

Abstract
Efficient control of Xanthomonas axonopodis pv. dieffenbachiae, the causal agent of anthurium bacterial blight, requires a sensitive and reliable diagnostic tool. A nested PCR test was developed from a sequence-characterized amplified region marker identified by randomly amplified polymorphic DNA PCR for the detection of X. axonopodis pv. dieffenbachiae. Serological and pathogenicity tests were performed concurrently with the nested PCR test with a large collection of X. axonopodis pv. dieffenbachiae strains that were isolated worldwide and are pathogenic to anthurium and/or other aroids. The internal primer pair directed amplification of the expected product (785 bp) for all 70 X. axonopodis pv. dieffenbachiae strains pathogenic to anthurium tested and for isolates originating from syngonium and not pathogenic to anthurium. This finding is consistent with previous studies which indicated that there is a high level of relatedness between strains from anthurium and strains from syngonium. Strains originating from the two host genera can be distinguished by restriction analysis of the amplification product. No amplification product was obtained with 98 strains of unrelated phytopathogenic bacteria or saprophytic bacteria from the anthurium phyllosphere, except for a weak signal obtained for one X. axonopodis pv. allii strain. Nevertheless, restriction enzyme analysis permitted the two pathovars to be distinguished. The detection threshold obtained with pure cultures or plant extracts (103 CFU ml−1) allowed detection of the pathogen from symptomless contaminated plants. This test could be a useful diagnostic tool for screening propagation stock plant material and for monitoring international movement of X. axonopodis pv. dieffenbachiae.
Anthurium (Anthurium andreanum Linden ex André), the second largest crop in the world among tropical flowers (the value of world trade was more than $20 million in 2002 [http://www.tradenetsl.lk/Anthu/anthuriums.html]), is cultivated throughout the tropics, as well as in temperate areas. Production of anthurium in the world is threatened by anthurium bacterial blight (ABB), which is caused by Xanthomonas axonopodis pv. dieffenbachiae. ABB was first described in Hawaii (18) and subsequently has been found in the different producing regions, including Venezuela (16), Brazil (9), California (10), the Caribbean (34), the Philippines (24), Florida (29), Tahiti (23), and, more recently, Réunion Island (36) and Turkey (4). It has also been found sporadically in The Netherlands (the largest producer in the world) and Italy (35, 40). First described as a pathogen of Dieffenbachia species, X. axonopodis pv. dieffenbachiae can infect a broad range of plants in the family Araceae (aroids), including species and cultivars of Aglaonema, Alocasia, Anthurium, Caladium, Syngonium, and Xanthosoma (8, 25). The phenotypic and genetic diversity of strains originating from several aroids is related to geographic origin and host plant (5, 6, 8, 15). Pathogenicity for dieffenbachia is a characteristic shared by all X. axonopodis pv. dieffenbachiae isolates (21). Strains of X. axonopodis pv. dieffenbachiae isolated from anthurium can infect other aroids (8, 21). Strains isolated from syngonium have a narrow host range, and classification of these organisms as X. axonopodis pv. syngonii has been suggested; however, this is a controversial issue (8, 11, 19). In this work we refer to these strains as X. axonopodis pv. dieffenbachiae isolated from syngonium.
In the absence of effective chemical control, the management strategies for ABB consist principally of prevention, sanitation, and the use of axenically propagated plants. Diagnosis of X. axonopodis pv. dieffenbachiae infection is currently based on isolation of the pathogen, followed by biochemical identification, pathogenicity tests, or serological tests (7, 21, 25). Such tests require one to several weeks before final confirmation is obtained. The sensitivity of the enzyme-linked immunosorbent assay (ELISA) (around 106 CFU ml−1) is adequate only for detection in symptomatic plants (1). Enrichment of target bacteria on semiselective media before ELISA improves the sensitivity, but it is time-consuming (5 days) and the sensitivity can be affected by other bacteria present in the sample (26). Therefore, there is a need for rapid and sensitive methods for routinely indexing propagation stock and asymptomatic plant material in nurseries.
X. axonopodis pv. dieffenbachiae is considered a quarantine organism in the European Union (it is on the A2 list of the European Plant Protection Organization) and in major anthurium-producing countries that are still free of the pathogen (e.g., Mauritius, whose trade amounted to approximately $4 million in 1999 [http://ncb.intnet.mu/mida/mepzanth.htm]). This makes the availability of highly specific and sensitive PCR-based diagnostic tools developed for X. axonopodis pv. dieffenbachiae a priority. PCR-based techniques have been reported to be highly efficient for detecting and identifying xanthomonads, such as X. axonopodis pv. citri (17) or X. fragariae (31), from plant material. In nested PCR protocols, the sensitivity and specificity of detection are enhanced by performing a second round of PCR using primers internal to the first amplification product (17). In this study, a randomly amplified polymorphic DNA (RAPD)-based PCR technique was used to identify DNA fragments that were putatively specific to X. axonopodis pv. dieffenbachiae. These fragments were characterized further as sequence-characterized amplified region (SCAR) markers, a technique which has been used successfully to design specific primers for many bacterial taxa, including xanthomonads (22, 31, 37, 38). One of the SCARs was used for development of a nested PCR protocol in order to detect the pathogen in both pure culture and anthurium tissue extracts. The nested PCR was specifically developed for X. axonopodis pv. dieffenbachiae that is isolated from anthurium and is capable of infecting this plant genus. Nevertheless, X. axonopodis pv. dieffenbachiae strains isolated from a wide range of aroids and geographical regions were included in this study since strains isolated from anthurium are known to be pathogenic to a wide range of hosts.
MATERIALS AND METHODS
Bacterial strains, culture conditions, and DNA extraction.
