

Abstract
Exposure of Vibrio harveyi (strain VH1114) to V. harveyi siphovirus-like phage 1 (VHS1) resulted in the production of a low percentage of lysogenized clones of variable stability. These were retrieved most easily as small colonies within dot plaques. Analysis revealed that VHS1 prophage was most likely carried by VH1114 as an episome rather than integrated into the host chromosome. In the late exponential growth phase, lysogenized VH1114 continuously produced VHS1 but also gave rise to a large number of cured progeny. The absence of phage DNA in the cured progeny was confirmed by the absence of VHS1 DNA in Southern blot and PCR assays. Curiously, these very stable, cured subclones did not show the parental phenotype of clear plaques with VHS1 but instead showed turbid plaques, both in overlaid lawns and in dot plaque assays. This phenotypic difference from the original parental isolate suggested that transient lysogeny by VHS1 had resulted in a stable genetic change in the cured clones. Such clones may be called pseudolysogens (i.e., false lysogens), since they have undergone transient lysogeny and have retained some resistance to full lytic phage development, despite the loss of viable or detectable prophage.
Luminous vibriosis and luminescent bacterial disease are terms that are used commonly to describe mortality caused mostly by pathogenic strains of Vibrio harveyi in hatcheries or rearing ponds of Penaeus species (6, 10). In serious epizootics, moribund shrimp emit a greenish luminescence due to bacterial infection or colonization. There is good evidence that exotoxins are involved in mortality from luminescent vibriosis, since extracellular products of cultured V. harveyi can be toxic when injected into Penaeus monodon (7, 8) and since exotoxin production by V. harveyi is involved in shrimp larval disease (5).
Upon examining the genetic relationship among virulent isolates of V. harveyi, Pizzutto and Hirst (17) found no discernible associations and thus proposed that virulence factors were transmitted by mobile elements such as transposons, plasmids, or bacteriophages. This led to a search for bacteriophages in Australian isolates of V. harveyi and to the eventual discovery of V. harveyi myo-like phage from a highly virulent V. harveyi isolate derived from diseased shrimp larvae (11-13). Phage conversion leading to toxin production occurs with many human-pathogenic bacteria (1, 2), including Vibrio cholerae (4), but had not previously been proven for V. harveyi.
Association of V. harveyi virulence with a bacteriophage has also been reported in Thailand (14, 19). However, phage production could not be sustained in culture, and the phage was lost, preventing further study. Later, another bacteriophage was isolated from shrimp culture ponds in Thailand, together with a partner V. harveyi isolate, VH1114 (15). It was an unenveloped virus with a long, noncontractile tail (100 to 120 nm long) and an isometric head (approximately 60 nm in diameter) that morphologically resembled the earlier Thai bacteriophage associated with virulence for shrimp. The nucleic acid consisted of double-stranded DNA with a total length of approximately 80 kbp. These characteristics would place it in the order Caudovirales and family Siphoviridae (21), and it was called V. harveyi siphovirus-like phage 1 (VHS1). Preliminary results indicated that VHS1 could cause an increase in virulence of VH1114 for the black tiger shrimp Penaeus monodon (18).
The purpose of this study was to examine the interaction between VHS1 and VH1114 and to determine whether VHS1 could lysogenize VH1114 and whether it had potential to be used as a model to study the relationship between lysogeny and V. harveyi virulence.
MATERIALS AND METHODS
Media and reagents.
Restriction enzymes were acquired from Promega (Madison, WI), digoxigenin nonradioactive DNA labeling and detection kits from Boehringer Mannheim and Roche (Mannheim, Germany), Bradford protein assay reagents from Bio-Rad (Hercules, Calif.), and marker λ DNA cut with EcoRI-HindIII from GIBCO BRL (Gaithersburg, MD). PCR reagents were purchased from Perkin-Elmer (New Jersey). Mueller-Hinton broth (MHB) was obtained from Oxoid (Hampshire, England), and solid medium (Mueller-Hinton agar [MHA]) was produced by addition of Difco agar (Becton, Dickinson and Co., Franklin Lakes, NJ) at 1.5%. The media were prepared according to the manufacturers' instructions except that distilled water was replaced by artificial seawater (ASW) containing 400 mM NaCl, 100 mM MgSO4 · 7H2O, 20 mM KCl, and 20 mM CaCl2 · 2H2O.
Bacteria and VHS1.
VH1114 and its partner phage VHS1 were obtained from a Thai shrimp pond where black tiger prawns were being cultivated (15). All other bacterial isolates used were derived as subclones of this isolate. VH1114 and subclones were stored in MHB containing 20% glycerol at −80°C until revived for use. The process of revival consisted of thawing on ice followed by inoculation in appropriate broth culture medium overnight. VHS1 was stored in sterile ASW at 4°C.
Bacterial challenge test.
A preliminary bacterial challenge test was carried out using VH1114 and a subclone of the lysogen VHL5 (see “Preliminary bacterial challenge test” in Results). A group of 130 juvenile Penaeus monodon animals (selected by equal length and approximately 15 g each) were obtained from a local shrimp farm and acclimatized in the laboratory for 5 days as a single group in a rectangular concrete tank (1.5 by 2.0 m) filled to a depth of 40 cm with artificial seawater at 28 to 30°C and 15 ppt and constantly aerated by airlift filters set in a bed of crushed coral. They were fed once daily with a commercial pelleted feed equivalent to 1.5% of body weight (dry weight of feed versus wet weight of shrimp). They were then subdivided into 13 groups of 10 each and held in separate 100-liter aquaria at the same temperature and salinity for the challenge tests. Bacteria for the challenge tests were prepared using 50 μl of 24-h bacterial stock culture to inoculate 5 ml MHB. This was incubated at 30°C for 6 h (i.e., into exponential phase) to an optical density at 600 nm of 0.85 (2 × 108 CFU per ml). The culture fluid was then used directly or diluted in MHB to allow injection of each shrimp in the sixth abdominal musculature with 0.05 ml containing an appropriate number of bacterial cells. There were groups for injection at approximately 1 × 107 down to 1 × 102 CFU/shrimp in 10-fold steps. The control group was injected with 50 μl MHB. The number of surviving shrimp was observed for 24 h after injection in order to determine the 24-h 50% lethal dose.
