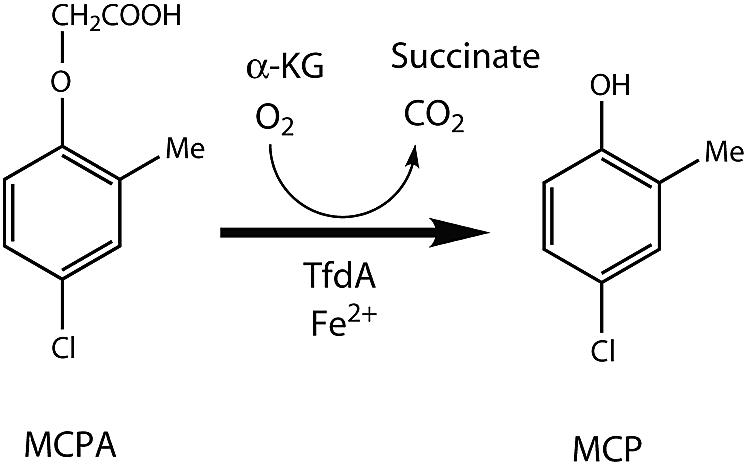
Catabolic activity of the TfdA enzyme in the degradation of MCPA. α-KG, α-ketoglutarate.
Numerous strains capable of degrading compounds such as 2,4-D and MCPA have been isolated from various environments and have been found to be distributed over many different phylogenetic groups (4, 13, 24, 25, 33, 35, 55, 57). Itoh et al. (25) suggested classifying the strains into three groups. The first group, which can be further divided into three classes (classes I to III) based on their tfdA gene sequence (40), consists of β- and γ-proteobacteria containing tfdA genes related to that of R. eutropha JMP134. The second group consists of α-proteobacteria that are closely related to Bradyrhizobium spp. and contains the tfdA-like gene tfdAα (60% identical to the canonical tfdA gene). The strains in this group were first isolated from pristine environments in Hawaii, Canada, and Chile (30) and subsequently from arable soil in Japan (24). Members of the third group are α-proteobacteria belonging to a species of the genus Sphingomonas. They are also characterized by using the tfdAα gene product during the initial step of the pathway instead of the TfdA enzyme (20). As well as this, several strains in the second and the third groups are known to contain the cadA gene, which is related to the tftA gene involved in the degradation of 2,4,5-trichlorophenoxyacetic acid (25).
The main objective of this paper was to describe the functional diversity of MCPA degraders in a profile of MCPA-treated Danish soil. Furthermore, PCR using primers targeting the three groups of phenoxy acid degradation genes was performed to investigate whether the genes were present and which primer set revealed the most specific amplification product. In light of this, one degenerated primer set (this work) targeting the canonical tfdA gene for real-time quantitative PCR was chosen. The relative increase in the tfdA gene concentration was monitored during a degradation experiment in microcosms. Denaturing gradient gel electrophoresis (DGGE) analyses of the tfdA amplification product were used to elucidate whether only one or several homologues of the tfdA gene were involved in the MCPA degradation. This revealed a number of clearly separated bands, which were sequenced and compared to known tfdA sequences. We further examined the mineralization kinetics of MCPA depending on the effects of the initial substrate and cell concentrations in top- and subsoil. The topsoil was suggested to contain a higher initial cell concentration than the subsoil. The initial cell concentration and the growth of MCPA degraders were determined and monitored during a degradation period by quantitative real-time PCR of the tfdA gene. Microbial growth during degradation of MCPA in natural soil has previously been suggested but has only been quantified using less accurate techniques (12, 41).
MATERIALS AND METHODS
Bacteria.
Ralstonia eutropha JMP134 (13), which carries the tfdA gene on the pJP4 plasmid, was used as a positive control and for standard curve preparation in the quantitative real-time PCR assay. Furthermore, it was used as a model organism in the DGGE analysis. DNA from Burkholderia cepacia DB01(pRO101), which carries the same tfdA gene as pJP4 (22), was used to create a 32P-labeled probe for Southern blot hybridization. Both strains were propagated using standard techniques (27).
Soils.
An organic topsoil sample (10 to 25 cm) and a subsoil sample (35 to 50 cm) from a sandy profile were used for the experiments. The profile was taken from the Danish agricultural experimental station in Jyndevad, located on the outwash plain in southern Jutland. The soil has been used for organic farming since 1995, and additionally, it has not been treated with any form of phenoxy acid herbicide since at least 1988. The soils were collected and mixed manually in October 2003 and stored in the dark for 2 months at 5°C before they were passed through a 2-mm sieve. The soils were thereafter stored in the dark at −18°C until 10 days before use. Defrosting and acclimatization were performed in the dark for 10 days at 10°C (41) (for further soil characteristics, see Table 1).
TABLE 1.
Soil characteristics
| Soil | Depth (cm) | pH | C (%) | Clay (%) | Silt (%) | Sand (%) | WHCa (% [vol/vol]) |
|---|---|---|---|---|---|---|---|
| Topsoil | 10-25 | 6.9 | 2.4 | 5 | 3 | 92 | 19.25 |
| Subsoil | 35-50 | 6.1 | 1.0 | 5 | 1 | 94 | 17.76 |
WHC, water-holding capacity, measured at a −log water potential of 2.0 cm.
Microcosm setup for degradation experiment.
Before experiment setup, stock solutions of MCPA were prepared. To ensure even moistening of the soils to a water content of approximately the water-holding capacity, stock solutions of 20, 174, and 23 mg/liter MCPA were required to obtain concentrations of 2.3 mg MCPA kg−1 (dry weight) soil in the topsoil (Top2.3), 20 mg MCPA kg−1 (dry weight) soil in the topsoil (Top20), and 2.3 mg MCPA kg−1 (dry weight) soil in the subsoil (Sub2.3), respectively. The stock solutions were prepared by dissolution of MCPA (97.5% purity; Dr. Ehrenstorfer GmbH, Augsburg, Germany) in MilliQ water (Millipore, Birkerød, Denmark). To ensure the complete dissolution of MCPA and to avoid disturbance of the soil pH, the pH in the solutions was adjusted to 6.5 with NaOH.
Subsequently, 20 g (dry weight) of the topsoil and 15 g (dry weight) of the subsoil were transferred to 250-ml Pyrex red cap bottles (airtight). The soils were spiked with 2.3 ml (for the topsoils) and 1.5 ml (for the subsoils) of the respective stock solutions. Stock solutions with MCPA were carefully mixed into the soils with a sterile glass pipette, and the microcosms were incubated at 10°C in the dark. Triplicate bottles were set up for each soil scenario for eight sampling points, and in order to prevent the development of anaerobic conditions in the microcosms, they were aerated by leaving the flasks open for 20 min in a sterile hood every 14 days.
Microcosm setup for mineralization experiment.
A mineralization experiment was set up with 14C-labeled MCPA at the same concentrations described above. Stock solutions were prepared as described above for the degradation experiment. [U-14C]MCPA (specific activity of 159.7 μCi/mg, radiochemical purity of >95%; Izotop, Budapest, Hungary) was then added to the stock solutions in trace amounts to achieve an initial radioactivity in each microcosm of 60,000 dpm. The mineralization experiment was performed essentially as described previously by Mortensen and Jacobsen (41), except that 10 g of soil was used and no soil was removed during the experiment. We have previously determined the mass balance for the experimental setup, and recoveries of 93% ± 1.5% were found (5, 9).
MCPA extraction and quantification.