Bacterial strains used in this study were obtained from international collections or were isolated from aroids grown on Réunion Island (see Tables SA to SC in the supplemental material). Strains were stored at −80°C on beads in cryovials (Microbank Prolab Diagnostics, Austin, TX) and also were freeze-dried for a long-term storage. The epiphytic bacteria isolated from anthurium (see Table SC in the supplemental material) were identified by Gram staining, classical biochemical tests (glucose oxidation or fermentation, oxidase reaction), API20E or API20NE strips (BioMérieux, France), and fatty acid methyl ester analysis (Microbial ID, Inc. Newark, DE). Cultures were routinely grown at 28°C on YPGA medium (7 g liter−1 yeast extract, 7 g liter−1 peptone, 7 g liter−1 glucose, 18 g liter−1 agar; pH 7.2). Twenty-four-hour-old bacterial cultures were used for PCR assays and inoculation tests. Isolation and enumeration of X. axonopodis pv. dieffenbachiae colonies from plant extracts were performed on the semiselective cellobiose-starch (CS) medium (25).
For DNA purification, bacteria were cultivated in liquid medium (7 g liter−1 yeast extract, 7 g liter−1 peptone; pH 7.2) for 16 h at 28°C with agitation. Genomic DNA was then extracted by the cetyltrimethylammonium bromide method (3). DNA concentrations were estimated by fluorometry (TKO 100 fluorometer; Hoefer, San Francisco, CA).
Pathogenicity tests.
All strains of X. axonopodis pv. dieffenbachiae were inoculated onto the hosts from which they were obtained (Anthurium andreanum ‘Carré’, Aglaonema commutatum ‘Sword Queen’, Alocasia wentii, Caladium candidum, Dieffenbachia maculata ‘Tropic Marianne’, Syngonium podophyllum ‘Robusta’, Xanthosoma lindenii). All strains were inoculated onto anthurium and, when appropriate, onto dieffenbachia and syngonium (see Table SA in the supplemental material). All tests were performed with 18-month-old plants as previously described (21) by using bacterial suspensions containing 105 CFU ml−1. Plants were maintained at a relative humidity of 95% ± 5% in a growth chamber with day and night temperatures of 30 ± 1°C and 26 ± 1°C, respectively. The light intensity was 30 μmol m−2 s−1 with a photoperiod of 12 h. These conditions are optimal for disease development in a susceptible cultivar (12). Inoculated plants were examined three times per week for development of symptoms (water soaking, chlorosis, and necrosis) for 2 months.
Serological tests.
Strains were characterized by an indirect ELISA using the Xcd108 monoclonal antibody (MAb) (Agdia Inc., Elkart, IN) raised against X. axonopodis pv. dieffenbachiae (28). Bacterial antigen suspensions were prepared as described previously (2), and the concentrations were adjusted spectrophotometrically to 108 CFU ml−1 in carbonate buffer (1.59 g liter−1 Na2CO3, 2.93 g liter−1 NaHCO3; pH 9.6). The ELISA procedure was performed according to the recommendations of Agdia Inc., except for coating (microtitration plates coated with the bacterial suspensions were maintained at 4°C for 12 h prior to the next step).
PCRs. (i) RAPD-PCR.
Total DNA from five strains (see Table SA in the supplemental material) of the pathogen isolated from various geographical locations was used. Amplification was carried out in 25-μl mixtures that contained 25 ng of bacterial genomic DNA, 3 mM MgCl2, 0.4 μM primer, 2.5 U of Taq DNA polymerase (Invitrogen, Merelbeke, Belgium), and each deoxynucleoside triphosphate (dNTP) (Roche Diagnostics France SA, Meylan, France) at a concentration of 100 μM in 20 mM Tris-HCl-50 mM KCl buffer (pH 8.4). The following amplification conditions were used: initial denaturation at 94°C for 7 min, followed by 40 cycles of denaturation at 94°C for 1 min, annealing at 35°C for 1 min, and extension at 72°C for 2 min and a final extension step of 72°C for 5 min in a GeneAmp PCR system 9700 (Applied Biosystems, Courtaboeuf, France). One hundred arbitrary primers consisting of 10-base oligonucleotides from kits 80, 70, and 60 purchased from Sigma Genosys (Saint-Quentin, France) were tested. PCR amplification products were detected by electrophoresis in 2% NuSieve agarose (FMC Bioproducts, Rockland, ME) and were stained with ethidium bromide. Amplification products produced with primer 70-25 (5′-GCACCGAACG-3′) from a large number of X. axonopodis pv. dieffenbachiae strains and other xanthomonads (see Tables SA and SB in the supplemental material) were examined.
(ii) Nested PCR.
X. axonopodis pv. dieffenbachiae-specific nested PCR was performed with different templates, including purified bacterial genomic DNA (1 ng μl−1) and suspensions from pure bacterial cultures (a single colony in 1 ml of deionized water, boiled for 1 min). Plant extracts used as templates for PCR were prepared as described below.
Primers were selected from the SCAR sequence of X. axonopodis pv. dieffenbachiae strain LMG 695. They were designed with the OLIGO 5.1 software (National Biosciences, 1999) and were synthesized by Sequentia S.A. (Evry, France). The primers used for the first round of amplification were PXadU (5′-AGGGCTCCCCATGCCGGAAT-3′) and PXadL (5′-ACGCAATGCGCAGGGGAAAT-3′), which complemented bases 12 to 32 and bases 1562 to 1582, respectively, of the SCAR sequence. The amplification program included denaturation at 94°C for 3 min, 35 cycles consisting of 94°C for 30 s, 70°C for 30 s, and 72°C for 2 min, and an extra extension step at 72°C for 10 min. The primers used in the second round were NXadU (5′-AGCGCGGTACATTGTTGTTCGT-3′) and NXadL (5′-GCGGATCCTGACTGAGCAAAG-3′), which complemented bases 629 to 651 and 1393 to 1413, respectively, of the SCAR sequence. For the second round of PCR, 1 μl from the first reaction mixture was used as the template and the amplification program consisted of denaturation at 94°C for 3 min, 20 cycles of 94°C for 30 s, 70°C for 30 s, and 72°C for 30 s, and a final extension at 72°C for 5 min.
