

Abstract
The diagnosis of tuberculous meningitis (TBM) remains a complex issue because the most widely used conventional diagnostic tools, such as culture and PCR assay for cerebrospinal fluid (CSF) samples, are unable to rapidly detect Mycobacterium tuberculosis with sufficient sensitivity in the acute phase of TBM. Based on TaqMan PCR, we designed a novel technique consisting of an internally controlled quantitative nested real-time (QNRT) PCR assay that provided a marked improvement in detection sensitivity and quantification. We applied this novel technique to quantitatively detect M. tuberculosis DNA in CSF samples from patients with suspected TBM. For use as the internal control in the measurement of the M. tuberculosis DNA copy numbers in the QNRT-PCR assay, the original mutation (M) plasmid, which included an artificial random 22-nucleotide sequence within an inserted DNA fragment of the MPB64 gene of M. tuberculosis, was prepared. The QNRT-PCR assay showed high sensitivity and specificity that were approximately equivalent to those of the conventional nested PCR assay. Moreover, the QNRT-PCR assay made it possible to precisely and quantitatively detect the initial copy number of M. tuberculosis DNA in CSF samples. Therefore, compared to the conventional PCR assay, the QNRT-PCR assay can be considered a more useful and advanced technique for the rapid and accurate diagnosis of TBM. To establish the superiority of this novel technique in TBM diagnosis, it will be necessary to accumulate data from a larger number of patients with suspected TBM.
Recently, in the diagnosis of tuberculous meningitis (TBM), the detection of Mycobacterium tuberculosis DNA in cerebrospinal fluid (CSF) samples using PCR has been widely used as a more rapid, sensitive, and specific diagnostic method than the conventional bacteriological detection methods, such as direct smear for acid-fast bacilli (AFB) and culture for M. tuberculosis (7, 8, 12, 17, 18). Use of the nested PCR assay has provided a remarkable increase in sensitivity and specificity of DNA amplification compared to the conventional single-step PCR assay (8, 12, 17, 20). However, the nested PCR assay using CSF samples has yet to be widely used in TBM diagnosis, due to its laborious and time-consuming procedure, which carries a high risk of sample contamination (8, 12, 17, 20). Recently, real-time PCR assays have been applied in routine diagnostic laboratory testing (1, 10, 14, 16, 22). In addition to conventional qualitative analysis, real-time PCR assays make it possible to perform accurate quantitative analyses with a high degree of reproducibility (3, 4, 5, 6, 9, 15, 19, 21, 23).
In this study, we attempted to develop a novel technique of internally controlled quantitative nested real-time (QNRT) PCR assay based on TaqMan PCR (Applied Biosystems). This novel technique combines the high sensitivity of nested PCR with the accurate quantification of real-time PCR. Using the QNRT-PCR assay, we examined the ability to detect M. tuberculosis DNA in CSF samples and evaluated the clinical usefulness of this technique with regard to the rapid and accurate diagnosis of TBM compared to other conventional methods.
MATERIALS AND METHODS
Subjects and clinical samples.
This study was approved by the Nihon University Institutional Review Board.
Nine serial patients with suspected TBM and 20 non-TBM control patients were selected from patients who were admitted to our hospital between July 2000 and April 2004. All nine suspected TBM patients met the previously established clinical criteria and had supporting evidence of TBM (7, 8, 11, 12, 17, 18, 20) (see Table 3) and were classified as two “confirmed” cases (positive CSF culture or AFB smear) and seven “highly probable” cases (meeting all the clinical criteria and with three types of positive supporting evidence but without any bacterial isolation) (7, 8, 11, 12, 17, 18, 20). The CSF samples were collected from these nine suspected TBM patients upon admission (before antituberculosis treatment [ATT]). In addition, the extracted DNA specimen from the M. tuberculosis standard strain H37Rv (ATCC 25618) was used as the positive control in this study.
TABLE 3.
Summary of clinical features, CSE findings, PCR assay results, and outcomes for 9 patients with suspected TBMa
| Parameter | Confirmed cases
|
Highly probable cases
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | |
| Age (yr)/sex | 73/M | 76/M | 35/F | 65/F | 52/M | 24/F | 44/F | 59/F | 44/M |
| Past medical history (background disease) | Diabetes mellitus, renal cancer, hypertension | CML, atrial fibrillation, hypertension | SLE | Diabetes mellitus | Alcoholism, alcoholic cirrhosis, diabetes mellitus | SLE, lupus nephritis, APS | RPGN, CRF (hemodialysis) | MS, hyperlipidemia | ATL |
| Administration of corticosteroid (mg/day) | PSL:20 | PSL:20 | PSL:20 | PSL:30 | |||||
| Clinical stage upon admissionb | III | III | II | I | II | I | III | II | III |
| CSF findings upon admission (before treatment) | |||||||||
| Cells/μl [M:P] | 288 [75:141] | 165 [462:34] | 208 [170:455] | 107 [161:0] | 18 [27:26] | 30 [69:21] | 60 [41:138] | 40 [121:0] | 117 [345:7] |
| Protein (mg/dl) | 299 | 569 | 300 | 70 | 135 | 25 | 70 | 359 | 87 |
| Glucose (mg/dl) | 13 | 46 | 13 | 48 | 54 | 30 | 52 | 78 | 48 |
| Cl (mEq/l) | 96 | 97 | 94 | 122 | 96 | 118 | 116 | 125 | 130 |
| ADA (IU/l) | 23.4 | 12.3 | 16.3 | 7.8 | 8.6 | 4.4 | N.D. | 3.7 | 3.9 |
| AFB smear | − | − | − | − | − | − | − | − | − |
| Tb culture | + | + | − | − | − | − | − | − | − |
| Single PCR assay | + | + | − | − | − | − | − | − | − |
| Nested PCR assay | + | + | + | + | + | + | + | + | + |
| Copy number by QNRT-PCR assay (copies/250 μl CSF) | 2.6 × 104 | 1.6 × 104 | 9.0 × 102 | 1.4 × 103 | 6.7 × 102 | 4.8 × 103 | 3.8 × 103 | 1.4 × 103 | 5.6 × 103 |
| Cranial MRI findings | |||||||||
| Meningeal enhancement | + | + | + | − | + | − | − | − | + |
| Hydrocephalus | + | − | + | − | + | − | − | + | − |
| Cerebrovascular disorder | + | + | + | − | + | − | + | − | − |
| IFM | + | − | + | − | + | + | − | − | − |
| M. tuberculosis outside CNS | |||||||||
| Pulmonary (sputum or ball) | + | + | − | − | + | − | − | − | + |
| Gastric aspirate | + | − | − | − | + | − | − | − | + |
| Urine | − | − | − | − | − | − | − | − | − |