On days 0, 6, 12, 22, 33, 50, 68, and 115 after incubation, triplicates of each soil-MCPA degradation series (Top2.3, Top20, and Sub2.3) were extracted by pressurized liquid extraction for the determination of MCPA amounts. The extractions were performed with an ASE 200 system (Dionex, Sunnyvale, CA). The soils from the degradation assay were packed in 33-ml extraction cells sufficient to contain 40 to 50 g soil. To assist the extractions, Ottawa sand (Fisher scientific, Loughborough, United Kingdom), sufficient to fill out the remaining volume (approximately 1:1 [wt/wt]), was mixed into the soil samples. The entire extraction process was carried out at 50°C. The solvent used for the extractions was a 50:50 (vol/vol) mixture of methanol-water (high-performance liquid chromatography-grade methanol; Fisher Scientific, Loughborough, United Kingdom). Other extraction conditions were as follows: a 40% solvent volume was used for a 10-min static time in one extraction cycle and a 60-s purge time with nitrogen at 1,500 lb/in2 pressure (1 lb/in2 = 6,894.76 Pa). The extraction method was optimized for soil samples spiked with MCPA, resulting in extraction efficiencies of 80% ± 2% for the topsoil and 92% ± 4% for the subsoil.
A soil extract of 15 to 20 ml was obtained in 50-ml glass vials. After being thoroughly mixed, an aliquot was filtered through a 0.45-μm polytetrafluoroethylene filter (Titan2; Sun-Sri) to remove particles. To avoid overloading of the liquid chromatography (LC) mass spectrometry system, the filtrates were diluted with a methanol-water (50:50) solution to obtain MCPA concentrations in the range of 50 to 150 μg/liter.
The quantification of MCPA in the soil extracts was performed using an LC tandem mass spectrometry (MS/MS) system. Twenty microliters of the samples was injected, and the compounds were separated on a 100- by 2.1-mm Xterra RP18 column (3.5-μm particle size) from Waters (Milford, MA) using a Waters Alliance model 2695 high-performance liquid chromatography system. A mobile phase composed of methanol-water-0.2% acetic acid (78:20:2) at 25°C with a flow rate of 0.18 ml/min was used. For detection and quantification of the MCPA compound, a Quattro Ultima triple quadrupole mass spectrometer from Micromass (Manchester, United Kingdom) equipped with an electrospray ionization unit was used. Electrospray ionization in the negative mode was used, while further conditions were as follows: capillary voltage was 3.20 kV, cone voltage was 80 V, and collision energy for MS/MS was 20 eV. Furthermore, nitrogen was used as the desolvation gas at a flow rate of 600 liters/h. A limit of detection of 1 μg MCPA liter−1 was determined for the LC-MS/MS system.
DNA extraction.
Soils for DNA extractions were set up parallel to the degradation experiment. Red cap bottles with 20 g (dry weight) of soil amended with 0, 2.3, and 20 mg MCPA kg−1 (dry weight) soil for the topsoil (Top0, Top2.3, and Top20) and 0 and 2.3 mg MCPA kg−1 (dry weight) soil for the subsoil (Sub0 and Sub2.3) were set up in triplicate. The amount of soil sampled for DNA extractions was 0.5 g (wet weight) to ∼0.4 g (dry weight), which was accomplished using a sterile stainless steel sampler designed to collect a composite sample of 0.5 g soil from 5 to 10 distinct spots in the bottle. The same bottle was used throughout the experiment and was aerated parallel to the bottles in the degradation experiment as well as for sample collection on the same days as in the degradation experiment. Whole-community DNA was extracted from the 0.5-g wet soil samples with the FastDNA SPIN kit for soil (Bio101, Inc., Carlsbad, CA). The protocol recommended by the manufacturer was followed, except that the bead-beating step was modified and a freeze-thaw step was included. The beat-beating step was modified to four 30-s pulses at speed 4 instead of one 30-s pulse at speed 5.5 in the FastPrep FP 120 instrument (Bio101), and the freeze-thaw steps were performed for 1 h at −80°C and subsequently for 30 min at 37°C. DNA was finally eluted in 100 μl DNase/RNase-free water (Bio101, Carlsbad, CA) and stored at −80°C.
Primer testing.
All PCRs were carried out as real-time PCR assays in an iCycler iQ (Bio-Rad, Hercules, CA). In order to select the most effective primer set, five primer sets were tested (Table 2) using two different commercial PCR master mix kits. tfdA* and tfdA** were the most intensively tested primers. tfdA* yielded a 307-bp fragment, while the tfdA** yielded a 210-bp fragment. tfdA** was designed in connection with this work by BLAST alignment analysis (3) of 22 known tfdA gene sequences found with R. eutropha JMP134 as the search sequence. The GenBank accession numbers for the 22 tfdA genes were as follows: M16730, AY238497, AY238496, U43197, U43276, AF439768, AY078159, AF029344, U32188, U25717, U87394, U22499, AF181982, AF182758, AF176240, U65531, U43196, AY238495, AY238494, AY238493, AY238492, and AB074491. Primers were designed to attach to most conserved regions, and since the primer set was designed on the basis of tfdA gene sequences belonging to class I and class III, it was expected that it was highly specific for these genes. Primers were obtained from MWG Biotech (Ebersberg, Germany). The first of the two commercial SYBR green PCR master mix kits we tested was iQ SYBR green supermix (Bio-Rad, Hercules, CA) containing PCR buffer, 0.4 mM of each deoxynucleoside triphosphate, 50 U/ml of iTaq DNA polymerase, and 6 mM MgCl2. This kit was used mainly with the tfdA* primer set and through the testing of tfdAα*, tfdAα**, and cadA primer sets. The other kit we tested was the QuantiTect SYBR green PCR kit (QIAGEN, Crawley, United Kingdom) containing deoxynucleoside triphosphate mix, HotStar Taq DNA polymerase, PCR buffer, Rox, and 2.5 mM MgCl2. This kit was only used for PCR with the tfdA* and tfdA** primer sets. The 25-μl reaction mixtures contained 0.4 μM of each primers, 12.5 μl of the respective SYBR green mix, 25.5 μg bovine serum albumin (Amersham Bioscience, Buckinghamshire, United Kingdom), 2.5 μl of 1:10-diluted DNA extract, and RNase/DNase-free water to complete the 25-μl volume. The DNA extracts were diluted 1:10 in RNase/DNase-free water to reduce the influence of PCR-disturbing factors from the soil, such as humic acids.
TABLE 2.
PCR primers
| Primer set | Model gene(s) | Primer (5′-3′)e | Optimal annealing temp used (°C)c | Characteristicf | Reference or source |
|---|---|---|---|---|---|
| tfdA* | R. eutropha JMP134/Burkholderia sp. strain RASC | F, AACGCAGCGRTTRTCCCAa R, ACGGAGTTCTGYGAYATGa | 58 | Unspecific binding; qPCR not possible | 59 |
| tfdA-intern | R. eutropha JMP134 | F, GCATACGACGCGCTGCCTCG R, CTTCGGCCACCGGAAGGCCT | 65 | Used for probe preperation for Southern blot hybridization | This work |
| tfdA** | Sequence alignment of 23 known tfdA genes | F, GAGCACTACGCRCTGAAYTCCCGa,b R, CTTCGGCCACCGGAAGGCCT | 64 | One band on agarose gel (approx. 220 bp); qPCR possible | This work |
| tfdAα* | Alignment of RD5-C2, HWK12, and HW13 (tfdAα genes) | F, CCGGCGTCGATCTGCGCAAG R, GTTGACGACGCGCGCCGA | NDd | No clear band on the agarose gel; fluffy PCR products | 51 |
| tfdAα** | Alignment of HW13, HWK12, and BTH (tfdAα genes) | F, ACSGAGTTCKSCGACATGCGa R, GCGGTTGTCCCACATCAC | 65 | One band (approx. 350 bp); qPCR may be possible | 25 |
| cadA | HW13 (cadA gene) | F, AAGCTGCARTTTGARAAYa | 58 | No PCR product | 24 |
| R, MGGATTGAAGAAATCCTGRTAa |
R, G/A; Y, T/C; S, G/C; K, G/T; M, A/C.
AGC clamp was added to the 5′ end of the primer for the PCR/DGGE (5′-CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGG-3′).
The optimal annealing temperature is based on gradient ramping (58 to 69°C).
ND, no optimal annealing temperature could be determined.
F, forward; R, reverse.
qPCR, quantitative PCR.