VHS1 preparation.
Phage preparation for morphological study or for use in amplification experiments consisted of harvesting from surface plaques formed after lysis of V. harveyi growing on MHA and overlaid with a phage extract or picked from individual plaques using a sterile wooden toothpick. The total wash liquid (sterile ASW) was centrifuged at 7,000 × g for 10 min to pellet the bacteria. The supernatant solution was filtered sequentially through 0.45-μm and 0.2-μm disposable membrane filters (Sartorius) and stored at 4°C. To confirm that the filtrates were free from bacterial cells, aliquots were streaked on MHA plates and incubated overnight at 30°C.
VHS1 quantitation by dot plaque assay.
VHS1 was quantified by the dot plaque technique. Briefly, an overnight culture of VH1114 (0.1 ml) was spread on MHA and allowed to absorb completely into the agar. Filtered phage solution was diluted in 10-fold steps and dropped as 10-μl spots onto the agar. After overnight incubation, the final dilution that gave a clearly visible plaque was considered equivalent to one phage particle. The concentration of phage was then calculated according to the dilution (i.e., phage concentration = 1 × 10n × 100 PFU/ml, where n is the dilution number).
Qualitative assessment of phage DNA.
VHS1 was concentrated from phage preparations by ultracentrifugation at 100,000 × g for 4 h, and pellets were resuspended in a minimal volume of ASW. These pellets were used for DNA extraction (QIAamp DNA minikit; QIAGEN GmbH, Germany), followed by restriction enzyme digestion, agarose gel electrophoresis, ethidium bromide staining, and visualization by UV transillumination. Some gels were used for Southern blot hybridization.
Purification of VHS1 phage particles.
VHS1 stocks were purified by either Urografin gradient separation or polyethylene glycol precipitation. For the latter, phage lysate after ultrafiltration (i.e., free of bacterial contamination) was mixed with one-fourth volume of a solution containing 20% polyethylene glycol 6000 and 10% NaCl and centrifuged at 12,000 × g to precipitate the phage particles, which were then resuspended in ASW. VHS1 concentrated by ultracentrifugation was layered over discontinuous Urografin (Schering AG, Berlin, Germany) gradients (10, 20, 30, and 40% Urografin diluted with ASW) and centrifuged to equilibrium at 100,000 × g for 90 min at 4°C. The band at the 20 to 30% interface was removed for DNA extraction (QIAGEN) and for transmission electron microscopy using negative staining (15).
Potential lysogens from liquid cultures.
VH1114 (overnight culture in MHB) was mixed 1:10 with bacteriophage (109 PFU/ml in ASW) and incubated at room temperature for 1 h. This mixture was then centrifuged at 7,000 × g for 10 min. The supernatant was discarded, and the pellet was washed three times with 3 ml of ASW. The suspension was streaked on MHA, and individual colonies were selected for testing as lysogenized clones.
Potential lysogens from solid medium cultures.
Bacteria were isolated from overnight growth around or within dot plaques of VH1114 (see above) and streaked on MHA so that individual clones could be selected for testing as lysogenized clones. The percentage of lysogenized cells was also estimated by counting the number of colonies within the plaques with a stereomicroscope at 10 to 15 h after dotting and dividing this number by the number of original bacteria calculated by dividing the area of the plaque by the area of the plate, multiplied by the number of cells used to prepare the original lawn times 100 (for percentage).
Identification of putative lysogens by resistance to VHS1 lysis.
Single colonies were picked from MHB culture streaks on MHA or from steaks of cells within dot plaques on MHA. These were transferred to MHB and cultivated overnight with shaking. Aliquots of the overnight cultures were used for dot plaque assays as described above. A similarly treated culture of VH1114 served as a positive control. After incubation at 30°C overnight, plates without plaque formation were considered to contain putative lysogens, while those with plaques were considered to contain nonlysogenized clones. The absence of prophage in nonlysogenized subclones was subsequently confirmed by failure to produce VHS1 and a negative PCR test for VHS1.
Confirmation of putative lysogens.
Overnight MHB cultures of putative lysogens were centrifuged at 7,000 rpm for 10 min. The culture supernatant was assessed for the presence of infectious VHS1 phage particles by dot plaque assay using VH1114. Pellets were washed three times with 1 ml of ASW and then extracted for genomic DNA using a QIAamp DNA minikit (QIAGEN) or for plasmid DNA using a QIAprep spin miniprep kit (QIAGEN) (silica based), a HiSpeed plasmid midikit (QIAGEN) for anion-exchange-based extraction of plasmids larger than 50 kb, or a QAIGEN large-construct kit for extraction of very large constructs. Genomic or plasmid extracts were used for PCR, dot blot, and Southern blot hybridization tests designed to detect the presence of VHS1 genomic material.
Stability of confirmed lysogens.
Broth cultures of subclones confirmed to be lysogens by PCR, Southern blotting, and phage production were restreaked on MHA, followed by incubation at 30°C overnight. Single colonies were then selected for dot plaque assay with VHS1 as described above. Lawns without plaque formation were considered to be lysogens, while those with turbid plaques were considered to be cured subclones.
Detection of VHS1 DNA in host V. harveyi by PCR.