| Peripheral blood | − | − | − | − | − | − | − | − | − |
| Treatment | |||||||||
| INH (mg/day) | 500 | 600 | 800 | 450 | 500 | 500 | 200 | 600 | 500 |
| Intrathecal administration (mg) | 100 (3 times/week) | ||||||||
| RFP (mg/day) | 600 | 600 | 600 | 450 | 450 | 450 | 225 | 450 | 450 |
| PZA (g/day) | 1.5 | 1.5 | 1.5 | 1.5 | 1 | 2 | 1.5 | 2 | 2 |
| EB (mg/day) | 750 | 750 | |||||||
| SM (g/day) | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| Anticonvulsant | Phenytoin | Phenytoin | Valproate sodium | Phenytoin Phenobarbital | |||||
| V-P shunt | + | − | + | − | − | − | − | − | |
| Complication | SIADH/hyponatremia, acute renal failure, DIC/ARDS | Drug-induced hepatopathy drug-induced paracusis | SIADH/hyponatremia, left facial palsy | − | Symptomatic epilepsy | − | Symptomatic epilepsy, Acute pancreatitis | − | Leukemic meningitis (Intrathecal administration of carcinostatics) |
| Outcomec | Death | Recovery | Recovery | Recovery | Recovery | Recovery | Recovery | Recovery | Death |
| Clinical criteria and supporting evidenced | All A and three B (1,2,3) | All A and all B | All A and three B (1,2,4) | All A, and three B (1,2,4) | All A and all B | All A and three B (1,2,4) | All A and three B (1,2,4) | All A and three B (1,2,4) | All A and three B (1,2,3) |
CML, chronic myelocytic leukemia; SLE, systemic lupus erythematosus; APS, antiphospholipid syndrome; RPGN, rapidly progressive glomerulonephritis; CRF, chronic renal failure; MS, multiple sclerosis; ATL, adult T-cell leukemia; M, mononuclear cell; P, polymorphonuclear cell; ADA, adenosine deaminase; Tb, tuberculosis; INH, isoniazid; RFP, rifampin; PZA, pyrazinamide; EB, ethambutol; SM, streptomycin sulfate; DX, dexamethasone; PSL, prednisolone; V-P shunt, ventricle-peritoneal shunt; IFM, intracranial focal mass; SIADH, syndrome of inappropriate secretion of antidiuretic hormone; DIC, disseminated intravascular coagulation; ARDS, adult respiratory distress syndrome. −, negative; +, positive.
According to the clinical stages defined by the British Medical Research Council (11): stage I, no definite neurological symptoms; II, signs of meningeal irritation with slight clouding of consciousness and neurologic defects; III, severe clouding of consciousness and neurologic defects.
Outcome classified as recovery with minor or no neurological impairment, severe neurologic impairment, or death.
A, the clinical criteria suggestive of TBM are fever, headache, and neck stiffness of more than 1 week duration. B, supporting evidence for TBM includes (i) compatible abnormal CSF findings that included increased white cell counts with lymphocytes predominating, hypoglycorrhachia, protein concentration of >100 mg/dl, adenosine deaminase (ADA) greater than 10 IU/liter and negative results for routine bacterial and fungal cultures; (ii) MRI findings suggesting tuberculous involvement of the CNS (basal exudates, hydrocephalus, and intracranial focal mass, etc.); (iii) presence of tuberculosis in the body outside of the CNS or a history of tuberculosis; and (iv) clinical response to antituberculosis therapy. The suspected TBM cases were classified as “confirmed” cases (having bacterial isolation of M. tuberculosis, such as CSF culture, or being AFB smear positive) or “highly probable” cases (meeting all the above clinical criteria and with all three types of supporting evidence positive).
The non-TBM control group consisted of four cases of bacterial meningitis, three of cryptococcal meningitis, three of viral meningitis, six of multiple sclerosis, and one each of central nervous system (CNS) lupus, CNS malignant lymphoma, hepatic insufficiency, and neuro Behçet's disease. The diagnoses for the non-TBM control cases were based on their specific clinical and laboratory findings. Moreover, to determine the analytical specificity and cross-reactivity of our assays, the extracted DNA specimens from six additional reference strains of non-M. tuberculosis species—Mycobacterium bovis BCG (ATCC 19274), Mycobacterium avium (ATCC 15769), Mycobacterium intracellulare (ATCC 15985), and clinically isolated Staphylococcus aureus, Klebsiella pneumoniae, and Pseudomonas aeruginosa—were tested.
Extraction and purification of DNA from CSF samples.
A 500-μl aliquot of original lysis buffer containing 20 mM Tris-HCl (pH 8.0), 300 mM NaCl, 0.8% (vol/vol) sodium dodecyl sulfate, and 0.5 mg of proteinase K was prepared. This lysis buffer was added to 500 μl of the CSF sample, followed by incubation in a water bath at 65°C overnight. After incubation, the total 1,000-μl suspension was divided into two 500-μl aliquots for use in the conventional nested PCR or the novel QNRT-PCR assay.
For DNA extraction and purification from the 500-μl aliquots, the conventional phenol-chloroform method and ethanol precipitation were used. To efficiently extract a small amount of DNA, a high-molecular-weight carrier, Ethachinmate (Nippon Gene, Tokyo, Japan), was used as a coprecipitating agent for the nucleotides in the ethanol precipitation. After complete vacuum desiccation, the extracted DNA specimen was resuspended in 20 μl of pure water and then stored at −20°C until it was used.
Principle and assay conditions of conventional nested PCR.
For use in the two subsequent amplification steps of the nested PCR assay, two pairs of primers capable of specifically amplifying the gene sequence encoding the MPB64 protein of M. tuberculosis (MPT64; GenBank accession no. NC_000962) were prepared. The sequences and positions of the outer forward (F-1) and reverse (R-1) primers, as well as the inner forward (F-2) and reverse (R-2) primers, are shown in Table 1 and Fig. 1. An 18-μl mixture of the PCR solution containing 10 mM Tris-HCl (pH 8.0), 50 mM KCl, 1.5 mM MgCl2, 400 μM of each deoxynucleoside triphosphate mixture, 20 pM (each) of the primers F-1 and R-1 at the first-step (single) PCR or the primers F-2 and R-2 at the second-step (nested) PCR, and 2.5 U of Taq DNA polymerase was prepared. As the template, 2 μl of the extracted DNA specimen at the first step or 2 μl of the single PCR product at the second step was added to the PCR solution mixture (each total reaction volume was 20 μl). These preparations were subjected to two subsequent PCR amplification protocols as shown in Table 2.
TABLE 1.