The conditions for the real-time PCR were as follows: 6 min at 95°C for enzyme activation; 50 cycles of 45 s at 94°C for denaturation, 30 s at the annealing temperature specified in Table 2, and 2 min at 72°C for elongation; and a final 6-min step at 72°C for extension. To obtain a specific melting profile for the real-time PCR product, an 80-cycle protocol of 45 s at 58°C; 30 s at 58°C to 98°C, increasing 0.5°C every cycle; and 45 s at 58°C was used after the real-time PCR. The melting profile was used to determine the presence of the specific product and to determine if the PCR product was composed of DNA sequences of more than one length.
Further investigations of the PCR products were done by gel electrophoresis of 8-μl PCR products on a 1.5% agarose gel in 1× Tris-acetate-EDTA buffer. The gels were stained in ethidium bromide and visualized under UV light.
In case the gels revealed more than one band, a Southern blot hybridization was performed as described previously by Jacobsen (26) and Sambrook and Russell (49), except that the digestion reaction was excluded. The 32P tfdA probe for the hybridization was generated using [32P]dCTP (Amersham, United Kingdom) and the Random Primed DNA Labeling kit (Roche Applied Science, Penzberg, Germany) based on the method described previously by Feinberg and Vogelstein (16). A PCR product derived from DNA of the tfdA-containing strain B. cepacia DB01(pRO101) with the tfdA-intern primer set (Table 2) was used as a template.
Quantitative real-time PCR.
After optimization of primers, quantitative real-time PCR was performed only with the tfdA** primer set as described above on the triplicate samples from days 0, 6, 12, 22, 33, 50, 68, and 115. All quantitative real-time PCRs were set up in triplicate and included three negative control samples in each PCR setup.
Standards for the quantitative PCR were prepared using the well-characterized phenoxy acid degrader R. eutropha JMP134. After inoculation into 0.5 g of the topsoil in amounts of 8 × 106, 8 × 105, 8 × 104, 8 × 103, 8 × 102, and 8 × 101 cells g−1 soil, the DNA was extracted from the soils as described above, and the extracts were diluted 10-fold to reduce the effect of humic acid disturbances. R. eutropha JMP134 was used as the positive control strain as well.
DGGE analysis.
PCR for the DGGE analysis was performed as described above with the QuantiTect mastermix kit. The tfdA** primer set was used with a GC clamp attached to the 5′ end of the forward primer (Table 2). DGGE analyses were performed on real triplicates of DNA extracts from days 0, 6, 12, 33, and 68 in order to secure enough wells for one data series on one gel. The technique described previously by Muyzer et al. (42) was used for separating and visualizing the tfdA PCR products of the DNA extracts. The specific details of the method used are as follows. Gels contained 8% acrylamide and a urea-formamide gradient of 55 to 70% (where 100% denaturant contained 7 M urea and 40% formamide). Electrophoresis was carried out at 70 V for 17 h in 1× Tris-acetate-EDTA buffer using a D-code apparatus (Bio-Rad, Hercules, CA). After electrophoresis, the gels were stained with SYBR gold (Molecular Probes, Eugene, OR) for 45 min, followed by a brief rinse with MilliQ water, and then visualized under UV light and finally photographed. Bands that appeared to have a unique pattern for the soil scenarios were stabbed with sterile pipette tips, which were placed in 20 μl RNase/DNase-free water and rinsed repeatedly. These served as templates in further PCRs using the tfdA** primer set (Table 2) without the GC clamp to obtain enough DNA material for sequencing. The nucleotide sequencings were performed by MWG Biotech (Ebersberg, Germany).
Data analysis.
The amounts of extracted MCPA were corrected for the extraction efficiency, after which linear regression analyses were performed on the steep segment of the degradation curves to fit it to zero-order kinetics with the following equation:
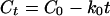 |
(1) |
where Ct and C0 are the amounts of herbicide remaining (percentage of initial concentration) at time t and at time zero, respectively; t is the incubation period (days); and k0 is the zero-order rate constant. The linear regressions were performed only for the steep segment of the degradation curve excluding sampling points from the lag phase and points where MCPA concentrations were below the detection limit.
For the results from the mineralization experiment, the calculated cumulative mineralization values were corrected for background radioactivity. After this, nonlinear regression analyses were performed as described previously by Mortensen and Jacobsen (41).
Nucleotide sequence accession numbers.
The GenBank accession numbers for the DNA sequences reported in this study are DQ272405, DQ272406, DQ272407, DQ272408, DQ272409, DQ272410, DQ272411, DQ272412, DQ272413, DQ272414, DQ272415, and DQ272416 for A1, A2, A3, A4, A5, A6, A7, A8, A9, B1, B2, and B3, respectively.
RESULTS
MCPA degradation.
The form of the curves for the disappearance of MCPA can be divided into three segments: first, a lag phase, where no or little degradation took place; second, a steep part with relatively fast degradation of MCPA (equation 1); and finally, the last segment, where almost all MCPA (>98%) had disappeared. The lag phases for Top20 and Sub2.3 both had durations of 12 days, while in Top2.3, no lag phase was observed. The steep part of the curve, where MCPA disappeared, shows a large degree of linearity. Therefore, this part was fitted to zero-order kinetics by means of linear regression (Fig. 2), and zero-order rate constants along with DT50 values were determined (Table 3). Degradation was fast and resulted in time to 50% degradation (DT50) values of 16, 33, and 37 days for Top2.3, Top20, and Sub2.3, respectively. However, MCPA could be detected in the soil for rather longer times, until days 33, 50, and 68 for Top2.3, Top20, and Sub2.3, respectively. In addition to detection and quantification of MCPA, we also detected contents of the first degradation product, MCP. In Top2.3 at days 0 to 22 and in Top20 at days 22 to 33, we found MCP in concentrations up to 5% of the initial MCPA concentration (data not shown). MCP was not detected in Sub2.3 and was not detected after days 22 and 33 for Top2.3 and Top20, respectively.
FIG. 2.

Degradation of MCPA in the three soil scenarios measured by LC-MS/MS. Linear regression lines of the steep segments are shown. Solid line, Top2.3; dashed line, Top20; dotted line, Sub2.3. Error bars indicate standard errors.
TABLE 3.
Parameter estimates from the zero-order kinetics modela of the steep part of the degradation curves and calculated DT50 values
| Soil | MCPA (mg/kg) | R2 | k0 (% × day−1) | DT50 (days) |
|---|---|---|---|---|
| Topsoil | 2.3 | 0.990 | −2.75 | 16.2 |
| Topsoil | 20 | 0.941 | −1.83 | 33 |
| Subsoil | 2.3 | 0.711 | −1.45 | 37 |
See equation 1.
Mineralization studies.
The mineralization curves showing accumulated amounts of 14CO2 released from mineralization of 14C-MCPA versus time were all sigmoid, and, like the degradation curves, they could be divided into three segments: a lag phase, a steep segment, and a plateau (Fig. 3A). The lag phase was significantly longer for Top20 and Sub2.3 than for Top2.3. Furthermore, the lag phase was longer for Sub2.3 than for Top20, a pattern similar to that observed in the degradation curves (Fig. 2). An F test (P < 0.05) showed that the mineralization curves for Top2.3 and Top20 were best fitted to the exponential growth form of the 3/2-order model (7) indicating growth during the degradation. The plateau of the curves for Top2.3 and Top20 occurred after approximately days 37 and 54, respectively, where 55% and 61% of the initially added 14C had been mineralized. Furthermore, the F test and other model parameters (data not shown) showed that Top20 fitted better to the model than Top2.3.
FIG. 3.

Mineralization of 14C-MCPA in the three soil scenarios. (A) Curves show triplicate cumulative data from Top2.3, Top20, and Sub2.3. The dashed line shows a nonlinear fit to the exponential form of the 3/2-order model. Due to the large standard deviation, the plot of the triplicates for Sub2.3 was not fitted. (B) Single plots of the Sub2.3 replicates. The dotted line shows nonlinear fits to the linear form of 3/2-order model, and the dashed line shows a nonlinear fit to the exponential form of the 3/2-order model (plots 2 and 3). Plot 1 was not satisfactorily fitted to any model. Error bars in A indicate standard errors.
