

Abstract
Mammalian glycolipid transfer proteins (GLTPs) facilitate the selective transfer of glycolipids between lipid vesicles in vitro. Recent structural determinations of the apo- and glycolipid-liganded forms of human GLTP have provided the first insights into the molecular architecture of the protein and its glycolipid binding site (Malinina, L., Malakhova, M. L., Brown, R. E., and Patel, D. J. (2004) Nature 430, 1048–1053). In the present study, we have evaluated the functional consequences of point mutation of the glycolipid liganding site of human GLTP within the context of a carrier-based mechanism of glycolipid intermembrane transfer. Different approaches were developed to rapidly and efficiently assess the uptake and release of glycolipid by GLTP. They included the use of glass-immobilized, glycolipid films to load GLTP with glycolipid and separation of GLTP/glycolipid complexes from vesicles containing glycolipid (galactosylceramide or lactosylceramide) or from monosialoganglioside dispersions by employing nickel-nitrilotriacetic acid-based affinity or gel filtration strategies. Point mutants of the sugar headgroup recognition center (Trp-96, Asp-48, Asn-52) and of the ceramide-accommodating hydrophobic tunnel (Phe-148, Phe-183, Leu-136) were analyzed for their ability to acquire and release glycolipid ligand. Two manifestations of point mutation within the liganding site were apparent: (i) impaired formation of the GLTP/glycolipid complex; (ii) impaired acquisition and release of bound glycolipid by GLTP. The results are consistent with a carrier-based mode of GLTP action to accomplish the intermembrane transfer of glycolipid. Also noteworthy was the inefficient release of glycolipid by wtGLTP into phosphatidylcholine acceptor vesicles, raising the possibility of a function other than intermembrane glycolipid transfer in vivo.
Glycolipid transfer proteins (GLTPs)1 are ∼24 kDa proteins that selectively accelerate the intermembrane transfer of glycolipids in vitro (1–7). Investigations of purified GLTPs from bovine spleen, porcine, and bovine brain as well as of cloned GLTP cDNAs from various mammalian cells indicate that the protein is highly conserved and ubiquitous among mammals (1–10). Protein specificity is directed to glycolipids in which the initial sugar head group (glucose, galactose, or mannose) is β-linked to either a ceramide or diglyceride hydrophobic moieties (11, 12). Purified recombinant GLTPs from human skin fibroblasts or from bovine and porcine brain are fully active upon expression in Escherichia coli (8–10, 13).
Mammalian GLTPs2 differ distinctly in both sequence and structure (13) from many other soluble glycolipid-binding proteins, such as sterol carrier protein 2 (14–16) and sphingolipid activator proteins (17–21) that facilitate hydrolysis of glycosphingolipids and delivery of glycolipid antigens to CD1 proteins during immune presentation processes (22, 23). However, a GLTP-like sequence occurs in the carboxyl-terminal region of phosphoinositol 4-phosphate adaptor protein-2 and related human FAPP2 proteins, which localize to the trans-Golgi network at exit sites where transport carriers destined for the plasma membrane are formed (24, 25). GLTP orthologs in other eukaryotes have been implicated in the activation of stress-induced, programmed cell death in plants (Arabidopsis) (26) as well as in a self-destructive death process triggered by cell-cell incompatibility in filamentous fungi (Podospora anserina) (27). The findings suggest that GLTP is a member of an emerging new protein family involved in important cellular processes.