PCR detection of VHS1 in bacterial host cells was carried out as previously described (15). Briefly, bacteria were cultured individually in MHB overnight, followed by centrifugation and washing three times with ASW before resuspension. Total DNA was extracted using QIAamp DNA minikits according to the manufacturer's handbook (QIAGEN, Hilden, Germany). Extracts were used as the template for PCR amplification with a pair of VHS1-specific primers, PH102 F8/2 (5′-AAA CGA CTT CGC GCA TGT T-3′) and PH102 R2/2 (5′-GAC TCG CTT TTA ACT GCT A-3′) reported for the VHS1 DNA polymerase gene (15). The PCR was performed in a final volume of 25 μl containing 1× PCR buffer II (500 mM KCl, 1.5 mM MgCl2, and 100 mM Tris-HCl, pH 8.3); 200 μM each of dATP, dTTP, dCTP, and dGTP; 200 nM of each primer; 100 ng of DNA template; and 1.25 units of Ampli Taq DNA polymerase. The amplification protocol consisted of preheating at 94°C for 5 min followed by 35 cycles of denaturation at 94°C for 1 min, annealing at 60°C for 1 min, and extension at 72°C for 2 min, except for a final extension at 72°C for 7 min. Positive controls used for each PCR assay consisted of VHS1 genomic DNA, while negative controls consisted of deionized water or VH1114 genomic DNA. PCR products were confirmed to be derived from VHS1 phage by Southern blot hybridization using digoxigenin-labeled VHS1 DNA as a probe under highly stringent conditions (20).
Southern blot hybridizations.
VHS1 genomic DNA was cut with EcoRI/XbaI, λ marker DNA was cut with HindIII/DraI, and the digests were labeled with digoxigenin 11-dUTP using a random prime labeling kit (Boehringer, Mannheim, Germany) according to the manufacturer's instructions. Labeled DNA (20 μl) was subsequently precipitated and separated from unincorporated nucleotides by addition of 2.5 ml of LiCl and 75 ml of prechilled (−20°C) absolute ethanol, gentle mixing, and incubation at −80°C for 30 min. Thereafter, the DNA pellets were collected by centrifugation at 12,000 rpm for 10 min at 4°C. The absolute ethanol was decanted. The DNA pellets were washed with 100 ml of prechilled (−20°C) 70% ethanol and recentrifuged at 12,000 rpm for 10 min at 4°C. The ethanol was decanted, and the labeled DNA pellets were dried and dissolved in 50 μl Tris-EDTA buffer. The sample DNA template (1 to 3 μg) was heat denatured in a boiling water bath for 10 min and immediately chilled on ice for 5 min. DNA test samples were digested completely with EcoRI/XbaI or Hind III/DraI for at least 3 h at 37°C and separated by 1% agarose gel electrophoresis. Gels were photographed and then processed for Southern blot hybridization using a mix of the labeled probes for λ DNA and VHS1 DNA (20). Preliminary tests had shown that there was no cross hybridization and that the two probes could be used simultaneously. In the case of dot blots, the whole, undigested genomic solution mixed with 0.75 N NaOH (1:1) was incubated at room temperature for 5 min before dotting on membranes and probing with the labeled VHS1 DNA probe only.
RESULTS
Preliminary bacterial challenge test.
The results from the challenge test (Fig. 1; see also Table S1 in the supplemental material) indicated that the 24-h 50% lethal dose of the lysogen VHL5 (6.3 × 103 cells/shrimp) was approximately 100 times lower than that of VH1114 (5 × 105 cells/shrimp).
FIG. 1.

Graph showing regression lines for shrimp mortality versus log CFU for injection of VH1114 and VHL5 into juvenile P. monodon. LD50, 50% lethal dose.
Dynamics of VHS1 bacteriophage infection in VH1114.
When agar cultures of VH1114 were exposed to bacteriophage VHS1 by dot plaque assay, small colonies always appeared in the centers of the clear plaques. These could be counted and selected easily using a stereomicroscope in the interval of 10 to 15 h after initiation of the plaque assay (see Fig. S1 in the supplemental material). Longer periods of incubation resulted in merged colonies (see, e.g., Fig. 7, type 0). As shown in Table 1, 20 clones removed from the centers of such plaques were all (100%) putative lysogens. All were refractory to lysis by VHS1 in dot plaque assays, and all produced VHS1 particles (i.e., their culture supernatants produced clear dot plaques in lawns of VH1114). Using rough calculations based on plaque diameter (1 cm), total spread diameter (8 cm), and number of bacteria in 0.1 ml of spread plate inoculum (1 × 108), a rough estimate of lysogenic conversion was 0.002%. From liquid cultures exposed to VHS1, 4 of 50 isolates (8%) were putative lysogens, while the other 46 (92%) gave clear plaques, identical to those of VH1114. Candidate clones from both sources were used for confirmatory tests to detect the presence of VHS1 nucleic acid. Three of the putative lysogens gave strong colony blot hybridization signals with the digoxigenin-labeled VHS1 probe. They were designated VHL5, VHL8, and VHL10 and were used together with the VH1114 parental strain to study the nature and stability of the relationship between VHS1 and V. harveyi.
FIG. 7.

Examples of VHS1 overlay assay results for VH1114 (type 0) and a representative type 2 revertant of a lysogen. Note that the type 2 subclone shows turbid plaques that differ from those of original parental isolate.
TABLE 1.
Screening of colonies obtained after VHS1 exposure
| Exposure medium | No. of putative lysogens/total | % Putative lysogens |
|---|---|---|
| Solid-state dot plaque | 20/1.6 × 106 | 0.002 |
| Liquid state | 4/50 | 8 |
Tests for lysogeny in VHL5, VHL8, and VHL10 by PCR and phage production assays.
Culture supernatants of VHL5, VHL8, and VHL10 revealed the presence of VHS1 bacteriophage DNA both by PCR assay using primers PH 102 F8/R2 (Fig. 2) and by restriction enzyme digestion of DNA extracts from ultracentrifuge pellets of culture supernatants (Fig. 3). The VHL supernatants also produced clear dot plaques in lawns of VH1114. By contrast, supernatants of VH1114 cultures were negative for these tests (not shown). Southern blot assays of restriction enzyme digests of total DNA extracts directly from subcultured cells of VHL5, -8, and -10 initially gave positive results for the presence of VHS1 DNA but later gave negative results, even at high loading levels.