Sequences of primers and TaqMan probes for PCR assays
| Objective | Type | Sequencea |
|---|---|---|
| Conventional nested PCR assay | ||
| First-step PCR | Outer forward primer (F-1) | 5′-ATCCGCTGCCAGTCGTCTTCC-3′ |
| Outer reverse primer (R-1) | 5′-CTCGCGAGTCTAGGCCAGCAT-3′ | |
| Second-step PCR | Inner forward primer (F-2) | 5′-CATTGTGCAAGGTGAACTGAGC-3′ |
| Inner reverse primer (R-2) | 5′-AGCATCGAGTCGATCGCGGAA-3′ | |
| Internal control | Human β-globin forward primer (HBB-I) | 5′-GGCAGACTTCTCCTCAGGAGTC-3′ |
| Human β-globin reverse primer (HBB-R) | 5′-CTTAGACCTCACCCTGTGGAGC-3′ | |
| QNRT-PCR assay | TaqMan forward primer (TqMn-F) | 5′-GTGAACTGAGCAAGCAGACCG-3′ |
| TaqMan reverse primer (TqMn-R) | 5′-GTTCTGATAATTCACCGGGTCC-3′ | |
| TaqMan probe-wild-VIC (TqMn-wild-VIC) | 5′-VIC-TATCGATAGCGCCGAATGCCGG-TAMRA-3′ (total 22 nucleotides, A:5, T:4, G:7, C:6 [GC%, 59]) | |
| TaqMan probe-mutation-FAM (TqMn-mut-FAM) | 5′-FAM-ATGGGACGGCTAGCAATCCGTC-TAMRA-3′ (total 22 nucleotides, A:5, T:4, G:7, C:6 [GC%, 59]) | |
| Creating “mutation” plasmid | NheI random-forward primer (Nhe1-F) | 5′-ATGCTAGCAATCCGTCCTTGGACCCGGTGA-3′ |
| NheI random-reverse primer (Nhe1-R) | 5′-ATGCTAGCCGTCCCATCCTGTTGTCCGGTC-3′ |
Underline, artificial sequence; double underline, Nhe1 site.
FIG. 1.

Schema demonstrating the positions of primers and TaqMan probes in the MPB64 protein-encoding gene (MPT64). ⋆, fluorescent reporter dye VIC; ★, fluorescent reporter dye FAM; ○, quencher dye TAMRA.
TABLE 2.
PCR assay conditions
| PCR assay conditions | Value
|
|
|---|---|---|
| First-step PCR | Second-step PCRa | |
| Conventional nested PCR | ||
| Initial denature | 96.0°C, 3 min | 96.0°C, 3 min |
| Amplificationb | ||
| Denature | 95.0°C, 30 s | 94.0°C, 30 s |
| Annealing | 60.0°C, 30 s | 55.0°C, 30 s |
| Extension | 72.0°C, 1 min | 72.0°C, 1 min |
| Final extension | 72.0°C, 10 min | 72.0°C, 10 min |
| PCR product | 239 bp | 194 bp |
| QNRT PCR | ||
| Initial denature | 96.0°C, 3 min | |
| Amplificationc | ||
| Denature | 95.0°C, 30 s | |
| Annealing | 60.0°C, 30 s | |
| Extension | 72.0°C, 1 min | |
| Final extension | 72.0°C, 10 min | |
| Incubation | 50.0°C, 2 min | |
| Initial denature | 95.0°C, 10 min | |
| Amplificationd | ||
| Denature | 95.0°C, 15 s | |
| Annealing-extension | 60.0°C, 1 min | |
| PCR for creating “mutation” plasmid | ||
| Initial denature | 96.0°C, 3 min | |
| Amplificatione | ||
| Denature | 98.0°C, 25 s | |
| Annealing | 55.0°C, 30 s | |
| Extension | 72.0°C, 5 min | |
| Final extension | 72.0°C, 10 min | |
| PCR product | 4,168 bp | |
Real-time TaqMan for QNRT-PCR.
35 cycles for first-step PCR, 25 cycles for second-step PCR.
35 cycles.
40 cycles.
35 cycles.
In addition, all assays were simultaneously monitored for extraction efficiency from CSF samples and for the presence of PCR inhibitors by amplifying a fragment of human chromosomal DNA as the internal control. A pair of primers that were specific for the human β-globin gene (HBB; GenBank accession no. L48217) was prepared (Table 1). The 196-bp HBB fragment, as the internal control, was amplified in another tube under the same assay conditions as the single PCR for M. tuberculosis DNA.
Principle of QNRT-PCR (i) Assay conditions.
Similar to the conventional nested PCR assay, the novel QNRT-PCR assay also consists of two consecutive PCR amplification steps. However, the second step of the QNRT-PCR assay is changed to the real-time (TaqMan) PCR for quantitative analysis.
In the first-step PCR, 2 μl of the extracted DNA specimen as the template was amplified by using the same outer primers, F-1 and R-1, under the same assay conditions as in the conventional single PCR (Table 2). For the second-step PCR, a new pair of inner primers that were also specific for MPT64 was prepared. The sequences and positions of these new inner primers (TaqMan forward primer [TqMn-F] and TaqMan reverse primer [TqMn-R]) are shown in Table 1 and Fig. 1. In addition, two specific 22-nucleotide TaqMan probes, which were labeled with the fluorescent reporter dye VIC or 6-carboxyfluorescein (FAM), were prepared. The sequences and positions of these two TaqMan probes (TaqMan probe-wild-VIC [TqMn-wild-VIC] and TaqMan probe-mutation-FAM [TqMn-mut-FAM]) are also shown in Table 1 and Fig. 1. The 23-μl mixture for each of the PCR solutions, containing 12.5 μl TaqMan Universal PCR Master Mix, 0.9 μM (each) of the inner primers (TqMn-F and TqMn-R), and 0.2 μM TaqMan probe (TqMn-wild-VIC or TqMn-mut-FAM), was prepared. As the template, 2 μl of the first-step PCR product was added to this PCR solution mixture (each total reaction volume was 25 μl). This preparation was subjected to the protocol shown in Table 2, and the procedure used the ABI PRISM 7700 sequence detector system (PE Applied Biosystems, Foster City, CA).
(ii) Preparation of the two types of the original plasmid.
For the quantitative detection of M. tuberculosis DNA in CSF samples, two types of original plasmid, “wild” (W) and “mutation” (M) plasmids, were created in our laboratory.
The original W plasmid was prepared for use as the standard template in the second step of the QNRT-PCR assay and included a 239-bp DNA fragment of MPT64. A purified first-step PCR product obtained from the positive control strain (M. tuberculosis H37Rv) was inserted into the pCR2.1 vector and then cloned using the TA cloning kit (Invitrogen Corp., San Diego, CA).
The original M plasmid was generated based on the W plasmid for use as the internal control (“ruler”) in the QNRT-PCR assay. The M plasmid included 22 artificial random nucleotides within the inserted 239-bp DNA fragment of MPT64 (Fig. 1). The sequence of these 22 artificial random nucleotides was set so that it had the same nucleotide composition as the W plasmid. To generate the M plasmid, a pair of primers, which were specific for MPT64 and contained an additional 16 artificial nucleotides at the 5′ end, was prepared. The sequences and positions of these primers (Nhe-1F and Nhe-1R) are shown in Table 1 and Fig. 1. The additional artificial nucleotides of the each primer included the NheI restriction site (5′-GCTAGC-3′) at the 5′ end (Table 1). The PCR was performed using these primers and 2 μl of the W-plasmid preparation as the template and was subjected to the protocol shown in Table 2. The PCR product was digested at both ends by the restriction enzyme NheI and was then ligated. Since there was no NheI site within the pCR2.1 vector, it was possible to accurately ligate both ends of the PCR product. This ligation product (i.e., M plasmid) was also cloned using the TA cloning kit (Invitrogen Corp.).