Human rGLTP, as recently revealed by x-ray diffraction, is characterized by a novel folding motif among proteins that transfer or bind lipids (13). The structural data show that complexation of lactosylceramide (LacCer) by GLTP involves a single glycolipid liganding site. The glycolipid liganding site of GLTP is composed of a surface recognition center for the sugar headgroup and a molded-to-fit, hydrophobic tunnel that accommodates the hydrocarbon chains of the ceramide moiety via a cleft-like conformational gating mechanism. Mapping of the recognition center and hydrophobic tunnel of GLTP has enabled identification of amino acid side chains that hydrogen-bond and make hydrophobic contacts with the bound glycolipid (13). Point mutation analyses indicated that several of the residues lining the liganding site are essential for GLTP to maintain maximum capacity to transfer glycolipids between membranes quickly and efficiently. However, from kinetic measurements of the intermembrane glycolipid transfer activity of the point mutants, it is not possible to discern whether the reduced activity occurs because of an impaired ability of GLTP to acquire glycolipid from the donor vesicles or to release the bound glycolipid to acceptor phospholipid membranes.
The goal of the present work was to elucidate the molecular basis of the decreased transfer activity of GLTPs containing point mutations in their liganding sites by directly assessing their relative glycolipid binding capacities as well as the ability of the resulting GLTP/glycolipid complexes to release glycolipid to acceptor membranes. To accomplish this goal, approaches were developed for the rapid isolation and functional analysis of soluble wtGLTP (and point mutants) complexed with various glycolipids (GalCer, LacCer, and ganglioside GM1). The experimental strategy not only provided further evidence for a wild type GLTP/glycolipid complex acting as a soluble intermediate that moves between membranes to accomplish glycolipid transfer but yielded novel insights into the functional consequences of point mutation within the glycolipid complexation site of GLTP with respect to complex solubility, stability, and functionality.
EXPERIMENTAL PROCEDURES
Materials—Porcine galactosylceramide (GalCer) and 1-palmitoyl-2-oleyl phosphatidylcholine (POPC) were purchased from Avanti Polar Lipids (Alabaster, Al), and monosialoganglioside (GM1) was purchased from Sigma. Radioactive GalCer [6-3H]galactose (20 Ci/mmol) was obtained from American Radiolabeled Chemicals Inc., and l-α-1-palmitoyl-2-oleoyl-[oleoyl-1-14C]phosphatidylcholine (52.6 mCi/mmol) was purchased from PerkinElmer Life Sciences. [3H]GM1 was synthesized using galactose oxidase as described previously (28, 29), and [3H]LacCer 16:0 was synthesized as described below. The purity of radioactive lipids was confirmed by TLC.
SUV Preparation—Lipids of the desired composition including either [3H]GalCer or [14C]POPC were mixed in chloroform/methanol and dried under a stream of nitrogen. Residual solvent was removed under high vacuum for 4 h. The lipid films were hydrated in 150 mm NaCl, 20 mm
Tris (or 50 mm NaH2PO4), pH 8.0, by vigorous vortexing and brief bath sonication. SUVs were produced by probe sonication (ultrasonic processor W-225) followed by ultracentrifugation (100,000 × g, 45 min) to remove contaminating titanium particles from the probe and partially sonicated lipid aggregates (30). Typical recovery of lipid in the supernatant as SUVs was ∼80%. SUVs comprised of 99 mol % POPC and 1 mol % GalCer (1–2 mm final total lipid concentration) were used in Ni-NTA affinity and size exclusion chromatography (SEC) experiments. Pure POPC SUVs, prepared similarly but at 10–20 mm concentration, served as acceptor vesicles.