FIG. 2.
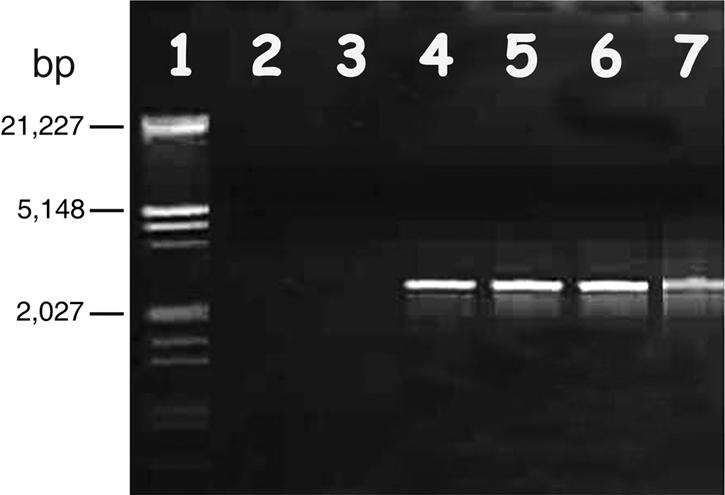
Agarose electrophoresis of PCR products amplified by the primers PH102 F8/R2 from total DNA extract templates of phage pellets from supernatant solutions of VH, VHL5, VHL8, and VHL10. Lane 1, λ DNA EcoRI/HindIII markers; lane 2, distilled water (negative control); lane 3, supernatant from V. harveyi; lanes 4 to 6, supernatants from VHL5, -8, and -10, respectively; lane 7, VHS1 (positive control).
FIG. 3.
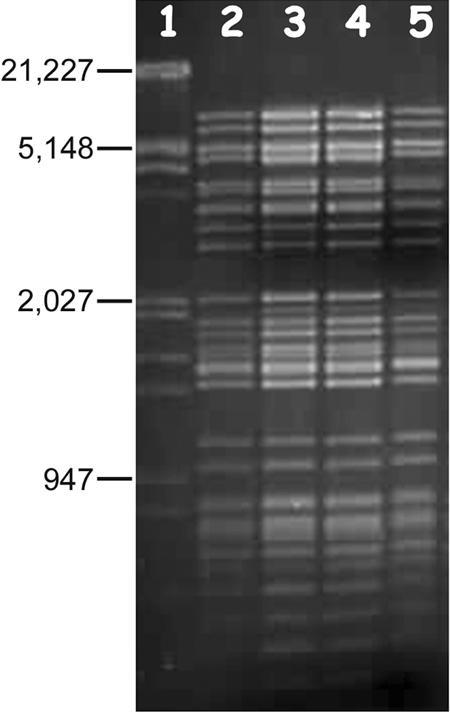
Agarose gel (1%) electrophoresis of total DNAs extracted from supernatant pellets of VHL5, VHL8, and VHL10 digested with EcoRI/XbaI. Lane 1, λ DNA EcoRI/HindIII marker; lane 2, VHS1 DNA; lanes 3 to 5, DNAs from supernatants of VHL5, -8, and -10, respectively.
Tests for stability of lysogeny in VHL5.
Since preliminary Southern blot assays for the presence of VHS1 genetic material in total DNA extracts of VHL5, VHL8, and VHL10 cells gave variable results, VHL5 was chosen for detailed tests on the percentage of lysogenized cells upon successive subculture. It is important to realize that this VHL5 culture had already undergone several subcultures by broth transfer during a few months of maintenance in the laboratory. Thirteen single colonies (subclones) were selected from an agar plating of a washed cell suspension from an exponential culture of VHL5 and tested individually for the presence of VHS1 by PCR using primers PH 102 F8/2-R2/2. Only one subclone showed the presence of VHS1 by positive PCR for VHS1 (i.e., by resistance to VHS1 lysis and by production of VHS1 [109 PFU/ml] in culture supernatant by dot plaque assay using VH1114). According to veterinary sampling tables (3), this would indicate with 95% confidence a maximum prevalence of approximately 31% putative lysogenic cells in the sampled population of VHL5. A second test carried out using 20 individual subclones of VHL5 (named VHL5.1 to VHL5.20) again showed only one subclone that was positive for VHS1 by PCR and by dot plaque assay. All others gave turbid plaques (see Fig. 6 and 7) and were negative for VHS1 production. According to veterinary sampling tables (3), this would indicate with 95% confidence a maximum prevalence of approximately 21% putative lysogenic strains in the sampled population of VHL5. Thus, for the two tests, the mean estimated maximum prevalence of putative lysogenic cells in the population of VHL5 was 26% ± 7%. All other subclones were negative for VHS1 by PCR, were negative for VHS1 production, and produced turbid dot-plaques with VHS1. Together, the results indicated that the majority (∼74%) of the cells in the VHL5 population were nonlysogenic (i.e., cured).
FIG. 6.

Three types of dot plaque assay results obtained when using VHS1 to challenge VH1114 and its subclones.
Assays for VHS1 DNA in VHL5.12 and VHL5.18.