The nucleotide sequences of the DNA fragments inserted into the W and M plasmids were confirmed by direct sequencing in both strands. Concentrations of the W- and M-plasmid DNAs were measured at least three times by a UV spectrophotometer at 260 nm, and then 11 serial sets of 10-fold dilutions were prepared (1 to 1010 copies/2 μ1). The seven serial sets of the 10-fold dilutions of the W and M plasmids (104 to 1010 copies/2 μ1) were used as the standard templates to construct the two specific standard curves. In addition, 103 copies of the M plasmid were adopted as the internal control (“ruler”). This copy number was determined by preliminary experiment (described below).
(iii) Quantitative detection of M. tuberculosis DNA.
In the second step of the QNRT-PCR assay, TqMn-wild-VIC specifically anneals to the natural sequence of MPT64 in wild-type M. tuberculosis, whereas TqMn-mut-FAM specifically anneals to only the artificial 22 random nucleotides of the M plasmid for use as the internal control. TqMn-mut-FAM has exactly the same nucleotide composition as TqMn-wild-VIC but has a different and random sequence that is complementary to the 22 artificial random nucleotides of the M plasmid (Table 1). Therefore, the annealing efficiencies of these two TaqMan probes to the template can be regarded as the same.
In the QNRT-PCR assay, both M. tuberculosis DNA and the internal control (M plasmid) were extracted simultaneously from the CSF samples. In addition, they were simultaneously amplified by the two common pairs of primers under the same assay conditions, followed by detection using the two TaqMan probes having the same annealing efficiency. Therefore, the efficiencies of extraction, amplification, and detection for both the M. tuberculosis DNA and the internal control can be regarded as being equivalent throughout the extraction and two steps of the PCR amplification procedure (Fig. 2). In order to calculate the initial copy number of M. tuberculosis DNA in CSF samples before passing them through the extraction and the two subsequent PCR amplification steps, we formulated equation 1, based on the above hypothesis.
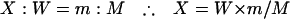 |
(1) |
In equation 1, X represents the initial copy number of M. tuberculosis DNA in the 250 μl of CSF sample; m represents the initial copy number of the internal control (103 copies of M plasmid); and W and M represent the copy numbers of M. tuberculosis DNA and the internal control, respectively, after they have passed through the extraction and the PCR amplification procedures.
FIG. 2.

Schema demonstrating the hypothesis used to construct the principle of the novel QNRT-PCR assay. M.Tb, M. tuberculosis.
Clinical application of the QNRT-PCR assay.
In this study, the QNRT-PCR assay was used to examine a total of 29 clinical CSF samples that were collected from 9 patients with suspected TBM and 20 non-TBM control patients. In addition, six reference strains of non-M. tuberculosis species were also examined. The data obtained from the QNRT-PCR assays were used in equation 1 to calculate the initial copy number of M. tuberculosis DNA for each sample.
Evaluation of the precision of the standard curves.
For real-time (TaqMan) PCR, the standard curve for the quantitative analysis is constructed automatically by plotting each of the threshold cycle numbers (CT values) against the log of the starting copy numbers for the standard templates.
For the QNRT-PCR assay, two specific standard curves for the quantitative detection of M. tuberculosis DNA and the internal control, respectively, are needed. To statistically evaluate the precision of these two specific standard curves, the following preliminary experiments were performed. Samples (2 μl) of the seven serial sets of the 10-fold dilutions of the W and M plasmids (104 to 1010 copies/2 μl) were used as the standard templates. The real-time PCR to construct two specific standard curves was performed in duplicate under the same assay conditions as the second step of the QNRT-PCR assay. This preliminary experiment was repeated five times.
All of the CT value data collected from the preliminary experiments were statistically analyzed by a simple regression analysis and a two-way analysis of variance (ANOVA).
Optimization of QNRT-PCR assay conditions.
For the QNRT-PCR assay, three important parameters may affect the assay conditions: the amplification cycle number for the first-step PCR, the copy number of the internal control, and the primer concentration for the second-step PCR. These were determined by the following series of preliminary experiments.
In the preliminary experiments, 500 μl of “imitative” CSF samples containing the W plasmids (1 to 105 copies) instead of the actual M. tuberculosis DNA were prepared (see Fig. 5A). In addition, 103, 104, and 105 copies of the M plasmids were used as the internal control (see Fig. 4A). Under various different assay conditions, two subsequent PCR amplification steps were performed using 2 μl of the extracted products from the “imitative” CSF samples as the template. In the second-step PCR, the CT value data against the starting copy numbers of the W and M plasmids were collected. These preliminary experiments were performed in duplicate and repeated five times.
FIG. 5.

Results of the QNRT-PCR assays for the nine patients upon admission (before treatment). (A) Amplification curves of M. tuberculosis DNA for the single-step real-time PCR assay (analyzed by VIC). (B) Amplification curves of M. tuberculosis DNA for the QNRT-PCR assay (analyzed by VIC). (C) Amplification curves of the internal controls (M plasmid) for the QNRT-PCR assay (analyzed by FAM). In the single-step real-time PCR assay, there was a slight amount of specific PCR amplification of M. tuberculosis DNA found only in the two “confirmed” cases (cases 1 and 2). In contrast, the QNRT-PCR assay demonstrated sufficient specific amplification for both M. tuberculosis DNA and the internal control.
FIG. 4.

Statistical evaluation of the CT value data collected from the preliminary experiments. (A) Schema for preparing the “imitative” CSF samples. (B) Result of the Tukey-Kramer test against the CT values of the W plasmids when performing 35 cycles of the first-step PCR. (C) Result of the Tukey-Kramer test against the CT values of the W plasmids when using 103 copies of the M plasmid as the internal control. (D) Result of the one-way ANOVA against the CT values of 103 copies of the M plasmid as the internal control.
To determine the optimal amplification cycle number for the first-step PCR, it was performed every 5 cycles from 20 to 35 cycles. The CT value data for each starting copy number of the W plasmids were statistically analyzed at each cycle number for the first-step PCR using a multiplex comparison test (Tukey-Kramer test).
To determine the optimal copy number for the internal control, 103, 104, and 105 copies of the M plasmids were examined. The CT value data for each starting copy number of the W plasmids were also statistically analyzed at each copy number of the internal control using the Tukey-Kramer test. In contrast, the CT value data for each starting copy number of the internal control were statistically analyzed using a one-way ANOVA at each 1, 10, and 100 copies of the W plasmids.