Protein Expression and Purification—Human GLTP was cloned and expressed in E. coli using pET-30 vector as described previously (13). The purification procedure was modified slightly as follows. Transformed E. coli BL21(DE3) cells (Novagen) were grown at 37 °C in LB medium (750 ml). Cells were induced by 0.1 mm isopropyl-β-d-galactopyranoside at A600 = 0.9–1.1, and bacterial growth was continued at 15 °C for 20 h. The cell pellet was resuspended in 30 ml of washing buffer comprised of 10 mm imidazole, 150 mm NaCl, 50 mm NaH2PO4, pH 8.0, 10% glycerol including 1 mg/ml lysozyme, and 10 mm β-mercaptoethanol. After brief sonication and centrifugation, clarified cell lysate was loaded on 1–1.5 ml of Ni-NTA affinity resin (Qiagen). The majority of protein was soluble. The column was thoroughly washed with washing buffer. The protein was released by stepwise elution with buffer containing increasing concentrations of imidazole (60, 100, and 200 mm) in 150 mm NaCl, 50 mm NaH2PO4, pH 8.0, and 5% glycerol. To remove imidazole, fractions were combined and loaded on desalting columns (Econo-Pac 10 DG; Bio-Rad) equilibrated with 150 mm NaCl and 50 mm NaH2PO4, pH 8.0. Protein fractions were then concentrated to 2–6 mg/ml using Centriplus centrifugical filter devices YM-10 (Amicon). Glycerol was added at 10% final concentration to stabilize and protect the protein from freeze-thaw damage. Protein purity was analyzed by 15% SDS-PAGE and Coomassie Brilliant Blue staining. Protein concentration was measured by the Bradford assay (Bio-Rad protein assay) using bovine serum albumin as standard. To remove the His tags and S-tags, rGLTP (pET-30) was incubated with Factor Xa (Novagen) for 16 h at room temperature. Then, the protein was purified by fast protein liquid chromatography SEC using a HiLoad 16/60 Superdex-75 prep grade column (Amersham Biosciences) equilibrated with 150 mm NaCl, 20 mm Tris, pH 8.0. Protein fractions were concentrated to 2–6 mg/ml and stored in buffer containing 10% glycerol. Site-directed mutants were obtained using the pET-30/wtGLTP construct vector and QuikChange® site-directed mutagenesis kit (Stratagene). All mutant plasmid constructs were verified by DNA-sequencing performed at the Mayo Molecular Biology Core Facility (Rochester, MN) with an Applied Biosystems 377 sequencer using thermocycler protocols and fluorescent dye terminators. Expressed mutants were affinity-purified as described for wtGTLP. The yields of purified mutant proteins were similar to wtGLTP (∼10 mg/liter culture). Near-UV CD and far-UV CD spectra confirmed the absence of global changes in the folding of the mutants (13). Because control experiments revealed that the liganding data were not affected by the presence of the amino-terminal fusion tag, we routinely used His fusion GLTP mutants to avoid the more difficult and lengthy purification of nonfusion proteins. The relative ease of producing high protein amounts by recombinant approaches enabled adequate protein to be loaded on SEC columns to avoid the poor protein recoveries reported for porcine GLTP (4).
GLTP/Glycolipid Complex Formation Using Immobilized Glycolipid Films—GalCer, dissolved in chloroform:methanol (9:1, 250 μl) along with [3H]GalCer, was dried under nitrogen while vortexing in 1-ml conical glass vials (Kimble catalog number 60702-1, VWR International). The vials containing the glycolipid films were then placed under high vacuum for 1.5 h. The protein solution (0.1–1 mg in 0.5 ml) was added to the dry lipid film, immobilized on the glass wall of the vial, and gently incubated at room temperature. Aliquots (50 μl) were taken at different time points and counted (Beckman Instruments LS 3801) in scintillation mixture (ICN EcoLume). Protein recovery after 1 h of incubation was greater than 98% as determined by Bradford analyses. Because of well known deviations from classical enzymatic and liganding behavior that occur when either the protein or the ligand is immobilized or insoluble (56, 57), conditions were established to optimize detection of GLTP/glycolipid complexation. The deviations originate from the inherent insolubility of lipid ligands and the diffusional and partitioning limitations associated with immobilized ligands and proteins. With glycolipid films, simply increasing the total glycolipid ligand to substantial excess relative to protein did not guarantee that more ligand becomes available for interaction with the protein. We found that a 10-fold excess of ligand (as compared with protein) only slightly increased the wtGLTP acquisition rate (∼1.2-fold; data not shown) as compared with a 2-fold excess of ligand. It appeared that higher glycolipid amounts resulted in thicker immobilized films and only marginally increased glycolipid availability to protein in solution. Nonetheless, the relative differences in the time-dependent acquisition of glycolipid by wtGTLP and the point mutants were found to be unaffected by the changes in ligand-to-protein ratio, even when protein molar ratio exceeded glycolipid (e.g. 3:1).