Due to large standard errors, the average mineralization curve of the triplicates for Sub2.3 was not fitted to any kinetic model. The plot of the individual replicates (Fig. 3B) shows three different mineralization curves, only two of which (plots 2 and 3) had sigmoid forms and could be fitted as the topsoil experiments. According to the F test (P < 0.05) (48), plot 2 was best fitted to the exponential form of the 3/2-order model as for the topsoil scenario, while plot 3 was best fitted to the linear 3/2-order model (7), indicating no or slight growth. Plot 1 showed no sigmoid curvature and was therefore not fitted to any model.
Primer testing.
To make the real-time PCR quantification technique as accurate as possible, the choice of primer set was critical. Therefore, a well-characterized primer set covering a broad range of known tfdA sequences was chosen (59). Unfortunately, we found that this primer set was likely to generate diverse bonds, indicating that the PCR product contained genes of more than one length (Fig. 4A). One band (band 2) had the same length, approximately 350 bp, as the positive control strain, R. eutropha JMP134 (band 3).
FIG. 4.
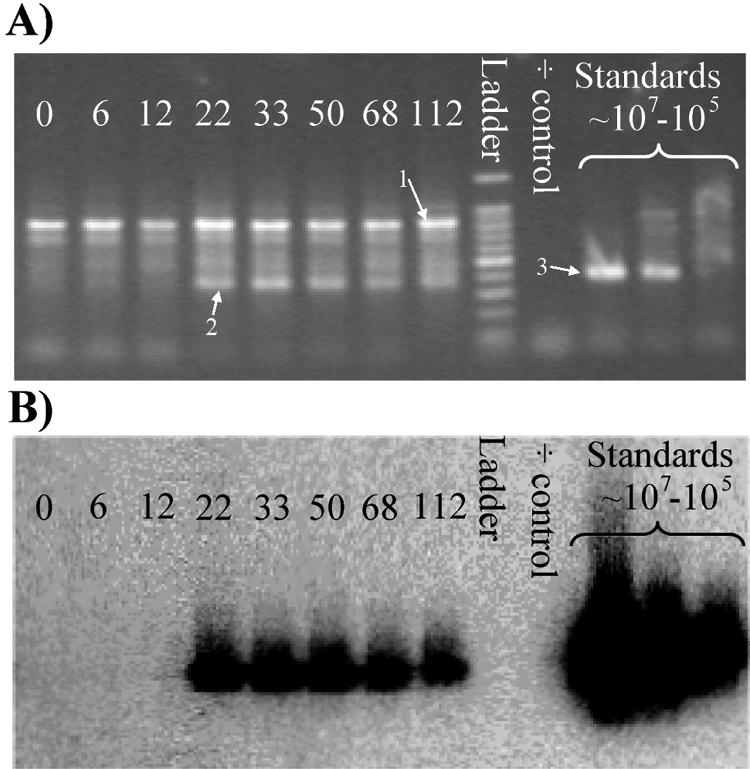
Primer optimization. (A) Standard agarose gel electrophoresis of real-time PCR amplification products using the tfdA* primer; the iQ SYBR green supermix and soil DNA from days 0, 6, 12, 33, 50, 68, and 115 of Top20 were used as a template. The standard is R. eutropha JMP134 inoculated into the soil in concentrations of 8 × 106, 8 × 105, and 8 × 104 cells/g soil, with 8 × 106 cells/g soil shown to the left. Bands 1 and 2 represent the most characteristic bands, as described in the text. Band 3 represents the strain known to contain the tfdA gene R. eutropha JMP134. The ladder is a 100-bp DNA ladder. (B) Southern hybridization analysis of the agarose gel shown in A using a tfdA probe. Similarity can be detected for bands 2 and 3.
In comparison to the topsoil, the subsoil showed no clear pattern in the bands from the PCR product, even though there were several unspecific bands on the gel (data not shown). To determine which bands were related to the tfdA gene, a Southern blot assay was performed with the gels (Fig. 4B). This revealed that only bands 2 and 3 contained DNA that matched the tfdA probe designed from the R. eutropha JMP134 strain. Several optimization experiments, including increasing annealing temperature, were made to get rid of the unspecific amplification products. Among these, we tried to use another commercial real-time PCR master mix kit, which decreased the unspecific bands (data not shown), but still not satisfactorily, which was expressed by a high detection limit and low R2 values (<0.90) for the standard curve. Other optimization steps included concentration of MgCl2 and concentration of the primers in the reaction mixture, but we still had no success (data not shown). Therefore, we designed a new set of primers, tfdA** (Table 2), which revealed PCR products with no unspecific bindings and a decrease in the cycle threshold values during real-time PCR. Lower cycle threshold values indicate a more specific and effective PCR. The detection limit in the real-time PCR assay for this primer was rather low (800 genes g−1 soil). Furthermore, the R2 values for the standard curves were high (0.999 to 0.990).
Consequently, we used the tfdA** primer set for further quantitative real-time PCR assays. Primer testing of primer sets tfdAα* (51), tfdAα** (24), and cadA (25) were performed as well (Table 2). Only tfdAα** amplified a PCR product that may be possible to use for quantitative real-time PCR (data not shown).
Quantitative real-time PCR analysis.
The quantitative real-time PCR analysis indicated that growth occurred in all three soil scenarios during the degradation period (Fig. 5). In Top2.3, the tfdA concentration increased to a maximum of 3.0 × 104 genes g−1 soil at day 12 (Fig. 5A), while the maximum gene concentrations in Top20 and Sub2.3 were 7.0 × 105 and 2.6 × 104 genes g−1 soil, obtained at days 22 to 33 and 50 to 68, respectively (Fig. 5B and C). The number of genes is based on the assumption that R. eutropha JMP134(pJP4) grown on rich media and used for real-time PCR standard curve preparation only contains one copy of the tfdA gene. The increase in the number of tfdA genes is inversely related to the concentration of MCPA in the three soils (Fig. 5A to C). Lag phases were clearly expressed and were shortest for Top2.3 and Top20 and longest for Sub2.3. It is interesting, however, that the increase in the number of tfdA genes occurred before the exponential mineralization phases did. It is also interesting that the maximum concentrations of the tfdA genes were reached while half of the MCPA remained and that a decrease in concentrations of tfdA genes was subsequently observed in the topsoils.
FIG. 5.

Nonfitted mineralization and degradation curves compared with the log number of tfdA genes using the tfdA** primer set in the soil. (A) Top2.3; (B) Top20; (C) Sub2.3. Error bars indicate standard errors.
Functional diversity of tfdA genes.
Two analyses of the functional diversity of the tfdA genes were performed, and both indicated a shift during the degradation period. First, an analysis of the melting profiles of the real-time PCR amplification products was performed. A shift in the temperature optimum of the melting profile was observed when the concentration of tfdA genes increased. This was observed as a melting optimum of 90°C at day 0 and a melting optimum of 87°C at day 22 (Fig. 6). Therefore, a DGGE analysis of PCR products using the GC-clamped tfdA** primer set (Table 2) was performed to obtain more detailed information about the shift in the degrader population with time. This analysis revealed several different bands, which grew up and grew stronger during the degradation experiment. In the Top2.3 and Top20 analyses, the overall pattern was about the same, with at least five dominating bands (Fig. 7A and B). However, the variation of band intensity differed slightly between treatments. In Top2.3, the bands grew up between days 6 and 12, while in Top20, the bands were weak at day 12 and first became strong at day 33, corresponding to the longer lag phase in Top20. For comparison, the melting optimum of the DNA amplified from the standards containing R. eutropha strain JMP134 was 90°C. The same changes in melting profiles were observed for all degradation experiments. The melting profile for day 12 in Top20, however, seemed to evolve a double top, which indicated a beginning shift in the composition of the degrader population (Fig. 6 and see Fig. 8). Moreover, the intensities of the bands in Top2.3 were strongest at day 12, after which the intensity declined throughout the experiment; however, this was not seen in Top20.