VHL5.12 was a putative lysogen that was refractory to lysis by VHS1, positive for VHS1 by PCR assay, and capable of producing VHS1. By contrast, VHL5.18 gave turbid dot plaques with VHS1, was negative for VHS1 by PCR, and did not produce VHS1. When the lysogen VHL5.12 was subjected to DNA extraction and plasmid extraction followed by digestion with EcoRI/XbaI, electrophoresis, and Southern blot hybridization using digoxigenin-labeled VHS1 DNA, VHS1 DNA was not clearly visible in the ethidium bromide-stained gels (Fig. 4), probably because of the overwhelming quantity of host DNA present. However, the Southern blot revealed a strong signal for VHS1 DNA (Fig. 4). Since the hybridization bands were identical to those for VHS1 (i.e., there were no band shifts), the results suggested that the phage did not integrate into the host chromosome. Similar tests for VHS1 DNA in VHL5.18 were all negative.
FIG. 4.

A 1% agarose electrophoresis gel and its Southern blot for EcoRI/XbaI-digested chromosomal and plasmid DNA extracts from VH1114, VHL5.18, and VHL5.12 probed with a digoxigenin-labeled mix of VHS1 DNA and λ DNA EcoRI/HindIII marker. Lanes 1 and 10, λ DNA EcoRI/HindIII marker; lanes 2 and 6, VHS1 DNA; lanes 3, 4, and 5, chromosomal DNAs of VH1114, VHL5.18 and VHL5.12, respectively; lanes 7, 8, and 9, minikit plasmid DNA extracts of VH1114, VHL5.18, and VHL5.12, respectively.
Plasmid extraction tests.
An additional Southern blot assay for the presence of VHS1 DNA in VHL5.12 by using DNA obtained with a midiprep plasmid extraction kit gave a positive hybridization smear with high loading of the chromosomal DNA fraction but no hybridization for the plasmid fraction, even at high loading (Fig. 5, top panel). In addition, neither fraction was positive for VHS1 by PCR assay. Identical results were obtained using a miniplasmid extraction kit except that the chromosomal fraction was positive for VHS1 by PCR (data not shown) (see Fig. S2 in the supplemental material). This suggested that the kits were unable to effectively separate phage DNA from chromosomal DNA and that the phage DNA was degraded during the plasmid extraction procedure. Similar tests using high loading of the plasmid fraction from a large-construction extraction kit gave clear positive hybridization with VHS1 for an undigested DNA band and a restriction enzyme digest smear (Fig. 5, bottom panel). By contrast, only faint hybridization signals were obtained with the chromosomal fraction (Fig. 5, bottom panel). Thus, the large-construction kit gave better separation of the phage DNA from chromosomal DNA. This was supported by positive PCR results for VHS1 in the plasmid DNA fractions and negative results for VHS1 in the chromosomal DNA fractions. However, treatment of the plasmid DNA fractions with the kit exonuclease resulted in total digestion (Fig. 5, bottom panel) and loss of all hybridization signals (not shown).
FIG.5.


Attempted VHS1 episome extraction tests. (Top panel) Electrophoresis gels and Southern blots of chromosomal and plasmid DNA fractions from a plasmid midiextraction kit. Lanes 1 and 3 were loaded with 500 ng undigested DNA from each fraction, while lanes 2 and 4 were loaded with all the remaining DNA digested with EcoRI/XbaI. Note the lack of positive hybridization signals with labeled VHS1 in the plasmid fractions. (Bottom panel) Similar to top panel but with a large-construct extraction kit, excluding the exonuclease treatment step and showing the strongest hybridization signal in the plasmid fractions. Lanes 1, 3, and 5 were loaded with 500 ng DNA from each fraction, while lanes 2, 4, and 6 were loaded with all the remaining DNA digested with EcoRI/XbaI. The gel on the far right (lanes 7 to 10) parallels the gel of plasmid fractions on the left (lanes 3 to 6, respectively) except that it was treated with the kit exonuclease prior to running the gel. Note that all DNA in the plasmid fraction was digested by the exonuclease. M, molecular size marker; Ph, restriction enzyme digest of VHS1 DNA (positive control).
Plaque morphology.
Three different results were obtained from the VHS1 dot plaque assays carried out in this work (Fig. 6). A type 0 result occurred with VH1114 that gave clear lysis together with included colonies that arose from lysogenic clones. A type 1 result occurred with putative lysogens (e.g., VHL5, VHL5.12, VHL8, VHL10, etc.) that showed no lysis. A type 2 result (e.g., VHL5.1 to -11, etc.) occurred as turbid plaques that arose with cured subclones from type 2 clones. Subclones that gave a type 2 reaction also showed turbid plaques in overlay assays (Fig. 7). Because subclones giving a type 2 result showed some characteristics of lysogenic clones (i.e., some resistance to lysis by VHS1) but contained no detectable phage DNA (i.e., by PCR or hybridization assays) and produced no VHS1, we refer to them as “false lysogens” or “pseudolysogens.” These could be defined as subclones of lysogens that had lost prophage DNA but retained some phenotypic characteristics of a lysogen. Based on these results, a type 0 dot plaque result was used to define a parental-type clone (PTC), while a type 1 result was used to define a true lysogenic clone (TLC) and type 2 was used to define a pseudolysogenic clone (PLC). These differences are summarized in Table 2.
TABLE 2.
Types of clones and their properties
| Type | Definition | Phage production | Immunity to VHS1 | Molecular probe for VHS1 |
|---|---|---|---|---|
| 0 | Parental-type clone | − | − | − |
| 1 | True lysogenic clone | + | + | + |
| 2 | Pseudolysogenic clone | − | +/− | − |
Stability of subclones.