PCRs were performed in 25-μl reaction mixtures containing 1.1 mM MgCl2, each dNTP at a concentration of 100 μM, each primer (PXadU and PXadL or NXadU and NXadL) at a concentration of 0.2 μM, 1 μl template DNA or 2 μl of plant extract, and 1 U Taq polymerase (Invitrogen, Merelbeke, Belgium) in 20 mM Tris-HCl-50 mM KCl buffer (pH 8.4).
Amplicons from the nested PCRs obtained for five X. axonopodis strains (see Tables SA and SB in the supplemental material) were digested with 18 restriction endonucleases, including EcoRI, HincII, AflIII, MseI, SacI, BspMI, BstUI, Sau96I, and HaeII (New England Biolabs, OZYME, St. Quentin en Yvelines, France), used according to the conditions specified by the manufacturer.
Fragment extraction from agarose gels, cloning, and sequencing.
SCARs were cut from the RAPD fingerprints of two strains (see Table SA in the supplemental material) after electrophoresis in an agarose gel. The DNA fragments were cleaned using a QIAquick gel extraction kit (QIAGEN S. A., France) and reamplified by using the PCR amplification program described above for RAPD analysis with an elongation temperature of 68°C instead of 72°C. The reaction mixtures (25 μl) contained 4 μl of template DNA, 0.4 μM primer, 0.9 U of Expand Long Template polymerase (Roche Diagnostics France SA, Meylan, France), and each dNTP at a concentration of 100 μM. The amplified fragments were cloned by direct ligation into the pGEM-T Easy vector as described by the manufacturer (Promega Corp., Madison, WI). Cloned fragments were sequenced by Genome Express S.A. (Meylan, France) (single-pass double-stranded analysis).
Southern blot hybridization. (i) Specificity of the 70-25 SCAR.
DNAs from nine X. axonopodis pv. dieffenbachiae isolates from anthurium (see Table SA in the supplemental material) and 17 other Xanthomonas strains (see Table SB in the supplemental material) were digested with EcoRI according to the supplier's instructions (New England BioLabs, OZYME, St. Quentin en Yvelines, France). Separation of DNA fragments and gel blotting were performed as described previously (14). The 1.6-kb SCAR70-25 fragment amplified from LMG 695 was used as the probe. Probe labeling, hybridization, and detection were performed with an ECL kit (Amersham Pharmacia Biotech Inc., Piscataway, NJ) used as recommended by the manufacturer.
(ii) Determination of the copy number of SCAR70-25.
The labeled purified 1,570-bp DNA fragment amplified after the first round of the nested PCR from LMG 695 was used as a probe against the DNA of eight X. axonopodis pv. dieffenbachiae strains obtained from different countries (see Table SA in the supplemental material) digested separately with EcoRI, EcoRV, HindIII, KpnI, or SmaI according to the supplier's instructions (New England BioLabs, OZYME, St. Quentin en Yvelines, France). Fragment separation, gel blotting, hybridization, and detection were performed as described above.
In planta detection of X. axonopodis pv. dieffenbachiae. (i) Neutralization of PCR-inhibiting plant substances.
Pieces (approximately 2 by 2 cm) cut from healthy anthurium leaves were placed in a blender (Waring Blendor, New Hartford, CT) containing 10 mM Tris buffer (pH 7.2) (20 ml g of leaf material−1) and homogenized for 1 min. After maceration for 30 min at room temperature, one of the following buffers was added (1:1) to plant extract aliquots: 10 mM Tris buffer pH 7.2 (control), PP buffer (8.5 mM K2HPO4-7.5 mM KH2PO4 [pH 7.0] with 2% or 5% polyvinylpyrrolidone [PVP] [Sigma]), PPP buffer (8.5 mM K2HPO4-7.5 mM KH2PO4 [pH 7.0] with 2% or 5% polyvinylpolypyrrolidone [Sigma]), TENP buffer (50 mM Tris-20 mM EDTA [pH 8.0]-100 mM NaCl with 2% or 5% PVP), or TENPP buffer (50 mM Tris-20 mM EDTA [pH 8.0]-100 mM NaCl with 2% or 5% polyvinylpolypyrrolidone) (32). Suspensions of strain LMG 695 were then added to each plant mixture to obtain a final concentration of 106 CFU ml−1. The mixtures were boiled for 1 min, and 2-μl aliquots were used as templates for nested PCR. Bacterial suspensions (106 CFU ml−1) in Tris buffer were used as a positive control, and X. axonopodis pv. dieffenbachiae-free plant extracts were used as a negative control.
(ii) Detection of X. axonopodis pv. dieffenbachiae on spray-infected anthuriums.
Seventy-two anthurium plants were inoculated as described previously (13) with an inoculum consisting of suspensions in 10 mM Tris buffer (pH 7.2) prepared from strain LMG 695 and containing 107 CFU ml−1. A control set of 36 plants remained intact during the experiment and was used for daily visual assessment of disease. Plants from the second set were sampled every 2 days as follows. Three inoculated leaves were collected from each of two randomly chosen plants. The leaves were surface sterilized by briefly wiping them with 70% (vol/vol) ethanol, and the entire perimeter was removed (width, 1.5 cm) and cut into pieces (length, 6 cm). The number of sections tested varied depending on the circumference of the leaf. The average number of sections was 30.6 ± 4.4. The sections were homogenized as described above in 20 ml g−1 of 10 mM Tris buffer (pH 7.2). After maceration for 30 min at room temperature, an equal volume of 5% PP buffer was added to the macerated material. The suspensions were boiled, and 2 μl was used for nested PCR. Prior to addition of PP buffer, 50 μl was plated in duplicate on YPGA and CS agar plates with a spiral device (Interscience, Saint Nom la Bretèche, France) (20) to measure the concentration of culturable bacteria.
Sensitivity of detection.