FIG. 6.

Melting profiles of real-time PCR amplification products of soil DNA from days 0, 12, and 22 in Top20 using the tfdA-II primer set and the QuantiTect SYBR green PCR kit. Furthermore, the melting profile of the PCR amplification product from the tfdA-containing strain R. eutropha JMP134 is shown. The profile displays the negative first derivative of temperature versus relative fluorescence units (RFU) [−d(RFU)/dT] plotted against temperature.
FIG. 7.

Functional diversity of MCPA degrader genes using PCR/DGGE analysis with the tfdA** primer set. The gene product was separated using 55 to 70% urea gels. DNA extracts from days 0, 6, 12, 33, and 68 were analyzed in real triplicates. For the marker profile (M), DNA extracts from soil samples inoculated with the tfdA-containing strain R. eutropha JMP134 were used. A1 to A10 indicate bands selected for sequence analysis from topsoil DNA, and B1 to B3 indicate bands selected for sequence analysis from subsoil DNA. A shows the DGGE profile of DNA extracted from Top2.3, B shows the DGGE profile of DNA extracted from Top20, and C shows the DGGE profile of DNA extracted from Sub2.3. D shows the DGGE profile of DNA extracted from Top0, and E shows the DGGE profile of DNA extracted from Sub0.
FIG. 8.

Sequence alignment of the DNA obtained with the tfdA** primer set. The position number relates to GenBank accession number M16730 (R. eutropha JMP134). The class I tfdA gene sequence represented by R. eutropha JMP134 and the class III tfdA sequence represented by Burkholderia cepacia pIJB were both obtained from GenBank. A2 and A9 correspond to the sequences obtained at day 68 and day 0, respectively. These products were run on a DGGE gel and stabbed prior to sequencing as indicated in the legend of Fig. 7. A2 is highly homologous to bands A1, A3 to A7, A10, and B1 to B3, while A9 and A8 are homologous. Black shading shows highly conserved regions. Designations in brackets are GenBank database accession numbers.
The DGGE profiles of Top2.3 and Top20 from days 0 to 6 and 0 to 12, respectively, were very similar to those of the DNA extracts from the topsoils without MCPA (Fig. 7D). A number of bands were visible on the gel containing the Top0 DNA extracts, where some of them were common bands that were represented in all lanes throughout the experiment and where some of them were distinct bands represented in only single lanes. The overall pattern in Top0 was that no change in the population containing the tfdA gene occurred over time and that the soil had a natural content of tfdA genes that denaturized in the same way as the tfdA gene from R. eutropha strain JMP134. Interestingly, the organisms represented by this band did not grow up during the experiments when MCPA was applied; however, the intensity of the band remained the same or had a slight decrease (Fig. 7A and B). The DGGE patterns from the subsoil were different from those from the topsoil. First, there were no bands in Sub0 (Fig. 7E) or on days 0 to 33 for Sub2.3 (Fig. 7C). Second, the intensity of the bands from Sub2.3 at days 33 and 68 varied between the triplicates, especially for day 33. Furthermore, the intensity of the upper two bands, if any, was weak. Despite this, the other bands were present and had about the same intensity as those for Top2.3 (Fig. 7A).
The sequences obtained from the bands stabbed from the DGGE gels were compared to existing sequences encoding α-ketoglutarate-dioxygenase obtained from GenBank, and a phylogenetic tree was constructed (data not shown). The phylogenetic analysis showed that bands A1 to A7, A10, and B1 to B3 (Fig. 7) were located in the same cluster as the class III tfdA genes showing 96 to 99% homology to the indigenous genes in this class (Fig. 8). Bands A8 and A9, on the other hand, were located in the same cluster as the class I tfdA genes showing 98 and 99% homology, respectively, to the R. eutropha JMP134 tfdA gene (Fig. 8). Furthermore, the GC content of bands A1 to A7, A10, and B1 to B3 was approximately 63%, while it was 69% for bands A8 and A9.
DISCUSSION
The carbon added in the form of the MCPA substrate should be sufficient to sustain the growth of approximately 107 new cells g−1 soil in Top20 and 1.2 × 106 cells g−1 in Top2.3 and Sub2.3. This is based on the assumption that one new cell requires approximately 1 pg of carbon to evolve (2). In further calculations, we assumed that one cell (JMP134 as well as naturally occurring bacterial populations) only contains a single tfdA gene. As the tfdA gene is plasmid borne, a single cell can possibly hold more than one tfdA gene; this latter assumption may be quite untenable (58). Furthermore, different cells may require different amounts of carbon to evolve, and different substrates may cause populations to grow differently as well (1, 2). When the uncertainties caused by the assumptions described above are taken into account, the maximum concentrations of 3.0 × 104, 7.0 × 105, and 2.6 × 104 genes g−1 soil observed in Top2.3, Top20, and Sub2.3, respectively, correspond to less than 10% of the theoretical numbers. This may be explained in two ways. First, predation by protozoa and amoebae may be responsible for a large decrease of the cell number. This was indicated in a study reported previously by Thirup et al. (56), which showed that the number of culturable microorganisms first increased as a result of amendment with carbon substrate and subsequently decreased as a result of predation. A corresponding pattern was seen in the present study, where numbers of tfdA genes, especially in the topsoil experiments, decreased by 0.5 to 1 order of magnitude throughout the experiment after the maximum gene concentration was observed. Second, the primer set used may not have targeted the entire tfdA degrader population. Recently, Shaw and Burns (51) found that the canonical tfdA gene could not be detected using the tfdA* primer set on DNA extracted from a 2,4-D degradation experiment for which the soil had not been preexposed to phenoxy acid herbicides but that the tfdAα-I primer set yielded a PCR product for the same soil. This indicates that the latter group of phenoxy acid degraders may be involved in the degradation of 2,4-D and probably also in the degradation of MCPA, especially in soils that are not frequently exposed to the herbicide. However, during the test of primers targeting the canonical tfdA gene, the tfdAα gene, and the cadA gene, we observed that PCR products were yielded with the canonical tfdA primer and with the tfdAα primer but not with the cadA primer set. This indicates that organisms that use the tfdAα gene product in MCPA metabolism may be present as well.
Growth among bacterial cells and increases in catabolic genes during degradation of phenoxy acids have been reported elsewhere previously (see, e.g., references 12, 28, and 36), but only Lee et al. (36) used a highly accurate quantitative real-time PCR method. They reported concentrations of 2.0 × 107 tfdA genes g−1 in activated sludge sequentially amended with 300 mg 2,4-D kg−1. Concerning the higher amount of carbon substrate, this corresponds to what was found in the present study. de Lipthay et al. (12), however, reported slightly lower increasing concentrations of the tfdA gene in water samples amended with approximately 20 mg 2,4-D liter−1, as they observed maximum concentrations of ∼103 genes ml−1.
Analysis of the functional diversity of the indigenous MCPA degrader community by DGGE on the tfdA genes extracted has to our knowledge not previously been published. In this study, results from two different methods both indicated a change in the community of the bacteria containing the tfdA gene during a degradation period of 68 days. First, we saw a change in the maximum of the melting profiles of the PCR products from the quantitative PCR assay of the topsoil DNA from 90°C to 87.5°C. This indicates that the tfdA PCR products from the organisms grown on MCPA had a lower GC content and therefore were different from the genes present in the soil not exposed to MCPA. This corresponds with the results from the DGGE analyses in which the PCR products from the DNA sampled during the first days of the experiment had a tendency to denaturize at a higher denaturation grade. Interestingly, the maximum of the melting profiles from the first sampling days of the experiment were similar to that of the standard strain R. eutrophus JMP134. This was seen on the DGGE gels as well, where a major part of the DNA from the first sampling days seemed to denaturize in the lower part of the gels where DNA from the R. eutropha JMP134 also denaturized. The presence of tfdA genes in the control topsoil was interesting as well. The soil used for the experiments had not been exposed to phenoxy acid herbicides for at least 15 years; however, a background population of tfdA genes is maintained even in the absence of appropriate pesticide substrates. These tfdA genes did not increase in numbers during the degradation period, which indicates that they were placed in populations that did not grow actively.