Detailed tests on PTCs, TLCs, and PLCs are illustrated here using VH1114 as a PTC representative, subclone VHL5.12 as a TLC representative (type 1 result), and subclone VHL5.18 as a PLC representative (type 2 result). Of 20 subclones (VHL5.12.1 to VHL5.12.20) selected from agar plating of TLC representative VHL5.12, almost all (19/20; 95%) had characteristics of a TLC (i.e., positive for VHS1 using molecular probes, refractory to VHS1 lysis, and capable of producing VHS1 at concentrations ranging from 106 to 108 PFU/ml). Only one subclone (VHL5.12.11) had characteristics of a PLC. Further subcloning from two of the TLC subclones gave 17/20 (85%) TLC for one (VHL5.12.2) and 10/20 (50%) TLC for the other (VHL5.12.3), while all the remaining subclones were PLC. Thus, the three TLC subclones produced cured progeny (PLC) at prevalences ranging from 5% to 50%. This was less than the estimated maximum prevalence of cured clones (74%) for VHL5 described above. Further subcloning of PLC subclone VHL5.12.11 gave 20/20 (100%) PLC. Third-generation subclones (20 each) from PLC representatives VHL5.12.2.6, VHL5.12.2.16, and VHL5.12.2.20 all gave PLCs (60/60), as did subclones (20 each) for PLC representatives VHL5.12.3.1 and VHL5.12.3.6 (i.e., 40 PLCs).
Of 20 subclones (VHL5.18.1 to VHL5.18.20) selected from agar plating of PLC representative VHL5.18, all (100%) had characteristics of a PLC; that is, none showed the presence of VHS1 by use of molecular probes, none produced VHS1, and all gave turbid plaques with VHS1. The overall results are summarized in Fig. 8, where it can be seen that TLC clones gave rise to a mixture of TLC and PLC subclones, while PLC clones always gave PLC subclones. The original parental-type clone was never obtained from subclones of TLCs or PLCs. The presence or absence of VHS1 in all TLCs was confirmed by Southern blot analysis of selected isolates as previously described and by dot blot assays of all isolates. Total VH1114 DNA extracts were used as the negative control (Fig. 9). PLC representative clones VHL5.12.2.6, VHL5.12.16, VHL5.12.2.20, VHL5.12.3.1, and VHL5.12.3.6 were reexposed to VHS1 in dot plaque assays. Cells were picked from the turbid plaques and subcloned by streaking on MHA. Out of 100 subclones tested, 16 were TLCs and 84 remained as PLCs. In addition to these assays, 20 randomly selected colonies that arose in dot plaques of VHS1 on VH1114 were subcloned and tested in a similar manner for TLC, PLC, and PTC progeny (data not shown), and the same pattern shown in Fig. 8 was found.
FIG. 8.

Flow diagram illustrating the origin of parental-type clones, true lysogenic clones, and pseudolysogenic clones resulting from sequential exposure of VH1114 and its subclones to VHS1. This was a general phenomenon, since it was observed not only with subclones of VHL5 but also with 20 randomly selected subclones from the centers of VH1114 dot plaques.
FIG. 9.

Example of a DNA dot blot hybridization membrane using digoxigenin-labeled VHS1 DNA to probe various bacterial chromosomal DNA extracts. A1 to A10 and B1 to B10, VH1114 subclones 1 to 20, respectively; C1 to C10 and D1 to D10, VHL5.12.1 to VHL5.12.20, respectively; E1 to E10 and F1 to F10, VHL5.18.1 to VHL5.18.20, respectively; A11, VHS1 (positive control). Note that D1 (VHL5.12.11) was the only VHL5.12 subclone that was negative for VHS1.
Eliminating the possibility of a second phage.
Since many subclones derived from TLCs had characteristics of PLCs, there was a possibility that turbid plaques in the PLC subclones resulted from the presence of a second interacting phage. Several tests were carried out to eliminate this possibility. Tests with mitomycin C as described by Oakey and Owens (13) did not reveal the presence of a prophage in VH1114 or in selected PLCs. In addition, no supernatant from any PLC culture ever gave a plaque when dotted on a lawn of VH1114. Phage purification from supernatant solutions of VHS1-challenged VH1114 or PLC revealed the presence of a single phage resembling VHS1 by electron microscopy with negatively stained samples (data not shown). In addition, DNA extracts of these purified phages digested with EcoRI/XbaI and examined by agarose gel electrophoresis all showed patterns of fragments identical to those for VHS1.
DISCUSSION
Out of a population of VH1114 cells challenged with VHS1, we showed that only a relatively small number of subclones contained VHS1 (i.e., became lysogens). These clones were refractory to lysis by VHS1, and they began to produce bacteriophage particles spontaneously from the late exponential phase of growth. However, during early exponential growth, they did not produce phage particles. On the other hand, earlier work showed that they did produce a higher quantity of extracellular protein with a profile different from that of the parental clone VH1114, and we have shown that they are more virulent to shrimp (18). Clearly, VHS1 and VH1114 can exist in a balanced lysogenic state that results in a more virulent phenotype for shrimp. Although phage production from the late exponential phase is spontaneous, this does not imply an absence of lytic triggers that could be related to host physiology or to changes in a culture medium or a shrimp pond.
Positive hybridization of the VHS1 DNA probe to digested total DNA extracts of TLC clones clearly revealed the presence of VHS1 phage DNA. The fact that restriction fragment lengths were identical to those obtained with purified VHS1 DNA (i.e., no band shifts) suggested that VHS1 existed in the host as a free, plasmid-like episome. Since the cells used for total DNA extraction were derived from early-exponential-phase cells, prior to mature virion production, and since the cells were washed thoroughly before DNA extraction, it is not likely that the positive Southern blot hybridization result for VHS1 in whole DNA extracts arose from VHS1 viral particles attached to the outside of the bacterial cells.