The sensitivity of the nested PCR was determined for both pure cultures and plant extracts. Bacteria (LMG 695) from an overnight culture on YPGA medium plates were suspended and serially 10-fold diluted in 10 mM Tris buffer (pH 7.2). Fifty microliters of each sample was plated in duplicate on CS and YPGA medium plates with a spiral device (Interscience, Saint Nom la Bretèche, France) (20) to obtain direct data for the culturable population sizes added to each PCR mixture. After boiling, 1 μl of a serially diluted bacterial suspension was added to each PCR. For determination of the sensitivity in planta, the same procedure was performed with the same dilution series added to plant extracts treated with 5% PP buffer.
Nucleotide sequence accession number.
The DNA sequence of SCAR70-25 from strain LMG 695 has been deposited in the GenBank database under accession number DQ096647.
RESULTS
Identity of X. axonopodis pv. dieffenbachiae strains as determined by serological and pathogenicity tests.
Most strains were pathogenic to the host from which they were obtained (see Table SA in the supplemental material). The strains were divided into four groups based on serological and pathogenicity responses. Group I contained strains that were pathogenic to anthurium and reacted with MAb Xcd108. Group II contained three strains originating from syngonium that reacted with MAb Xcd108 and were pathogenic to syngonium but not to anthurium. Group III contained strains isolated from other aroid genera that tested negative with MAb Xcd108 and were pathogenic to the host from which they were obtained but not to anthurium. Group IV contained a few strains collected from anthurium that were not pathogenic to this host and did not react with MAb Xcd108; nevertheless, these strains were mildly pathogenic to Dieffenbachia maculata ‘Tropic Marianne,’ forming localized water-soaked lesions that became chlorotic and then necrotic in 3 weeks and multiplying in leaf tissues by approximately 3 logarithmic units over 21 days. Of the strains of unrelated phytopathogenic or saprophytic bacteria (see Tables SB and SC in the supplemental material), only strain CFBP 6380 (X. axonopodis pv. allii from onion) reacted with MAb Xcd108. No other X. axonopodis pv. allii strain (n = 7) exhibited this characteristic (data not shown). No lesions occurred on anthurium after inoculation of CFBP 6380. Mild symptoms were obtained when X. axonopodis pv. allii strain CFBP 6380 was inoculated onto dieffenbachia plants. The lesions, which were clearly different from a hypersensitive reaction (like the reaction observed when other xanthomonads are inoculated onto dieffenbachia), started as water-soaked areas around the infiltration sites 6 days after inoculation and became chlorotic zones and then necrotic zones 3 weeks after inoculation. CFBP 6380 multiplied in these lesions approximately 3 logarithmic units over 21 days. Nevertheless, CFBP 6380 did not become systemic compared to a positive control that was inoculated at the same time with LMG 695, in which secondary symptoms were observed along the veins.
RAPD analysis and selection of a DNA fragment specific to X. axonopodis pv. dieffenbachiae.
Similar reproducible PCR banding patterns were obtained with several primers for all five strains tested. When primer 70-25 was used, all X. axonopodis pv. dieffenbachiae strains pathogenic to anthurium exhibited a major 1.6-kb amplification product which was absent in the other Xanthomonas species tested (Fig. 1). When this amplification product was used as a hybridization probe against total DNA of several X. axonopodis pv. dieffenbachiae strains and other bacterial strains digested with EcoRI, bands at 2.4 kb and 4.2 kb were obtained for X. axonopodis pv. dieffenbachiae DNA. No hybridization (or a faint band at approximately 3.5 kb for CFBP 4643 and CFBP 2157) occurred with other xanthomonads and with saprophytic strains isolated from the anthurium phyllosphere. The 1.6-kb fragment (SCAR70-25) was cloned and sequenced for strains LMG 695 and JV589. Very similar 1,588-bp DNA sequences were obtained for the two strains (99.62% identity). Restriction fragment length polymorphism (RFLP) analysis using restriction enzymes with no cleavage site in SCAR70-25 suggested that a single copy of this fragment is probably present in the genome of X. axonopodis pv. dieffenbachiae from anthurium.
FIG. 1.
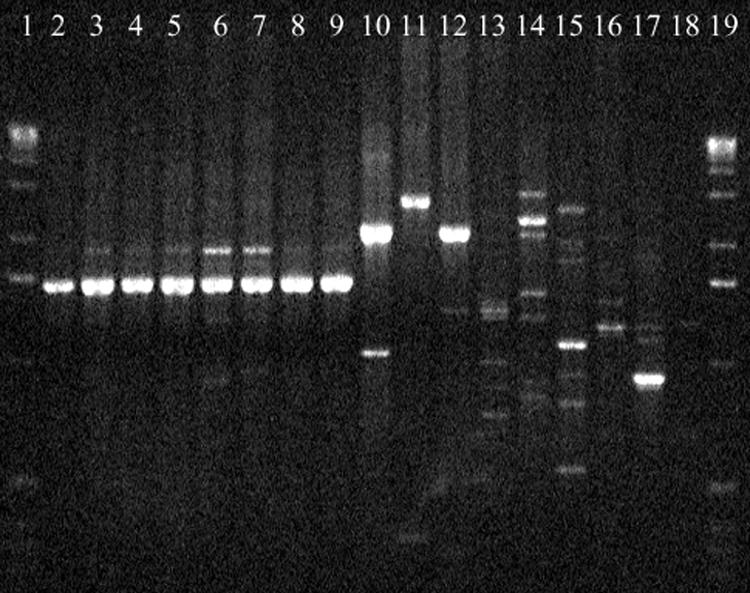
RAPD patterns of different Xanthomonas strains generated with primer 70-25. Lanes 1 and 19, 1-kb ladder (Gibco BRL); lanes 2 to 9, strains of X. axonopodis pv. dieffenbachiae from anthurium (LMG 12738, LMG 12739, LMG 12734, LMG 12724, LMG 12741, LMG 12743, LMG 695, and JS1007); lanes 10 to 17, strains belonging to several Xanthomonas species (X. axonopodis pv. vesicatoria JQ725, Xanthomonas sp. pathovar mangiferaeindicae JP740, X. axonopodis pv. allii CFBP 6364, X. hyacinthi CFBP 1156, X. fragariae CFBP 2157, X. arboricola pv. juglandis CFBP 2528, X. cucurbitae CFBP 2542, X. vasicola pv. holcicola CFBP 2543); lane 18, control reaction without DNA.