Phylogenetic analyses of nucleotide sequences stabbed from bands on the gels revealed a large degree of homology to what was seen on the melting profiles and on the DGGE gels. All the sequences stabbed from the DGGE gels belonged to the first group of phenoxy acid-degrading organisms. The sequences stabbed from the bands located at the same denaturation grade as R. eutropha strain JMP134 were closely related to this strain and other class I strains as well. In contrast, the sequences stabbed from the bands that increased during the degradation period were related to the class III tfdA genes. In general, the role of those organisms containing class I tfdA genes in this particular soil remains unclear. A likely and quite interesting explanation may be that they are organisms that can only perform one or a few steps of the degradation pathway, while the class III tfdA gene-containing strains are capable of degrading the compound completely. If the class I gene-containing organisms are only capable of degrading the first step, for instance, it may not be likely to grow as a result of the degradation process. If the class III gene-containing bacteria, on the other hand, are capable of degrading MCPA completely, they may be able to grow as a result of the degradation process. This explanation is further supported by the fact that we actually were able to detect the degradation product MCP at days 0 to 22 of the degradation experiment in Top2.3 and at days 22 and 33 in Top20. Neither this compound nor the class I tfdA gene was found in Sub2.3. Until now, the majority of the research on catabolic genes involved in phenoxyacetic acid degradation has been done with respect to 2,4-D, and in general, it has been accepted that organisms containing the class I tfdA gene were capable of degrading this particular compound completely. However, it may be suggested that minor differences in the metabolism between MCPA and 2,4-D have made the class I tfdA gene-containing bacteria in this particular soil incapable of degrading MCPA completely. Classes I to III were previously defined by Fulthorpe et al. (20) and Itoh et al. (24) based on homology and whether the tfdA gene is carried on a plasmid or not. The class III tfdA genes are 77% identical to the class I genes, whereas class II only consists of Burkholderia sp. strain RASC and a few strains that are 76% identical to the class I strains and 80% identical to the class III strains (24). As is the case for the strains containing the class I gene, the organisms containing the class III gene belong to a broad range of β- and γ-proteobacteria. Even though the diversity of the tfdA gene is not yet fully understood, the DGGE analysis indicated that up to five different tfdA genes were involved in the degradation of MCPA. However, no clear phylogenetic differences could be determined between those strains represented by the strong bands on the DGGE gels. This is probably due to comigration and other types of bias likely to be observed during DGGE analysis (43). Vallaeys et al. (60) reported that strains isolated from the same soil microcosm containing highly homologous tfdA genes were different based on their 16S rRNA genes. This may improve the hypothesis that several different species may contain the catabolic gene and be involved in the degradation and that the five bands sequenced from the DGGE gels represent tfdA genes from different strains of MCPA-degrading organisms.
The kinetics of mineralization and degradation observed in this study seem to be slightly slower, especially in the subsoil experiment, than what was reported previously by Caux et al. (8) and Mortensen and Jacobsen (41) but equal to what was reported previously by Helweg (23). This rather slow degradation/mineralization may be partly due to the high MCPA concentrations used, which, as seen in this study, results in a longer lag phase. As adaptation has been shown to increase the rate of mineralization (12), a more likely explanation may be the lack of exposure to MCPA during the last 15 years. Similar results were seen for the soil not previously exposed to phenoxy acid herbicides used by Helweg (23).
For the topsoils, the mineralization curve is shifted towards longer reaction times compared with the degradation curves, indicating metabolite formation during degradation. Even though the delay of mineralization compared to degradation was quite small, we detected the first degradation product, MCP, in Top2.3 and Top20 but not in Sub2.3. However, no proper accumulation of the metabolite was observed, as the compound was not detected after days 22 and 33 in Top2.3 and Top20, respectively. It was rather surprising that we did not detect the metabolite in the subsoil, as the delay of the mineralization was more pronounced for this scenario. It is possible that other metabolites evolve in the subsoil, as we only analyzed this particular compound. This indicates that the degradation of MCP may be rate limiting in topsoils. This is further supported by the increase in the number of tfdA genes, which occurred in advance of the exponential phase of the mineralization. Detection of MCP and other unidentified metabolites during the degradation of MCPA have been reported in recent studies as well (10, 29).
In general, the amounts of MCPA mineralized in topsoils did not exceed a maximum of 60 to 70% of the initial concentrations. The remaining 30 to 40% can be explained by the incorporation of 14C from MCPA into biomass and into soil organic matter. Mineralization potentials of this magnitude have been reported elsewhere previously (41, 61). Additionally, there is a tendency towards a slightly higher total mineralization in the subsoil experiment than in the topsoil, which can be explained by a larger degree of adsorption in the topsoil than in the subsoil. This was previously shown by Jensen et al. (29) to result in a larger bioavailability in the subsoil and thereby higher mineralization potentials, corresponding to the findings previously reported by Mortensen and Jacobsen (41) and Willems et al. (61).
Using kinetic modeling, we found that the mineralization in Top2.3 and Top20 was best described by the exponential form of the 3/2-order model, in agreement with results reported previously Mortensen and Jacobsen (41), who used the exponential form for soils from three depths amended with 1 mg MCPA kg−1 soil. Similarly, Reffstrup et al. (47) fitted the mineralization of the phenoxypropionic herbicide mecoprop amended at 5 to 500 mg kg−1 to the exponential form of the 3/2-order model as well, while experiments amended with 0.0005 to 0.5 mg mecoprop kg−1 were fitted to the linear form of the 3/2-order model. This indicates that the initial concentration of the substrate may be important for whether growth occurs or not. However, this was not observed in the present study, which may be due to substrate concentrations that were too high. The fact that Top20 fitted better to the model than Top2.3 may be a consequence of the relatively higher degree of growth in Top20 than in Top2.3 compared to the substrate concentration.
In the subsoil, the result of the present study differs from what was found previously by Fomsgaard (17) and Mortensen and Jacobsen (41). They found that growth was more likely to occur in subsoil than in topsoil because the initial population of degraders in topsoils may be able to degrade the pesticide without significant growth.
Conclusion.
In summary, we found that growth of microbial degraders occurs during the degradation of MCPA. The growth was more pronounced during the degradation of high concentrations (20 mg kg−1) than during the degradation of low concentrations (2.3 mg kg−1). This led to better fitting of mineralization data of the high-concentration scenario to a model taking exponential growth into account. In addition, we found that a shift in the degrader population occurred during the degradation period. Class I tfdA genes were found to be present among the naturally occurring microbial population in the topsoil, which had not been exposed to MCPA for 15 years. The concentration of these genes did not increase during the degradation period. Contrary to this, we found that degraders containing tfdA genes belonging to class III grew up and became dominant. We found that at least five different tfdA-containing genotypes grew up during the degradation period. Thus, the results of an analysis of functional genes present in soil prior to exposure with a contaminant might not necessary reflect the actual population carrying out the degradation. Last, in soils containing an initial load of tfdA class I genes, the metabolite MCP was detected during the degradation experiment, but no proper accumulation of metabolites from the MCPA degradation was observed.
Acknowledgments
The Danish Agricultural and Veterinary Research Council supported the present study in the framework of the SOUND project.
We thank M. Nicolaisen for phylogenetic analysis of tfdA genes, L. F. Nielsen for initial optimization of real-time PCR, M. Andersen for the valuable technical assistance, and A. Z. Nielsen and K. L. Demant for their useful suggestions on the manuscript.