The banding pattern in Southern blots of digested total DNA extracted from VHL clones (Fig. 4) suggested that they carried VHS1 as an episome. This was supported by partial separation of VHS1 DNA from bacterial chromosomal DNA by using a large-construct extraction kit (Fig. 5, bottom panel) and a positive VHS1 PCR only for the plasmid fraction. However, the contrast between the large amount of DNA in the gel and the low Southern blot hybridization signal (Fig. 5, bottom panel) indicated that a large quantity of other, nonhybridizing DNA (perhaps contaminating chromosomal DNA or DNA from other large constructs) was also present in the plasmid fraction. Curiously, all DNA in the plasmid fraction was digested by the kit exonuclease, suggesting that it contained no circular DNA. In addition, the restriction enzyme digest gave a smear rather than the characteristic VHS1 banding pattern. Together, the data suggested that VHS1 is present in its host as a large, fragile episome that is easily nicked and degraded during attempted plasmid isolations. The lack of plasmid bands in agarose gels of extracts from miniplasmid extraction kits (not shown) and midiplasmid extraction kits (Fig. 5, top panel) indicated that small plasmid constructs were not present in VH1114 and its subclones.
To understand the nature of bacteriophage-induced virulence, a good model system is needed, and a basic requirement is relatively stable lysogenic clones that can be produced and tested in experimental challenges with shrimp. The VHS1/VH1114 model partially satisfies these requirements, in that true lysogenic clones can be produced and easily isolated from the centers of dot plaques even though they occur in relatively small proportions in VHS1-challenged cultures. A disadvantage is that individual TLCs vary widely in stability and constantly generate large numbers (up to 50% or more) of cured clones that have lost their lysogenic status (i.e., PLCs). This drawback can be overcome by the careful selection of lysogenic subclones from streaks by using lysis resistance and PCR tests to verify TLC status before every experiment. Verified subclones can be cryopreserved in aliquots for later amplification and immediate use in tests or assays related to shrimp. However, it is important to recall that freezing and thawing of V. harveyi at −80°C may lead to loss of viability (22), so repeated freezing and thawing of a stock is not recommended. The instability of TLCs parallels the situation for some pathogenic field isolates of V. harveyi that rapidly lose their virulence upon serial subculture in the laboratory (19). It is possible that the phenomenon results from the loss of mobile genetic elements such as prophages.
The instability of TLCs and the rapid accumulation of high proportions of cured cells can also explain the failure of some of our early experiments to reveal the presence of VHS1 DNA by Southern blotting in serially subcultured stocks that produced VHS1 and were positive for it by PCR. These subcultures gave strong positive Southern blot results immediately after initial clonal isolation but not after serial subculture in the laboratory by broth transfer. Once the process of curing was understood, it was realized that the proportion of lysogenized cells in relation to cured cells in such broth cultures could drop rapidly. The problem was solved by single-colony selection combined with verification of lysogenic status by dot plaque assay before each Southern blot test. Since PCR assays are extremely sensitive, they can detect very minute quantities of VHS1 DNA even from a small portion of lysogenic cells in a mixed population dominated by cured cells. In such cases, the amount of viral DNA present would be small relative to the amount of host DNA and insufficient for its detection in Southern blots. In the future, those working with newly isolated phage-bacterial partners of a similar nature should be aware that lysogens may be lost easily if appropriate precautions are not taken during serial subculture.
A startling aspect of our work was the occurrence of cured clones of VHS1 lysogens that were characterized by loss of VHS1 genomic DNA but by retention of partial resistance to lysis as evident in the form of turbid dot plaques and overlay plaques. As far as could be determined, representatives of these clones were free of VHS1 and other bacteriophages. We have called these bacteria “pseudolysogens,” and our definition of the term differs from that used by others (9, 22). According to Los et al. (9), pseudolysogeny is characterized by halted but potentially productive phage development in nongrowing infected cells, where the phage “waits” until general environmental conditions are more favorable, while true lysogeny is characterized by a stably maintained prophage capable of causing host cell lysis only after specific environmental triggers. However, Paul and Jiang (16) proposed additional pseudolysogenic states, including those where bacteria produce phages continuously in culture in the same manner as VHS1. In our opinion, using the term “pseudolysogen” to describe quiescent bacteriophages or unstable lysogens such as VHS1 presents problems. For example, how can it be decided, from a continuous spectrum of potential bacterial phenotypes, what would be the point of separation between a lysogen and a pseudolysogen? By contrast, our definition is precise and leaves no ambiguity. It refers specifically to bacterial clones that have undergone transient lysogeny and retain some phenotypic characteristics of the lysogenic state despite the loss of functional prophage. Obviously, it would not be possible to apply the term without knowing the steps in derivation of the clone.
We are left with the question as to how pseudolysogens as we define them might arise. Although our PCR and Southern blot assays of VHS1 pseudolysogens revealed that prophage was not present, it was not possible to exclude the possibility that some small remnant of phage DNA may have been present that was too small in quantity to be detected in Southern blots and was devoid of the target sequence for our PCR primers. In addition, our experiments do not exclude the possibility of some kind of interaction, such as transient integration, crossover exchange, etc., between the host and prophage DNAs. Such interactions could leave very small changes (e.g., insertions or deletions) in the host chromosome that could alter gene expression but be very difficult to discover without detailed and sophisticated molecular analysis. The fact that liquid cultures of TLC subclones varied in the quantity of phage they produced and in the numbers of PLCs produced suggested the possibility of genetically regulated variation in the VHS1-host interaction. Whatever the nature of this interaction, it can clearly also produce stable PLCs of altered phenotype that do not contain detectable prophage. As far as we know, this phenomenon has not previously been reported for an episomal phage, but it may have important implications for environmental and medical bacteriology, if it is a common phenomenon.
In conclusion, the VHS1/VH1114 model has promise for studying the phenomenon of phage-mediated virulence in V. harveyi. With appropriate subcloning precautions, relatively stable TLCs can be maintained and the basis for the difference in virulence between TLCs and PTCs (18) can be examined in detail. In addition, the newly discovered PLCs can be tested for comparative virulence and examined in detail to discover the genetic basis for pseudolysogeny.
Supplementary Material
Acknowledgments
We thank the Thai National Center for Genetic Engineering and Biotechnology (BIOTEC) and Mahidol University for support to carry out this research. Thanks also go to the Asian Development Bank Program for Graduate Study and Research on Agricultural Biotechnology for support to Krit Khemayan and Orapim Puiprom.