Specificity and sensitivity of the nested PCR.
An amplicon that was the expected size (1,570 bp) was observed after the first round of amplification with the PXadU and PXadL primers for all 69 strains of X. axonopodis pv. dieffenbachiae isolated from anthurium (group I). In the second round of PCR (nested PCR), primers NXadL and NXadU directed amplification of a 785-bp product for all these strains. Identical amplification products (1,570 bp and 785 bp for the first and second rounds of nested PCR, respectively) were observed for four X. axonopodis pv. dieffenbachiae strains isolated from other aroid genera, including Caladium (strain JX31; group I) and Syngonium (strains JW148, JW188, and LMG 9055; group II). Strains originating from syngonium can be distinguished from strains originating from anthurium on the basis of restriction analysis of the amplification product of the second PCR round, using one of the following endonucleases: HincII, BspMI, Sau96I, and MseI. Interestingly, HincII and Sau96I did not cleave the amplification product generated from strains isolated from syngonium, while for strains isolated from anthurium two fragments (460 and 325 bp) and three fragments (385, 219, and 181 bp), respectively, were obtained. These differences were due to point mutations resulting in 98% identity with the amplified fragment from strain LMG 695 (data not shown). No amplification products were observed for group III and IV strains. No amplification was obtained for strains belonging to other bacterial genera, other Xanthomonas species or pathovars, and saprophytic bacteria isolated from anthurium (see Tables SB and SC in the supplemental material), except for X. axonopodis pv. allii strain CFBP 6380. For strain CFBP 6380, a 1,570-bp amplification product was obtained after the first round of nested PCR and weak amplification (785 bp) was observed after the second round in two of four replicates. There were differences in the endonuclease restriction patterns of nested PCR products between CFBP 6380 and strains from anthurium when the following restriction enzymes were used: EcoRI, AflIII, MseI, SacI, BspMI, BstUI, and HaeII. In particular, EcoRI and SacI generated two DNA fragments for strains from anthurium (536 and 249 bp and 222 and 563 bp, respectively), while no cleavage occurred for CFBP 6380. These differences in amplification and restriction were due to several point mutations that resulted in 93% identity with the sequence of the LMG 695 fragment (data not shown). The first round of PCR performed with dilution series of LMG 695 resulted in a detection sensitivity of approximately 106 CFU ml−1, as determined by dilution plating on both CS and YPGA media. The sensitivity was greatly improved with nested PCR, and the detection limit decreased to approximately 103 CFU ml−1 (one viable cell per reaction). A signal was sometimes obtained (two of five replicates) with suspensions containing approximately 102 CFU ml−1.
Detection of X. axonopodis pv. dieffenbachiae in plant extracts.
When cells of strain LMG 695 were added to healthy plant extracts (106 CFU ml−1), no amplification product was obtained even after two rounds of amplification, suggesting that inhibitory substances were present in plant extracts. Conversely, all 10 replicates were positive as determined by the nested PCR assay when PP buffer with 5% PVP was added to the plant extract, as were all 10 replicates of the positive control (bacterial cells in Tris buffer), and the band intensity was similar. Addition of PP buffer with 2% PVP resulted in detection in 8 of 10 replicates, and no or very weak bands were observed for the other buffers (Table 1). The detection threshold obtained in planta using PP buffer with 5% PVP was equivalent to that observed with fresh cultures of X. axonopodis pv. dieffenbachiae.
TABLE 1.
Effects of pretreatments applied to plant extracts on the frequency of detection of X. axonopodis pv. dieffenbachiae by nested PCR
| Treatment | Frequency of nested PCR detection (no. of positive samples/no. of samples tested)a |
|---|---|
| Tris buffer | 0/10 |
| PP buffer (2% PVP) | 8/10 |
| PP buffer (5% PVP) | 10/10 |
| PPP buffer (2% PVPP)b | 1/10c |
| PPP buffer (5% PVPP) | 4/10c |
| TENP buffer (2% PVP) | 1/9c |
| TENP buffer (5% PVP) | 9/10c |
| TENPP buffer (2% PVPP) | 2/10c |
| TENPP buffer (5% PVPP) | 4/10c |
| Positive controld | 10/10 |
Suspensions of X. axonopodis pv. dieffenbachiae LMG 695 were added to samples (final concentration, 106 CFU ml−1) containing healthy plant extracts mixed with different buffers (1:1) and were subjected to boiling and nested PCR as described in the text.
PVPP, polyvinylpolypyrrolidone.
The signals were weak.
LMG 695 suspension in 10 mM Tris buffer (final concentration, 106 CFU ml−1) and no plant extract.
Detection of X. axonopodis pv. dieffenbachiae on spray-infected anthurium: nested PCR, bacterial population sizes, and symptom development.
Although leaves were briefly sterilized, saprophytic bacteria were sometimes recovered on YPGA medium but not on the semiselective medium (CS medium). The plating efficiency on CS medium compared to YPGA medium for strain LMG 695 was 97.7% ± 0.3%. The first symptoms were water-soaked spots that were heterogeneously localized at the leaf margins and sometimes developed around macroscopic wounds. They appeared on anthurium plants 16 to 28 days after inoculation (Fig. 2). Isolation of X. axonopodis pv. dieffenbachiae from leaf margins on agar media was heterogeneous, and positive results were obtained for 7.1 to 58% of the leaf sections assayed. For leaf samples from which X. axonopodis pv. dieffenbachiae was isolated on agar media but not detected by nested PCR, the culturable population sizes were always less than 103 CFU ml−1 except for one sample collected 16 days after inoculation, for which no nested PCR signal was recorded and the culturable population density on YPGA medium was 1.6 × 103 CFU ml−1. Consistent detection of the inoculated strain from 100% of the leaves tested was obtained by nested PCR starting 4 days after inoculation (i.e., 12 days before the earliest lesions occurred in the control set) (Fig. 2). The samples which were positive after the first round of PCR were the samples with the highest population densities (1 × 106 to 1 × 107 CFU ml−1) and for which early symptoms were observed.