To determine the optimal primer concentration for the second-step PCR, 0.9 and 1.8 μM primer concentrations were examined. As the internal control, 103 and 104 copies of the M plasmids were used. The CT value data for the starting copy numbers of the W and M plasmids were statistically analyzed using an F test for each of the two primer concentrations.
A P value of <0.01 was considered statistically significant in all of the statistical analyses.
Precautions to avoid sample contamination.
To avoid contamination, the following original precautions were designed for this study. Screw-cap tubes were used for all sample preparations and reaction mixtures. One hundred preparations of the lysis buffer for the extraction of DNA from the CSF samples were provided simultaneously. The lysis buffer was divided into 500-μl aliquots per sample in new screw-cap tubes. Similarly, 100 preparations of the PCR mixture, except for the Taq DNA polymerase, were provided simultaneously and then divided into aliquots per five samples. Each step of the experimental procedure, consisting of reaction mixture preparation, DNA extraction from samples, PCR amplification, and analysis of PCR products, was performed in a separate room. In addition, we exclusively used disposable filter tips for each sample and changed tips for every step throughout the entire experimental procedure. To exclude carryover contamination, at least three negative controls, including no DNA template, were inspected together with the actual samples in each PCR assay.
RESULTS
Clinical features of the patients.
Table 3 summarizes the clinical features before the ATT (upon admission) and the outcomes for the nine serial patients, comprising the two “confirmed” and seven “highly probable” TBM cases. All of the patients were immunocompromised hosts with several background diseases. All nine patients presented with mild to severe neurological symptoms and demonstrated several different levels of abnormal findings in the CSF examination upon admission. The cranial magnetic resonance imaging (MRI) findings also demonstrated several abnormalities: four cases each of hydrocephalus (cases 1, 3, 5, and 8) and intracranial focal masses (cases 1, 3, 5, and 6). Seven patients (cases 2 to 8) demonstrated a good response to the ATT, but two patients (cases 1 and 9) died due to aggravation of the malignant background diseases (renal cancer and adult T-cell leukemia). Autopsies were not performed on these two patients, since permission could not be obtained from their families.
Conventional single and nested PCR assay results upon admission.
The single PCR assay revealed positive results in only the two “confirmed” (culture-positive) cases (cases 1 and 2) out of a total of nine patients (22.2%) (Table 3). In contrast, the nested PCR assay results were positive for all nine patients (100%) (Table 3). Negative results were found for all 20 of the patients in the non-TBM control group for both the single and nested PCR assays. In addition, positive results for the HBB-PCR as the internal control were found in all 29 of the CSF samples. These results indicated that the DNA was well extracted from the CSF samples and that no PCR inhibitors existed in the assay procedures.
Moreover, both the single and nested PCR assays revealed all negative results for the six reference strains of non-M. tuberculosis species. The analytical specificity of these assays was sufficiently demonstrated by these results.
Precision of the standard curves.
The two specific standard curves that were constructed based on CT value data collected from the preliminary experiments are shown in Fig. 3A and B. Both of these two standard curves demonstrated a significant linear relationship (value of fit for both curves, r = 0.999) between the CT values and the log of the starting copy numbers for each of the standard plasmids. In both of the standard curves, no significant differences were found among the plots constructed by the preliminary experiments that were repeated separately five times (F = 1.05 and P = 0.45). The precision of the two specific standard curves was sufficiently demonstrated by these results.
FIG. 3.

Two specific standard curves used for the QNRT-PCR assay. (A) Specific standard curve for use in quantitatively detecting M. tuberculosis DNA or the W plasmid. VIC (TqMn-wild-VIC) was used for the analysis. (B) Specific standard curve for use in quantitatively detecting the M plasmid as the internal control. FAM (TqMn-mut-FAM) was used for the analysis.
Optimization of QNRT-PCR assay conditions.
The CT value data (mean ± standard deviation [SD]) collected at different cycle numbers of the first-step PCR are shown in Table 4. When the first-step PCR was set at 35 cycles, the CT values demonstrated significant differences between all of the starting copy numbers of the W plasmids (1 to 105 copies) (P < 0.01) (Fig. 4B). Therefore, 35 cycles was adopted as the optimal cycle number in the first-step PCR.
TABLE 4.
CT value data collected for different first-step PCR cycle numbers
| First-step PCR cycle no.b |
CT valuea
|
|||
|---|---|---|---|---|
| 20 cycles | 25 cycles | 30 cycles | 35 cycles | |
| 105 | 14.43 ± 0.02 | 8.80 ± 0.03 | 6.13 ± 0.01 | 5.79 ± 0.01 |
| 104 | 19.36 ± 0.06 | 12.85 ± 0.3 | 7.67 ± 0.05 | 6.99 ± 0.05 |
| 103 | 22.20 ± 0.09 | 16.87 ± 0.09 | 11.80 ± 0.1 | 10.19 ± 0.05 |
| 102 | 23.33 ± 0.1 | 19.61 ± 0.09 | 14.36 ± 0.2 | 11.29 ± 0.07 |
| 10 | 23.37 ± 0.2 | 19.99 ± 0.04 | 15.64 ± 011 | 12.71 ± 0.09 |
| 1 | 23.66 ± 0.28 | 20.04 ± 0.24 | 15.45 ± 0.33 | 14.25 ± 0.05 |
The CT value data represent the means ± standard deviations in duplicate for five independent experiments.
W-plasmid starting copy number.
The CT value data (mean ± SD) collected for different copy numbers of the internal control are shown in Table 5. When 103 copies of the M plasmid were set as the internal control, the CT values demonstrated significant differences between all of the starting copy numbers of the W plasmids (P < 0.01) (Fig. 4C). In addition, the CT values for 103 copies of the M plasmid demonstrated the most uniform variance (no significant difference) between 1, 10, and 100 copies of the W plasmids (F = 1.19; P = 0.42) (Fig. 4D). Therefore, 103 copies of the M plasmid was adopted as the optimal copy number of the internal control.
TABLE 5.
CT value data collected under different copy numbers of internal control
| Copy no. with internal control reporter dyeb |
CT valuea
|
|||||
|---|---|---|---|---|---|---|
| M plasmid 105 copies
|
M plasmid 104 copies
|
M plasmid 103 copies
|
||||
| VIC | FAM | VIC | FAM | VIC | FAM | |
| 105 | 6.17 ± 0.05 | 10.25 ± 0.19 | 6.40 ± 0.04 | 23.94 ± 0.23 | 6.59 ± 0.01 | 25.27 ± 0.95 |
| 104 | 8.21 ± 0.03 | 8.91 ± 0.03 | 9.70 ± 0.23 | 14.31 ± 0.06 | 8.57 ± 0.04 | 16.78 ± 0.37 |
| 103 | 13.23 ± 0.14 | 7.22 ± 0.02 | 11.21 ± 0.09 | 10.09 ± 0.01 | 10.34 ± 0.08 | 14.03 ± 0.08 |
| 102 | 18.71 ± 0.34 | 7.25 ± 0.04 | 15.95 ± 0.08 | 10.04 ± 0.03 | 14.15 ± 0.03 | 13.39 ± 0.06 |
| 10 | 25.57 ± 0.63 | 7.21 ± 0.02 | 22.55 ± 0.37 | 9.98 ± 0.04 | 20.01 ± 0.16 | 13.37 ± 0.06 |
| 1 | 40.00 (NA)c | 7.26 ± 0.06 | 23.06 ± 0.18 | 10.16 ± 0.04 | 26.00 ± 0.11 | 13.32 ± 0.04 |
The CT value data represent the means ± standard deviations in duplicate for five independent experiments.