REFERENCES
- 1.Acea, M. J., and M. Alexander. 1988. Growth and survival of bacteria introduced into carbon amended soil. Soil Biol. Biochem. 20:703-709. [Google Scholar]
- 2.Acea, M. J., C. R. Moore, and M. Alexander. 1988. Survival and growth of bacteria introduced into soil. Soil Biol. Biochem. 20:509-515. [Google Scholar]
- 3.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 4.Amy, P. S., J. W. Schulke, L. M. Frazier, and R. J. Seidler. 1985. Characterization of aquatic bacteria and cloning of genes specifying partial degradation of 2,4-dichlorophenoxyacetic acid. Appl. Environ. Microbiol. 49:1237-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen, S. M., P. B. Hertz, T. Holst, R. Bossi, and C. S. Jacobsen. 2001. Mineralization studies of 14C-labelled metsulfuron-methyl, tribenuron-methyl, chlorsulfuron and thifensulfuron-methyl in one Danish soil and groundwater sediment profile. Chemosphere 45:775-782. [DOI] [PubMed] [Google Scholar]
- 6.Bollag, J. M., C. S. Helling, and M. Alexander. 1967. Metabolism of 4-chloro-2-methylphenoxyacetic acid by soil bacteria. Appl. Microbiol. 15:1393-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunner, W., and D. D. Focht. 1984. Deterministic three-half-order kinetic model for microbial degradation of added carbon substrates in soil. Appl. Environ. Microbiol. 47:167-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caux, P. Y., R. A. Kent, V. Bergeron, G. T. Fan, and D. D. Macdonald. 1995. Environmental fate and effects of MCPA—a Canadian perspective. Crit. Rev. Environ. Sci. Technol. 25:313-376. [Google Scholar]
- 9.Clausen, G. B., L. Larsen, K. Johnsen, J. R. de Lipthay, and J. Aamand. 2002. Quantification of the atrazine-degrading Pseudomonas sp. strain ADP in aquifer sediment by quantitative competitive polymerase chain reaction. FEMS Microbiol. Ecol. 41:221-229. [DOI] [PubMed] [Google Scholar]
- 10.Crespin, M. A., M. Gallego, M. Valcarcel, and J. L. Gonzalez. 2001. Study of the degradation of the herbicides 2,4-D and MCPA at different depths in contaminated agricultural soil. Environ. Sci. Technol. 35:4265-4270. [DOI] [PubMed] [Google Scholar]
- 11.Dejonghe, W., J. Goris, S. El Fantroussi, M. Hofte, P. De Vos, W. Verstraete, and E. M. Top. 2000. Effect of dissemination of 2,4-dichlorophenoxyacetic acid (2,4-D) degradation plasmids on 2,4-D degradation and on bacterial community structure in two different soil horizons. Appl. Environ. Microbiol. 66:3297-3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Lipthay, J. R., J. Aamand, and T. Barkay. 2002. Expression of tfdA genes in aquatic microbial communities during acclimation to 2,4-dichlorophenoxyacetic acid. FEMS Microbiol. Ecol. 40:205-214. [DOI] [PubMed] [Google Scholar]
- 13.Don, R. H., and J. M. Pemberton. 1981. Properties of 6 pesticide degradation plasmids isolated from Alcaligenes paradoxus and Alcaligenes eutrophus. J. Bacteriol. 145:681-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Don, R. H., and J. M. Pemberton. 1985. Genetic and physical map of the 2,4-dichlorophenoxyacetic acid degradative plasmid pJP4. J. Bacteriol. 161:466-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Don, R. H., A. J. Weightman, H. J. Knackmuss, and K. N. Timmis. 1985. Transposon mutagenesis and cloning analysis of the pathways for degradation of 2,4-dichlorophenoxyacetic acid and 3-chlorobenzoate in Alcaligenes eutrophus JMP134(pJP4). J. Bacteriol. 161:85-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feinberg, A. P., and B. Vogelstein. 1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 132:6-13. [DOI] [PubMed] [Google Scholar]
- 17.Fomsgaard, I. S. 1997. Modelling the mineralization kinetics for low concentrations of pesticides in surface and subsurface soil. Ecol. Model 102:175-208. [Google Scholar]
- 18.Fukumori, F., and R. P. Hausinger. 1993. Alcaligenes eutrophus JMP134 “2,4-dichlorophenoxyacetate monooxygenase” is an α-ketoglutarate-dependent dioxygenase. J. Bacteriol. 175:2083-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukumori, F., and R. P. Hausinger. 1993. Purification and characterization of 2,4-dichlorophenoxyacetate alpha-ketoglutarate dioxygenase. J. Biol. Chem. 268:24311-24317. [PubMed] [Google Scholar]
- 20.Fulthorpe, R. R., C. McGowan, O. V. Maltseva, W. E. Holben, and J. M. Tiedje. 1995. 2,4-Dichlorophenoxyacetic acid-degrading bacteria contain mosaics of catabolic genes. Appl. Environ. Microbiol. 61:3274-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaunt, J. K., and W. C. Evans. 1971. Metabolism of 4-chloro-2-methylphenoxyacetate by a soil pseudomonad—preliminary evidence for metabolic pathway. Biochem. J. 122:519-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harker, A. R., R. H. Olsen, and R. J. Seidler. 1989. Phenoxyacetic acid degradation by the 2,4-dichlorophenoxyacetic acid (tfd) pathway of plasmid pJP4: mapping and characterization of the tfd regulatory gene, tfdr. J. Bacteriol. 171:314-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helweg, A. 1987. Degradation and adsorption of C-14 MCPA in soil—influence of concentration, temperature and moisture content on degradation. Weed Res. 27:287-296. [Google Scholar]
- 24.Itoh, K., R. Kanda, Y. Sumita, H. Kim, Y. Kamagata, K. Suyama, H. Yamamoto, R. P. Hausinger, and J. M. Tiedje. 2002. tfdA-like genes in 2,4-dichlorophenoxyacetic acid-degrading bacteria belonging to the Bradyrhizobium-Agromonas-Nitrobacter-Afipia cluster in α-Proteobacteria. Appl. Environ. Microbiol. 68:3449-3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itoh, K., Y. Tashiro, K. Uobe, Y. Kamagata, K. Suyama, and H. Yamamoto. 2004. Root nodule Bradyrhizobium spp. harbor tfdAα and cadA, homologous with genes encoding 2,4-dichlorophenoxyacetic acid-degrading proteins. Appl. Environ. Microbiol. 70:2110-2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobsen, C. S. 2004. Detection and quantification of microbial DNA sequences in soil by Southern- and dot/slot blot hybridization, p. 333-344. In G. A. Kowalchuk (ed.), Molecular microbial ecology manual. Kluwer Academic, Dordrecht, The Netherlands.