FIG. 2.

Evaluation of the sensitivity of nested PCR and the delay between molecular detection and visual development of the disease. Leaf sections of infected anthurium were subjected to population size determination and nested PCR detection as described in the text. The symbols indicate means of the population sizes, and the error bars indicate standard deviations. Symbols: ○, X. axonopodis pv. dieffenbachiae (Xad) population sizes in leaf sections when no detection by nested PCR was observed; ▴, X. axonopodis pv. dieffenbachiae population sizes in leaf sections when there was nested PCR detection only after the second round of amplification; ▪, X. axonopodis pv. dieffenbachiae population sizes in leaf sections when there was nested PCR detection after the first and second rounds of amplification. The temporal progress of disease incidence is indicated by gray columns.
DISCUSSION
In this paper we describe the development of a reliable and sensitive method for the detection of X. axonopodis pv. dieffenbachiae pathogenic to anthurium, a highly destructive pathogen listed as a quarantine organism in the European Union. We developed a nested PCR assay using the sequence from an X. axonopodis pv. dieffenbachiae-specific fragment identified from RAPD-PCR analyses and subsequently characterized. The internal primer pair directed amplification of a 785-bp product that was obtained from all X. axonopodis pv. dieffenbachiae strains pathogenic to anthurium. No amplification product was obtained with bacterial strains not related to X. axonopodis pv. dieffenbachiae except for one strain belonging to X. axonopodis pv. allii. Nevertheless, the presence of restriction sites in X axonopodis pv. dieffenbachiae (EcoRI or SacI) which were absent in the X. axonopodis pv. allii strain can be used to discriminate these strains by RFLP analysis of nested PCR products.
The comparison of SCAR70-25 to sequences present in the GenBank database (BLASTN) revealed little sequence similarity with previously determined sequences. The closest match (72% of a 299-bp region) was obtained for a gene encoding a putative ABC transporter-type protein of Azoarcus sp. Moreover, BLAST searches with the predicted protein sequence (BLASTX) revealed similarity to a putative ABC type 2 transporter hydrophilic component encoded by wzt (67% identity and 82% homology) from the lipopolysaccharide cluster of X. oryzae pv. oryzicola. Very recently, Pagani and Ritchie (30) cloned and sequenced a DNA fragment from X. arboricola pv. pruni which encodes a putative protein exhibiting similarity to the ABC transporter family. This fragment allowed specific detection and identification of this bacterium. This gene family may be particularly suitable for elaboration of molecular detection tools for other xanthomonads.
A few strains isolated from other aroid genera reacted both to MAb Xcd108 and in nested PCR. JX31 is a strain isolated from caladium in Réunion Island which was pathogenic to anthurium. Moreover, this strain displayed the same AFLP pattern as anthurium-pathogenic strains of X. axonopodis pv. dieffenbachiae isolated in Réunion, and this AFLP pattern was closely related to that of strains from other regions that are pathogenic to anthurium (data not shown). These results are consistent with previous data (21) indicating that strains isolated from anthurium can infect (and be reisolated from) a broad range of aroid hosts. Strains isolated from syngonium also reacted both to MAb Xcd108 and in nested PCR but were not pathogenic to anthurium. This confirms results from a previous study which showed that strains from syngonium are serologically closely related to strains from anthurium and pathogenic to dieffenbachia but not to anthurium (21). RFLP analysis of nested PCR products revealed polymorphism at restriction sites which can be useful for strain discrimination (HincII or Sau96I). Most aroid strains which did not originate from anthurium did not produce amplicons with our nested PCR assay. The two populations (the population from anthurium and the population from other aroid genera) have been classified in different genetic groups within X. axonopodis by repetitive extragenic palindromic PCR and AFLP (33) and may represent populations that have distinct phylogenetic origins. A few strains isolated from anthurium and identified as xanthomonads (Biolog) were not pathogenic to this host species and did not respond to molecular and serological tests. They were mildly pathogenic to dieffenbachia when they were inoculated, but they clearly multiplied in dieffenbachia leaf tissue. These strains have characteristics similar to those of strains classified as serotype 11 and 12 strains by Norman and Alvarez (28). According to these authors, these strains, which were isolated from anthurium, are not pathogenic to this host species. Our nested PCR assay did not amplify DNA from these nonpathogenic strains. Thus, nested PCR-RFLP analysis can be used as a powerful screening tool to detect Xanthomonas populations that are virulent to anthurium. This PCR test can be used with plant samples without DNA extraction with a level of sensitivity equal to that obtained with pure bacterial cultures. This successful detection of the bacterium from plants was possible because vegetal PCR inhibitors were overcome by adding polyvinylpyrrolidone. The detection threshold (approximately 107 CFU ml−1) obtained with the first round of PCR (primers PXadU and PXadL) was adequate for detection of X. axonopodis pv. dieffenbachiae strains in heavily infected plant material but was not sensitive enough to detect early or latent infections. This relatively low sensitivity may be explained by the presence of a single copy of the target in the genome of X. axonopodis pv. dieffenbachiae. With a second round of PCR using the NXadL and NXadU internal primers, the detection threshold was lowered to approximately 103 CFU ml−1, which corresponds to one target DNA detected per reaction. This level of sensitivity, similar to the levels obtained in other studies (32, 39), is suitable for detecting the target bacterium in symptomless plants, as was shown with in planta experiments. The target bacterium was detected by PCR from symptomless plant tissue up to 12 days before the development of symptoms for populations sizes as low as 103 CFU ml−1. All experiments were performed with a highly susceptible cultivar (cultivar Carré) under environmental conditions that permit quick growth of X. axonopodis pv. dieffenbachiae and symptom development. In our study, the latent infection period ranged from 16 to 28 days, which is consistent with the latent period determined in a previous study in which the workers used four temperature regimens typical of anthurium growth in tropical environments (12). At lower temperatures, it is likely that the nested PCR assay could detect latent infections that last longer. Latent infections, which are thought to be involved in the spread of the pathogen within and between countries, have been reported to be present for more than 1 year in anthurium propagative material (27). Moreover, previous studies showed that some systemically infected cultivars that are considered resistant could harbor the pathogen at high densities without expression of symptoms for several months (13). The nested PCR assay should therefore be a very useful diagnostic tool for indexing propagation stock plant material in nurseries and for surveillance of international movement of X. axonopodis pv. dieffenbachiae on anthurium. This detection technique and the target sequence have been patented (33a; I. Robène-Soustrade, P. Laurent, and L. Gagnevin, 2005, French patent FR2848222 [pending]).