W-plasmid starting copy number.
NA, not amplified.
The CT value data (mean ± SD) collected at different primer concentrations for the second-step PCR are shown in Table 6. The CT value for the W plasmids and the internal controls (103 and 104 copies of the M plasmid) demonstrated no significant difference between the 0.9 and 1.8 μM primer concentrations by an F test (Table 6). Therefore, 0.9 μM was adopted as the sufficient and optimal primer concentration for the second-step PCR.
TABLE 6.
CT value data collected under different primer concentrations
| Copy no. for internal control reporter dyeb |
CT valuea
|
|||||||
|---|---|---|---|---|---|---|---|---|
| M plasmid 104 copies
|
M plasmid 103 copies
|
|||||||
| VICd
|
FAMe
|
VICf
|
FAMg
|
|||||
| 0.9 μM (20 pmol/μl)c | 1.8 μM (40 pmol/μl) | 0.9 μM (20 pmol/μl) | 1.8 μM (40 pmol/μl) | 0.9 μM (20 pmol/μl) | 1.8 μM (40 pmol/μl) | 0.9 μM (20 pmol/μl) | 1.8 μM (40 pmol/μl) | |
| 105 | 6.28 ± 0.03 | 6.34 ± 0.07 | 21.56 ± 0.20 | 20.81 ± 0.88 | 6.41 ± 0.01 | 6.27 ± 0.04 | 21.45 ± 0.30 | 20.63 ± 0.06 |
| 104 | 7.58 ± 0.13 | 7.52 ± 0.04 | 12.68 ± 0.60 | 12.37 ± 0.21 | 7.97 ± 0.18 | 7.51 ± 0.12 | 16.85 ± 0.25 | 17.12 ± 0.02 |
| 103 | 14.77 ± 0.11 | 14.50 ± 0.16 | 12.41 ± 0.10 | 12.48 ± 0.23 | 13.27 ± 0.26 | 12.98 ± 0.06 | 13.22 ± 0.18 | 13.21 ± 0.05 |
| 102 | 21.40 ± 0.04 | 20.30 ± 0.09 | 11.52 ± 0.03 | 11.54 ± 0.27 | 18.54 ± 0.3 | 18.05 ± 0.12 | 13.34 ± 0.26 | 13.30 ± 0.24 |
| 10 | 28.51 ± 0.51 | 27.67 ± 0.06 | 11.48 ± 0.35 | 11.17 ± 0.10 | 24.50 ± 0.25 | 23.98 ± 0.18 | 13.17 ± 0.10 | 13.50 ± 0.25 |
| 1 | 28.92 ± 0.19 | 27.57 ± 0.64 | 10.89 ± 0.16 | 11.22 ± 0.06 | 30.10 ± 0.25 | 31.02 ± 0.37 | 12.89 ± 0.19 | 13.01 ± 0.03 |
The CT value data represent the means ± standard deviations in duplicate for five independent experiments.
W-plasmid starting copy number.
Primer concentration.
F test: F = 1.12; P = 0.57.
F test: F = 1.16; P = 0.59.
F test: F = 0.94; P = 0.46.
F test: F = 1.21; P = 0.62.
QNRT-PCR assay results.
The results of the single-step real-time (TaqMan) PCR assay for the nine patients with suspected TBM are shown in Fig. 5A. In this single-step real-time PCR assay, 2 μl of the extracted DNA specimens from the CSF samples were directly used as the template. Although slight specific PCR amplification of M. tuberculosis DNA was found in only two “confirmed” cases (cases 1 and 2) out of the total of nine patients, both were insufficient for quantitative analysis. In the other seven “highly probable” patients (cases 3 to 9), no significant PCR amplification was found. These results were completely consistent with the results of the conventional single PCR assay (Table 3).
In contrast, sufficient PCR amplification of M. tuberculosis DNA for quantitative analysis was found for all nine patients (100%) for the QNRT-PCR assay after 35 cycles of the first-step PCR were performed (Fig. 5B). Further, the internal controls (M plasmids) were also amplified sufficiently and uniformly for all nine patients (Fig. 5C). The QNRT-PCR assay revealed no amplification at all for the CSF samples collected from the 20 patients in the non-TBM control group. Similarly, the QNRT-PCR assay revealed all negative results for the six reference strains of non-M. tuberculosis species. However, adequate amplification of the internal control was found for all of the non-TBM control cases and the non-M. tuberculosis reference strains.
Based on the data obtained from the QNRT-PCR assay, the initial copy number of M. tuberculosis DNA per 250 μl of CSF samples that were collected from the nine patients with suspected TBM upon admission was calculated by equation 1. These measured copy numbers of M. tuberculosis DNA are shown in Table 3.
DISCUSSION
We have developed a novel QNRT-PCR assay technique for the quantitative detection of M. tuberculosis DNA in CSF samples collected from patients with clinically suspected TBM. The novel and principal feature of this QNRT-PCR assay is the ability to calculate the initial copy number of M. tuberculosis DNA in CSF samples before they pass through the extraction and two subsequent PCR amplification steps. In the QNRT-PCR assay, the initial copy number of M. tuberculosis DNA was calculated from the amplification ratio of the specific internal control used as a “ruler.” For use as the specific internal control, the original M plasmid was designed so that it had equivalent amplification and detection efficiencies against the actual M. tuberculosis DNA. Based on a similar concept, two pairs of specific primers and specific TaqMan probes were prepared. Consequently, we were able to formulate equation 1, which can be used to determine the initial copy number.
To our knowledge, there have been no other reports in which accurate copy numbers of M. tuberculosis DNA have been measured by using a real-time (TaqMan) PCR technique (1, 10, 14, 16, 22). Although many previous studies have used a conventional single-step real-time PCR assay to quantitatively detect various infectious pathogens in different clinical samples, few have been able to describe accurate copy numbers of the causative pathogens (3, 4, 5, 6, 9, 15, 19, 21, 23). In many previous studies, even though various internal controls have been used for monitoring PCR assay conditions, they have regrettably never been used for correctly determining the copy numbers of the causative pathogens (5, 6, 9, 15, 19, 21, 23). To use an internal control as a “ruler” for correcting the copy number of a causative pathogen, it is necessary to construct a “new” specific internal control with equivalent amplification and detection efficiencies against the causative pathogen. In the current quantitative real-time PCR assay, which has a high sensitivity due to application of the nested PCR technique, we considered that such a “new” specific internal control would play a more important role. Therefore, we considered that the conventional internal controls used in many previous studies were inadequate for the purpose of correcting the copy numbers of causative pathogens in the current study (5, 6, 9, 15, 19, 21, 23).