- 27.Jacobsen, C. S. and J. C. Pedersen. 1992. Mineralization of 2,4-dichlorophenoxyacetic acid (2,4-D) in soil inoculated with DBO1 (pRO101), AEO106 (pRO101) and JMP134 (pJP4): effects of inoculation level and substrate concentration. Biodegradation 2:253-263. [DOI] [PubMed] [Google Scholar]
- 28.Jacobsen, C. S., and J. C. Pedersen. 1992. Growth and survival of Pseudomonas cepacia DB01 (pRO101) in soil amended with 2,4-dichlorophenoxyacetic acid. Biodegradation 2:245-252. [DOI] [PubMed] [Google Scholar]
- 29.Jensen, P. H., H. C. B. Hansen, J. Rasmussen, and O. S. Jacobsen. 2004. Sorption-controlled degradation kinetics of MCPA in soil. Environ. Sci. Technol. 38:6662-6668. [DOI] [PubMed] [Google Scholar]
- 30.Kamagata, Y., R. R. Fulthorpe, K. Tamura, H. Takami, L. J. Forney, and J. M. Tiedje. 1997. Pristine environments harbor a new group of oligotrophic 2,4-dichlorophenoxyacetic acid-degrading bacteria. Appl. Environ. Microbiol. 63:2266-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaphammer, B., J. J. Kukor, and R. H. Olsen. 1990. Regulation of tfdCDEF by tfdR of the 2,4-dichlorophenoxyacetic acid degradation plasmid pJP4. J. Bacteriol. 172:2280-2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaphammer, B., and R. H. Olsen. 1990. Cloning and characterization of tfdS, the repressor-activator gene of tfdB, from the 2,4-dichlorophenoxyacetic acid catabolic plasmid pJP4. J. Bacteriol. 172:5856-5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitagawa, W., S. Takami, K. Miyauchi, E. Masai, Y. Kamagata, J. M. Tiedje, and M. Fukuda. 2002. Novel 2,4-dichlorophenoxyacetic acid degradation genes from oligotrophic Bradyrhizobium sp. strain HW13 isolated from a pristine environment. J. Bacteriol. 184:509-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kukor, J. J., R. H. Olsen, and J. S. Siak. 1989. Recruitment of a chromosomally encoded maleylacetate reductase for degradation of 2,4-dichlorophenoxyacetic acid by plasmid pJP4. J. Bacteriol. 171:3385-3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laemmli, C. M., J. H. J. Leveau, A. J. B. Zehnder, and J. R. van der Meer. 2000. Characterization of a second tfd gene cluster for chlorophenol and chlorocatechol metabolism on plasmid pJP4 in Ralstonia eutropha JMP134(pJP4). J. Bacteriol. 182:4165-4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee, T. H., S. Kurata, C. H. Nakatsu, and Y. Kamagata. 2005. Molecular analysis of bacterial community based on 16S rDNA and functional genes in activated sludge enriched with 2,4-dichlorophenoxyacetic acid (2,4-D) under different cultural conditions. Microb. Ecol. 49:151-162. [DOI] [PubMed] [Google Scholar]
- 37.Leveau, J. H. J., and J. R. van der Meer. 1996. The tfdR gene product can successfully take over the role of the insertion element-inactivated TfdT protein as a transcriptional activator of the tfdCDEF gene cluster, which encodes chlorocatechol degradation in Ralstonia eutropha JMP134(pJP4). J. Bacteriol. 178:6824-6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loos, M. A. 1969. Phenoxyalkanoic acids, p. 1-128. In P. C. Kearney and D. D. Kaufman (ed.), Degradation of herbicides. Marcel Dekker, New York, N.Y.
- 39.Matrubutham, U., and A. R. Harker. 1994. Analysis of duplicated gene sequences associated with tfdR and tfdS in Alcaligenes eutrophus JMP134. J. Bacteriol. 176:2348-2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGowan, C., R. Fulthorpe, A. Wright, and J. M. Tiedje. 1998. Evidence for interspecies gene transfer in the evolution of 2,4-dichlorophenoxyacetic acid degraders. Appl. Environ. Microbiol. 64:4089-4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mortensen, S. K., and C. S. Jacobsen. 2004. Influence of frozen storage on herbicide degradation capacity in surface and subsurface sandy soils. Environ. Sci. Technol. 38:6625-6632. [DOI] [PubMed] [Google Scholar]
- 42.Muyzer, G., E. C. Dewaal, and A. G. Uitterlinden. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S ribosomal RNA. Appl. Environ. Microbiol. 59:695-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nikolausz, M., R. Sipos, S. Revesz, A. Szekely, and K. Marialigeti. 2005. Observation of bias associated with reamplification of DNA isolated from denaturing gradient gels. FEMS Microbiol. Lett. 244:385-390. [DOI] [PubMed] [Google Scholar]
- 44.Pemberton, J. M., B. Corney, and R. H. Don. 1979. Evolution and spread of pesticide degrading ability among soil microorganisms, p. 287-299. In K. N. Timmis and A. Pühler (ed.), Plasmids of medical, environmental and commercial importance. Elsevier, Amsterdam, The Netherlands.
- 45.Perkins, E. J., M. P. Gordon, O. Caceres, and P. F. Lurquin. 1990. Organization and sequence analysis of the 2,4-dichlorophenol hydroxylase and dichlorocatechol oxidative operons of plasmid pJP4. J. Bacteriol. 172:2351-2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pieper, D. H., W. Reineke, K. H. Engesser, and H. J. Knackmuss. 1988. Metabolism of 2,4-dichlorophenoxyacetic acid, 4-chloro-2-methylphenoxyacetic acid and 2-methylphenoxyacetic acid by Alcaligenes eutrophus JMP134. Arch. Microbiol. 150:95-102. [Google Scholar]
- 47.Reffstrup, T. K., H. Sorensen, and A. Helweg. 1998. Degradation of mecoprop at different concentrations in surface and subsurface soil. Pestic. Sci. 52:126-132. [Google Scholar]
- 48.Robinson, J. A. 1985. Determining microbial kinetic-parameters using nonlinear-regression analysis—advantages and limitations in microbial ecology. Adv. Microb. Ecol. 8:61-114. [Google Scholar]
- 49.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 50.Scheidleder, A. 2000. Groundwater quality and quantity in Europe. European Environment Agency, Copenhagen, Denmark.
- 51.Shaw, L. J., and R. G. Burns. 2004. Enhanced mineralization of [U-14C]2,4-dichlorophenoxyacetic acid in soil from the rhizosphere of Trifolium pratense. Appl. Environ. Microbiol. 70:4766-4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simkins, S., and M. Alexander. 1984. Models for mineralization kinetics with the variables of substrate concentration and population density. Appl. Environ. Microbiol. 47:1299-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smejkal, C. W., T. Vallaeys, S. K. Burton, and H. M. Lappin-Scott. 2001. Substrate specificity of chlorophenoxyalkanoic acid-degrading bacteria is not dependent upon phylogenetically related tfdA gene types. Biol. Fert. Soils 33:507-513. [Google Scholar]
- 54.Streber, W. R., K. N. Timmis, and M. H. Zenk. 1987. Analysis, cloning, and high-level expression of 2,4-dichlorophenoxyacetate monooxygenase gene tfdA of Alcaligenes eutrophus JMP134. J. Bacteriol. 169:2950-2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suwa, Y., A. D. Wright, F. Fukimori, K. A. Nummy, R. P. Hausinger, W. E. Holben, and L. J. Forney. 1996. Characterization of a chromosomally encoded 2,4-dichlorophenoxyacetic acid α-ketoglutarate dioxygenase from Burkholderia sp. strain RASC. Appl. Environ. Microbiol. 62:2464-2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thirup, L., F. Ekelund, K. Johnsen, and C. S. Jacobsen. 2000. Population dynamics of the fast-growing subpopulations of Pseudomonas and total bacteria, and their protozoan grazers, revealed by fenpropimorph treatment. Soil Biol. Biochem. 32:1615-1623. [Google Scholar]
- 57.Tonso, N. L., V. G. Matheson, and W. E. Holben. 1995. Polyphasic characterization of a suite of bacterial isolates capable of degrading 2,4-D. Microb. Ecol. 30:3-24. [DOI] [PubMed] [Google Scholar]
- 58.Trefault, N., P. Clement, M. Manzano, D. H. Pieper, and B. Gonzalez. 2002. The copy number of the catabolic plasmid pJP4 affects growth of Ralstonia eutropha JMP134 (pJP4) on 3-chlorobenzoate. FEMS Microbiol. Lett. 212:95-100. [DOI] [PubMed] [Google Scholar]
- 59.Vallaeys, T., R. R. Fulthorpe, A. M. Wright, and G. Soulas. 1996. The metabolic pathway of 2,4-dichlorophenoxyacetic acid degradation involves different families of tfdA and tfdB genes according to PCR-RFLP analysis. FEMS Microbiol. Ecol. 20:163-172. [Google Scholar]
- 60.Vallaeys, T., F. Parsello-Cartieaux, N. Rouard, C. Lors, G. Laguerre, and G. Soulas. 1997. PCR-RFLP analysis of 16S rRNA, tfdA and tfdB genes reveals a diversity of 2,4-D degraders in soil aggregates. FEMS Microbiol. Ecol. 24:269-278. [Google Scholar]
- 61.Willems, H. P. L., K. J. Lewis, J. S. Dyson, and F. J. Lewis. 1996. Mineralization of 2,4-D and atrazine in the unsaturated zone of a sandy loam soil. Soil Biol. Biochem. 28:989-996. [Google Scholar]