Supplementary Material
Acknowledgments
This work was funded by grants from ODEADOM and the European Union.
We thank A. Darrasse (INRA, Angers, France) for helpful discussions and A. Couteau and C. Boyer (CIRAD, La Réunion, France) for technical assistance.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Alvarez, A. M. 2004. Integrated approaches for detection of plant pathogenic bacteria and diagnosis of bacterial diseases. Annu. Rev. Phytopathol. 42:339-366. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez, A. M., A. A. Benedict, and C. Y. Mizumoto. 1985. Identification of xanthomonads and grouping of strains of Xanthomonas campestris pv. campestris with monoclonal antibodies. Phytopathology 75:722-728. [Google Scholar]
- 3.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl. 1991. Current protocols in molecular biology. John Whiley & Sons, New York, N.Y.
- 4.Aysan, Y., and F. Sahin. 2003. First report of bacterial blight of anthurium caused by Xanthomonas axonopodis pv. dieffenbachiae in Turkey. New Dis. Rep. [Online.] http://www.bspp.org.uk/ndr/july2003/2003-10.asp.
- 5.Berthier, Y., D. Thierry, M. Lemattre, and J. L. Guesdon. 1994. Isolation of an insertion sequence (IS1051) from Xanthomonas campestris pv. dieffenbachiae with potential use for strain identification and characterization. Appl. Environ. Microbiol. 60:377-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berthier, Y., V. Verdier, J. L. Guesdon, D. Chevrier, J. B. Denis, G. Decoux, and M. Lemattre. 1993. Characterization of Xanthomonas campestris pathovars by rRNA gene restriction patterns. Appl. Environ. Microbiol. 59:851-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berthier-Bayle, Y., J. P. Narcy, and M. Lemattre. 1990. DAS ELISA to detect Xanthomonas campestris pv. dieffenbachiae in anthurium propagative material, p. 925-933. In Z. Klement (ed.), Proceedings of the 7th Plant Pathogenic Bacteria Conference. Akadémiai Kiadó, Budapest, Hungary.
- 8.Chase, A. R., R. E. Stall, N. C. Hodge, and J. B. Jones. 1992. Characterization of Xanthomonas campestris strains from aroids using physiological, pathological, and fatty acid analysis. Phytopathology 82:754-759. [Google Scholar]
- 9.Coltri, P. P., E. R. Gonçalves, J. R. Neto, and Y. B. Rosato. 2000. Detecçao da diversidade em Xanthomonas axonopodis pv. dieffenbachiae por SDS-PAGE, RAPD e RFLP da regiao espaçadora 16S-23S. Summa Phytopathol. 26:399-406. [Google Scholar]
- 10.Cooksey, D. A. 1985. Xanthomonas blight of Anthurium andreanum in California. Plant Dis. 69:727. [Google Scholar]
- 11.Dickey, R. S., and C. H. Zumoff. 1987. Bacterial leaf blight of Syngonium caused by a pathovar of Xanthomonas campestris. Phytopathology 77:1257-1262. [Google Scholar]
- 12.Fukui, R., H. Fukui, and A. M. Alvarez. 1999. Effect of temperature on the incubation period and leaf colonization in bacterial blight of anthurium. Phytopathology 89:1007-1014. [DOI] [PubMed] [Google Scholar]
- 13.Fukui, R., H. Fukui, R. McElhaney, S. C. Nelson, and A. M. Alvarez. 1996. Relationship between symptom development and actual sites of infection in leaves of Anthurium inoculated with a bioluminescent strain of Xanthomonas campestris pv. dieffenbachiae. Appl. Environ. Microbiol. 62:1021-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gagnevin, L., J. E. Leach, and O. Pruvost. 1997. Genomic variability of the Xanthomonas pathovar mangiferaeindicae, agent of mango bacterial black spot. Appl. Environ. Microbiol. 63:246-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham, J. H., and A. R. Chase. 1992. Characterization of Xanthomonas campestris strains from aroids by genomic fingerprinting. Phytopathology 82:1124. [Google Scholar]
- 16.Guevara, Y. M., and E. Debrot. 1987. Plant Pathogenic Bacteria, College Park, Md., 2 to 7 June 1985.