The conventional methods of phenol-chloroform extraction and ethanol precipitation have been assumed to be inadequate for routine use in clinical examinations because of their laborious and time-consuming procedures (1, 3, 4, 5, 6, 7, 8, 9, 10, 12, 13, 14, 15, 16, 17, 18, 19, 21, 22, 23). Generally, commercial column extraction kits, such as the QIAmp Blood Kit (QIAGEN Inc., Valencia, CA) and the COBAS AMPLICOR respiratory-specimen preparation kit (Roche Diagnostic Systems Inc., Branchburg, NJ), are widely used for DNA extraction from various samples in many clinical laboratories (1, 3, 4, 5, 6, 9, 10, 13, 14, 15, 16, 19, 21, 22, 23). However, in the current study, it was impossible to extract sufficient M. tuberculosis DNA from CSF samples using commercial extraction kits, and therefore, these popular kits may be inadequate for extracting small amounts of M. tuberculosis DNA from CSF samples. A high-molecular-weight carrier (Ethachinmate) was used in the present study as a coprecipitating agent, together with conventional phenol-chloroform extraction and ethanol precipitation, making it possible to extract small amounts of M. tuberculosis DNA from the CSF samples more effectively and at a lower cost than with the commercial extraction kits.
Previously, the four major M. tuberculosis-specific sequences, including the regions of IS6110 insertion elements, 65-kDa heat shock protein antigen, 16S rRNA gene, and MPT64, were evaluated by PCR assays (1, 7, 8, 10, 11, 12, 14, 16, 17, 18, 20, 22). Of these four sequence regions, IS6110 and the 16S rRNA gene have been most frequently used as the target sequences for PCR of M. tuberculosis DNA in many clinical laboratories (1, 10, 12, 14, 16, 17, 22). In particular, the 16S rRNA gene is the target sequence of the COBAS AMPLICOR M. tuberculosis detection kit (Roche Diagnostic Systems Inc.) (16, 22). MPT64 has been reported to be the most specific and sensitive sequence of M. tuberculosis DNA for the PCR assay (7, 8, 18). Therefore, the specific primers and TaqMan probes for MPT64 were adopted in this study.
To quantitatively detect a small amount of M. tuberculosis DNA in CSF samples by the QNRT-PCR assay, it is extremely important that both M. tuberculosis DNA and the internal control be amplified with sufficient balance. Therefore, the precision of the two specific standard curves and the optimal assay conditions were strictly examined by statistical evaluation in a series of preliminary experiments. In particular, many investigators have reported that the precision of the standard curve is the principal factor for quantitative detection in real-time (TaqMan) PCR assays (2, 3, 4, 5, 6, 9, 15, 19, 21, 23). In the present study, the two specific standard curves demonstrated statistically significant precision (r = 0.999; F = 1.04). Therefore, we consider that any overall errors relating to the dilution procedure or within each experiment can be disregarded. In addition, three parameters were examined in detail as important factors that could affect the QNRT-PCR assay conditions. The optimal values of these three important parameters were determined by statistically analyzing the CT value data collected from the preliminary experiments.
Previously, the main argument against the use of the nested PCR protocol has been that, due to its highly increased sensitivity and the required additional amplification step, sample contamination could easily occur (3, 8, 12, 13, 17, 20). However, the possibility of sample contamination can be minimized by good laboratory practice. In this study, through the use of original precautions to avoid sample contamination, the incidence of sample contamination in the conventional nested PCR assay was markedly reduced from 52.3% to 3.2%. Moreover, its incidence in the QNRT-PCR assay fell below 1%. Therefore, we consider this novel technique to be a reliable assay method.
At present, although the gold standard for TBM diagnosis is bacterial isolation, several complex issues remain to be addressed (7, 8, 11, 12, 17, 18, 20). Therefore, in place of conventional bacteriological examinations, a new reliable diagnostic tool for TBM is needed (7, 8, 11, 12, 17, 18, 20). In this study, the conventional single-step PCR assays revealed positive results in only two “confirmed” cases (22.2%). These results may suggest that it is difficult to detect a small amount of M. tuberculosis DNA in CSF samples when using the conventional single-step PCR technique. In contrast, both the conventional nested PCR and the novel QNRT-PCR assays revealed positive results for all nine of the patients (100%). Therefore, the detection sensitivities of these two nested PCR techniques can be considered to be approximately equivalent and to be significantly higher than that of conventional single-step PCR. Since the nested PCR and the QNRT-PCR assays revealed negative results for all 20 CSF samples collected from the non-TBM control group and the six non-M. tuberculosis reference strains, the analytical specificity of both assays is also considered to be sufficiently high. This QNRT-PCR assay is a novel technique that can quantitatively detect a small amount of M. tuberculosis DNA with a low risk of sample contamination. Recently, we reported a diachronic study that examined the usefulness of the nested PCR assay for assessing the clinical course of TBM (20). Since the QNRT-PCR assay has the great advantage of being able to quantitatively detect the initial copy number of M. tuberculosis DNA in CSF samples as a numerical value, it may be more useful for assessing the clinical course and the ATT response of TBM patients than the conventional nested PCR assay. However, to establish the superiority of the QNRT-PCR assay in the diagnosis of TBM versus other, conventional methods, it will be necessary to accumulate data from a larger number of patients with suspected TBM.
In conclusion, the QNRT-PCR assay is a novel and advanced technique that combines the high sensitivity of nested PCR and the accurate quantification of real-time PCR. We speculate that if the QNRT-PCR assay is widely adopted within clinical practice, it will be a powerful tool for the rapid and accurate diagnosis of TBM.
Acknowledgments
We thank our colleagues within the Division of Neurology, Nihon University School of Medicine, for collecting the CSF samples and for providing the data for many of the patients.
This work was supported by a grant from the Ministry of Education, Science and Culture of Japan (High-Tech Research Center, Nihon University).