We also thank Willem Stevens for valuable comments on the manuscript.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Boyd, E. F., and H. Brussow. 2002. Common themes among bacteriophage-encoded virulence factors and diversity among the bacteriophages involved. Trends Microbiol. 10:521-529. [DOI] [PubMed] [Google Scholar]
- 2.Boyd, E. F., B. M. Davis, and B. Hochhut. 2001. Bacteriophage-bacteriophage interactions in the evolution of pathogenic bacteria. Trends Microbiol. 9:137-144. [DOI] [PubMed] [Google Scholar]
- 3.Cameron, A. 2002. Survey toolbox for aquatic animal diseases; a practical manual and software package. Australian Centre for International Agricultural Research, Canberra, Australia.
- 4.Faruque, S. M., M. J. Albert, and J. J. Mekalanos. 1998. Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol. Mol. Biol. Rev. 62:1301-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris, L., and L. Owens. 1999. Production of exotoxins by two luminous Vibrio harveyi strains known to be primary pathogens of Penaeus monodon larvae. Dis. Aquat. Org. 38:11-22. [Google Scholar]
- 6.Lavilla-Pitogo, C. R., M. C. L. Baticados, E. R. Cruz-Lacierda, and R. de la Pena. 1990. Occurence of luminous bacterial disease of Penaeus monodon larvae in the Philippines. Aquaculture 91:1-13. [Google Scholar]
- 7.Liu, P. C., and K. K. Lee. 1999. Cysteine protease is a major exotoxin of pathogenic luminous Vibrio harveyi in the tiger prawn, Penaeus monodon. Lett. Appl. Microbiol. 28:428-430. [DOI] [PubMed] [Google Scholar]
- 8.Liu, P. C., K. K. Lee, C. C. Tu, and S. N. Chen. 1997. Purification and characterization of a cysteine protease produced by pathogenic luminous Vibrio harveyi. Curr. Microbiol. 35:32-39. [DOI] [PubMed] [Google Scholar]
- 9.Los, M., G. Wegrzyn, and P. Neubauer. 2003. A role for bacteriophage T4 rI gene function in the control of phage development during pseudolysogeny and in slowly growing host cells. Res. Microbiol. 154:547-552. [DOI] [PubMed] [Google Scholar]
- 10.Muir, P. 1991. Factors affecting the survival of penaeid prawns in culture with particular reference to the larval stages. James Cook University of North Queensland, Townsville, Australia.
- 11.Munro, J., J. Oakey, E. Bromage, and L. Owens. 2003. Experimental bacteriophage-mediated virulence in strains of Vibrio harveyi. Dis. Aquat. Org. 54:187-194. [DOI] [PubMed] [Google Scholar]
- 12.Oakey, H. J., W. R. Cullen, and L. Owens. 2002. The complete nucleotide sequence of the Vibrio harveyi bacteriophage VHML. J. Appl. Microbiol. 93:1089-1098. [DOI] [PubMed] [Google Scholar]
- 13.Oakey, H. J., and L. Owens. 2000. A new bacteriophage, VHML, isolated from a toxin-producing strain of Vibrio harveyi in tropical Australia. J. Appl. Microbiol. 89:702-709. [DOI] [PubMed] [Google Scholar]
- 14.Pasharawipas, T., S. Sriurairatana, S. Direkbusarakom, Y. Donayadol, S. Thaikua, L. Ruangpan, and T. W. Flegel. 1998. Luminous Vibrio harveyi associated with tea brown gill syndrome in black tiger shrimp, p. 213-216. In T. W. Flegel (ed.), Advances in shrimp biotechnology. National Center for Genetic Engineering and Biotechnology, Bangkok, Thailand.
- 15.Pasharawipas, T., S. Thaikua, S. Sriurairatana, L. Ruangpan, S. Direkbusarakum, J. Manopvisetcharean, and T. W. Flegel. 2005. Partial characterization of a novel bacteriophage of Vibrio harveyi isolated from shrimp culture ponds in Thailand. Virus Res. 114:63-69. [DOI] [PubMed] [Google Scholar]
- 16.Paul, J. H., and S. C. Jiang. 2001. Lysogeny and transduction, p. 105-125. In J. H. Paul (ed.), Marine microbiology, vol. 30. Academic Press, San Diego, Calif. [Google Scholar]
- 17.Pizzutto, M., and R. G. Hirst. 1995. Classification of isolates of Vibrio harveyi virulent to Penaeus monodon larvae by protein profile analysis and M13 DNA fingerprinting. Dis. Aquat. Org. 21:61-68. [Google Scholar]
- 18.Puiprom, O. 2003. Characterization of a bacteriophage VHS1 that increases the virulence of Vibrio harveyi to shrimp. MSc. thesis. Mahidol Univesity, Bangkok, Thailand.
- 19.Ruangpan, L., Y. Danayadol, S. Direkbusarakom, S. Sriurairatana, and T. W. Flegel. 1999. Lethal toxicity of Vibrioharveyi to cultivated Penaeus monodon induced by a bacteriophage. Dis. Aquat. Org. 35:195-201. [Google Scholar]
- 20.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 21.Van Regenmortel, M. H. V., C. M. Fauquet, D. H. L. Bishop, E. B. Carstens, M. K. Estes, S. M. Lemon, J. Maniloff, M. A. Mayo, D. J. McGeoch, C. R. Pringle, and R. B. Wickner. 2000. Virus taxonomy: seventh report of the international committee on taxonomy of viruses. Academic Press, New York, N.Y.
- 22.Williamson, S. J., M. R. Mclaughlin, and J. H. Paul. 2001. Interaction of the FHSIC virus with its host: lysogeny or pseudolysogeny? Appl. Environ. Microbiol. 67:1682-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.