- 17.Hartung, J. S., O. P. Pruvost, I. Villemot, and A. Alvarez. 1996. Rapid and sensitive colorimetric detection of Xanthomonas axonopodis pv. citri by immunocapture and a nested-polymerase chain reaction assay. Phytopathology 86:95-101. [Google Scholar]
- 18.Hayward, A. C. 1972. A bacterial new disease of Anthurium andreanum in Hawaii. Plant Dis. Rep. 56:904-908. [Google Scholar]
- 19.Hodge, N. C., A. R. Chase, and R. E. Stall. 1990. Characterization of xanthomonads from Araceae by fatty acid analyses. Phytopathology 80:158. [Google Scholar]
- 20.Jalenques, F. 1988. Dénombrement rapide de colonies microbiennes par le “Système Spiral.” Infect. Tech. Biol. 1:13-16. [Google Scholar]
- 21.Lipp, R. L., A. M. Alvarez, A. A. Benedict, and J. Berestecky. 1992. Use of monoclonal antibodies and pathogenicity tests to characterize strains of Xanthomonas campestris pv. dieffenbachiae from aroids. Phytopathology 82:677-682. [Google Scholar]
- 22.Manulis, S., L. Valinsky, A. Lichter, and D. W. Gabriel. 1994. Sensitive and specific detection of Xanthomonas campestris pv. pelargonii with DNA primers and probes identified by random amplified polymorphic DNA analysis. Appl. Environ. Microbiol. 60:4094-4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mu, L. 1990. Anthurium culture and blight in Tahiti, p. 37-38. In A. M. Alvarez (ed.), Proceedings of the 3rd Anthurium Blight Conference, University of Hawaii, Hilo.
- 24.Natural, M. P. 1990. Anthurium blight in the Philippines, p. 38. In A. M. Alvarez (ed.), Proceedings of the 3rd Anthurium Blight Conference, University of Hawaii, Hilo.
- 25.Norman, D., and A. Alvarez. 1989. A rapid method for presumptive identification of Xanthomonas campestris pv. dieffenbachiae and other xanthomonads. Plant Dis. 73:654-658. [Google Scholar]
- 26.Norman, D., R. Lipp, A. Benedict, and A. Alvarez. 1992. Enhanced detection of Xanthomonas campestris pv. dieffenbachiae using a “miniplate system.” Phytopathology 82:1177. [Google Scholar]
- 27.Norman, D. J., and A. M. Alvarez. 1994. Latent infections of in vitro anthurium caused by Xanthomonas campestris pv. dieffenbachiae. Plant Cell Tissue Org. Cult. 39:55-61. [Google Scholar]
- 28.Norman, D. J., and A. M. Alvarez. 1994. Rapid detection of Xanthomonas campestris pv. dieffenbachiae in anthurium plants with a miniplate enrichment/ELISA system. Plant Dis. 78:954-958. [Google Scholar]
- 29.Norman, D. J., R. J. Henny, and J. M. F. Yuen. 1999. Resistance levels of pot anthurium cultivars to Xanthomonas campestris pv. dieffenbachiae. Hortscience 34:721-722. [Google Scholar]
- 30.Pagani, M. C., and C. F. Ritchie. 2003. A putative protein in Xanthomonas arboricola pv. pruni with similarity to the ABC transporter ATP-binding system. Phytopathology 93:S68. [Google Scholar]
- 31.Pooler, M. R., D. F. Ritchie, and J. S. Hartung. 1996. Genetic relationships among strains of Xanthomonas fragariae based on random amplified polymorphic DNA PCR, repetitive extragenic palindromic PCR, and enterobacterial repetitive intergenic consensus PCR data and generation of multiplexed PCR primers useful for the identification of this phytopathogen. Appl. Environ. Microbiol. 62:3121-3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poussier, S., and J. Luisetti. 2000. Specific detection of biovars of Ralstonia solanacearum in plant tissues by nested-PCR-RFLP. Eur. J. Plant Pathol. 106:255-265. [Google Scholar]
- 33.Rademaker, J. L. W., B. Hoste, F. J. Louws, K. Kersters, J. Swings, L. Vauterin, P. Vauterin, and F. J. De Bruijn. 2000. Comparison of AFLP and rep-PCR genomic fingerprinting with DNA-DNA homology studies: Xanthomonas as a model system. Int. J. Syst. Evol. Microbiol. 50:665-677. [DOI] [PubMed] [Google Scholar]
- 33a.Robène-Soustrade, I., P. Laurent, and L. Gagnevin. December 2003. Detection test of Xanthomonas axonopodis pv. dieffenbachiae. Dutch patent NL1024929.
- 34.Rott, P., and P. Prior. 1987. Un dépérissement bactérien de l'anthurium provoqué par Xanthomonas campestris pv. dieffenbachiae aux Antilles françaises. Agron. Trop. 42:61-68. [Google Scholar]
- 35.Sathyanarayana, N., O. R. Reddy, S. Latha, and R. L. Rajak. 1998. Interception of Xanthomonas campestris pv. dieffenbachiae on Anthurium plants from the Netherlands. Plant Dis. 82:262. [DOI] [PubMed] [Google Scholar]
- 36.Soustrade, I., L. Gagnevin, P. Roumagnac, O. Gambin, D. Guillaumin, and E. Jeuffrault. 2000. First report of anthurium blight caused by Xanthomonas axonopodis pv. dieffenbachiae in Reunion Island. Plant Dis. 84:1343. [DOI] [PubMed] [Google Scholar]
- 37.Toth, I. K., L. J. Hyman, R. Taylor, and P. R. J. Birch. 1998. PCR-based detection of Xanthomonas campestris pv. phaseoli var. fuscans in plant material and its differentiation from X. c. pv. phaseoli. J. Appl. Microbiol. 85:327-336. [Google Scholar]
- 38.Trebaol, G., C. Manceau, Y. Tirilly, and S. Boury. 2001. Assessment of the genetic diversity among strains of Xanthomonas cynarae by randomly amplified polymorphic DNA analysis and development of specific characterized amplified regions for the rapid identification of X. cynarae. Appl. Environ. Microbiol. 67:3379-3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verdier, V., S. Ojeda, and G. Mosquera. 2001. Methods for detecting the cassava bacterial blight pathogen: a practical approach for managing the disease. Euphytica 120:103-107. [Google Scholar]
- 40.Zoina, A., A. Raio, and A. Spasiano. 2000. First report of anthurium bacterial blight in Italy. J. Plant Pathol. 82:65. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.