REFERENCES
- 1.Aldous, W. K., J. I. Pounder, J. L. Cloud, and G. L. Woods. 2005. Comparison of six methods of extracting Mycobacterium tuberculosis DNA from processed sputum for testing by quantitative real-time PCR. J. Clin. Microbiol. 43:2471-2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heid, C. A., J. Stevens, K. J. Livak, and P. M. Williams. 1996. Real time quantitative PCR. Genome Res. 6:986-994. [DOI] [PubMed] [Google Scholar]
- 3.Kawada, J., H. Kimura, Y. Ito, Y. Hoshino, N. Tanaka-Kitajima, Y. Ando, M. Futamura, and T. Morishima. 2004. Comparison of real-time and nested PCR assays for detection of herpes simplex virus DNA. Microbiol. Immunol. 48:411-415. [DOI] [PubMed] [Google Scholar]
- 4.Kimura, H., Y. Ito, M. Futamura, Y. Ando, Y. Yabuta, Y. Hoshino, Y. Nishiyama, and T. Morishima. 2002. Quantitation of viral load in neonatal herpes simplex virus infection and comparison between type 1 and type 2. J. Med, Virol. 67:349-353. [DOI] [PubMed] [Google Scholar]
- 5.Kohmoto, M., M. Enomoto, Y. Yano, S. Otani, S. Minamitani, A. Tamori, D. Habu, T. Takeda, S. Shiomi, S. Seki, T. Arakawa, and S. Nishiguchi. 2003. Detection of serum hepatitis B virus DNA by real-time quantitative polymerase chain reaction (TaqMan PCR) during lamivudine treatment: comparison with three other assays. Hepatol. Res. 26:125-133. [DOI] [PubMed] [Google Scholar]
- 6.Larsen, H. H., H. Masur, J. A. Kovacs, V. J. Gill, V. A. Silcott, P. Kogulan, J. Maenza, M. Smith, D. R. Lucey, and S. H. Fischer. 2002. Development and evaluation of a quantitative, touch-down, real-time PCR assay for diagnosing Pneumocystis carinii pneumonia. J. Clin. Microbiol. 40:490-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee, B. W., J. A. Tan, S. C. Wong, C. B. Tan, H. K. Yap, P. S. Low, J. N. Chia, and J. S. Tay 1994. DNA amplification by the polymerase chain reaction for the rapid diagnosis of tuberculous meningitis. Comparison of protocols involving three mycobacterial DNA sequences, IS6110, 65 kDa antigen, and MPB64. J. Neurol. Sci. 123:173-179. [DOI] [PubMed] [Google Scholar]
- 8.Liu, P. Y., Z. Y. Shi, Y. J. Lau, and B. S. Hu. 1994. Rapid diagnosis of tuberculous meningitis by a simplified nested amplification protocol. Neurology 44:1161-1164. [DOI] [PubMed] [Google Scholar]
- 9.Locatelli, G., F. Santoro, F. Veglia, A. Gobbi, P. Lusso, and M. S. Malnati. 2000. Real-time quantitative PCR for human herpesvirus 6 DNA. J. Clin. Microbiol. 38:4042-4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marin, M., D. Garcia de Viedma, M. J. Ruiz-Serrano, and E. Bouza. 2004. Rapid direct detection of multiple rifampin and isoniazid resistance mutations in Mycobacterium tuberculosis in respiratory samples by real-time PCR. Antimicrob. Agents Chemother. 48:4293-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medical Research Council. 1948. Streptomycin treatment of tuberculous meningitis. Lancet i:582-596. [PubMed] [Google Scholar]
- 12.Nakajima, H., K. Ashida, H. Yamasaki, K. Shinoda, and N. Ohsawa. 1995. Intracranial tuberculoma with spontaneous recovery. Rinsho Shinkeigaku 35:521-525. (in Japanese.) [PubMed] [Google Scholar]
- 13.O'Neill, H. J., D. E. Wyatt, P. V. Coyle, C. McCaughey, and F. Mitchell. 2003. Real-time nested multiplex PCR for the detection of herpes simplex virus types 1 and 2 and varicella zoster virus. J. Med. Virol. 71:557-560. [DOI] [PubMed] [Google Scholar]
- 14.Rindi, L., N. Lari, D. Bonanni, and C. Garzelli. 2004. Detection of Mycobacterium tuberculosis genotypic groups by a duplex real-time PCR targeting the katG and gyrA genes. J. Microbiol. Methods 59:283-287. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez-Lazaro, D., M. Hernandez, and M. Pla. 2004. Simultaneous quantitative detection of Listeria spp. and Listeria monocytogenes using a duplex real-time PCR-based assay. FEMS Microbiol. Lett. 233:257-267. [DOI] [PubMed] [Google Scholar]
- 16.Ruiz, M., M. J. Torres, A. C. Llanos, A. Arroyo, J. C. Palomares, and J. Aznar. 2004. Direct detection of rifampin- and isoniazid-resistant Mycobacterium tuberculosis in auramine-rhodamine-positive sputum specimens by real-time PCR. J. Clin. Microbiol. 42:1585-1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scarpellini, P., S. Racca, P. Cinque, F. Delfanti, N. Gianotti, M. R. Terreni, L. Vago, and A. Lazzarin. 1995. Nested polymerase chain reaction for diagnosis and monitoring treatment response in AIDS patients with tuberculous meningitis. AIDS 9:895-900. [DOI] [PubMed] [Google Scholar]
- 18.Shankar, P., N. Manjunath, K. K. Mohan, K. Prasad, M. Behari, Shriniwas, and G. K. Ahuja. 1991. Rapid diagnosis of tuberculous meningitis by polymerase chain reaction. Lancet 337:5-7. [DOI] [PubMed] [Google Scholar]
- 19.Stranska, R., R. Schuurman, M. de Vos, and A. M. van Loon. 2004. Routine use of a highly automated and internally controlled real-time PCR assay for the diagnosis of herpes simplex and varicella-zoster virus infections. J. Clin. Virol. 30:39-44. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi, T., T. Nakayama, M. Tamura, K. Ogawa, H. Tsuda, A. Morita, M. Hara, M. Togo, H. Shiota, Y. Suzuki, M. Minami, H. Ishikawa, K. Miki, E. Shikata, S. Takahashi, T. Kuragano, K. Matsumoto, S. Sawada, and T. Mizutani. 2005. Nested polymerase chain reaction for assessing the clinical course of tuberculous meningitis. Neurology 64:1789-1793. [DOI] [PubMed] [Google Scholar]
- 21.Templeton, K. E., S. A. Scheltinga, A. W. Graffelman, J. M. Van Schie, J. W. Crielaard, P. Sillekens, P. J. Van Den Broek, H. Goossens, M. F. Beersma, and E. C. Claas. 2003. Comparison and evaluation of real-time PCR, real-time nucleic acid sequence-based amplification, conventional PCR, and serology for diagnosis of Mycoplasma pneumoniae. J. Clin. Microbiol. 41:4366-4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wada, T., S. Maeda, A. Tamaru, S. Imai, A. Hase, and K. Kobayashi. 2004. Dual-probe assay for rapid detection of drug-resistant Mycobacterium tuberculosis by real-time PCR. J. Clin. Microbiol. 42:5277-5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whiley, D. M., I. M. Mackay, M. W. Syrmis, M. J. Witt, and T. P. Sloots. 2004. Detection and differentiation of herpes simplex virus types 1 and 2 by a duplex LightCycler PCR that incorporates an internal control PCR reaction. J. Clin. Virol. 30:32-38. [DOI] [PubMed] [Google Scholar]