Intermembrane Transfer Activity of rGLTP—An intervesicular transfer assay involving radiolabeled GalCer was modified slightly as follows (5, 7, 31). POPC donor vesicles (1 mm total phospholipid) containing 2 mol % GalCer supplemented with [3H]GalCer (50 nCi/ml) and 10 mol % negatively charged dipalmitoyl phosphatidic acid were prepared by sonication as described above. 100 mol % POPC vesicles (10 mm lipid concentration) served as acceptor membranes. GLTP (1 μg) was preincubated for 5 min at 37 °C with donor vesicles (50 μl) before initiation of the transfer reaction by adding a 10-fold excess of POPC acceptors vesicles (50 μl). The reaction buffer consisted of 150 mm NaCl, 20 mm
Tris (pH 8.0). After incubation at 37 °C, separation of the charged donor and neutral acceptor vesicles was achieved by rapid elution over DEAE Sephacel (Amersham Biosciences) minicolumns (2 ml). Control experiments without protein enabled correction for leakage of unbound donor vesicles (∼2% of total counts). Control experiments without acceptor vesicles revealed no counts above background in the eluant unless protein levels exceeded ∼50 μg.
Transfer Activity of GLTP/GalCer Complex—The DEAE chromatographic approach was also used to assess GalCer transfer from the GLTP/GalCer complex to acceptor vesicles because small GLTP amounts (<50 μg) were completely retained by DEAE resin (2 ml). Protein (0.1 mg; ∼4 nmol) was incubated for 2 h with glass-adsorbed GalCer film comprised of [3H]GalCer (16 nCi) and unlabeled GalCer (5 nmol). To assess glycolipid release, aliquots of the resulting GLTP complex (1–5 μg) were incubated with POPC SUVs (50 μl; 10 mm) at 37 °C for the specified time and then separated on a DEAE minicolumn by washing with buffer (2 ml). Glycolipid release to eluted acceptor vesicles was quantitated by liquid scintillation counting. In the absence of acceptor vesicles, background levels of radioactivity were recovered, suggesting that the complex is stable and requires membrane for glycolipid release.
Ni-NTA Affinity Chromatography—Ni-NTA resin (0.2 ml) was equilibrated with washing buffer comprised of 10 mm imidazole, 150 mm NaCl, 50 mm NaH2PO4, pH 8.0, 10% glycerol. The amount of resin was based on its established binding capacity, i.e. 0.2 ml of Ni-NTA resin binds 1 mg of protein (Qiagen). The protein/vesicle mixtures were incubated with resin by shaking in Eppendorf tubes for 20–30 min. The mixtures were briefly centrifuged (14,000 rpm, 10 s) to separate the resin and supernatant fractions. Complete binding of GLTP to the resin was confirmed by monitoring the protein concentration in the supernatant. The resin was washed several times with 0.4 ml of washing buffer until radioactivity of [3H]GalCer or [14C]POPC dropped to background level. Then, protein was eluted by increasing the imidazole concentration stepwise to 200 mm and then to 1 m in 150 mm NaCl, 50 mm NaH2PO4, 10% glycerol, pH 8.0. Maximal elution of protein was achieved by incubating for 10 min before spinning. The protein was analyzed by SDS-PAGE and Bradford assay. Radioactivity of all fractions was measured by liquid scintillation counting. Control experiments showed that radioactive vesicles did not bind to the resin (data not shown).
Gel Filtration Chromatography—To separate protein from SUVs, Sephacryl S-300 resin (Amersham Biosciences) was chosen because of its known capability for resolving lipid vesicles and glycolipid micelles (29). A Sephacryl S-300 column (1.5 × 20 cm, 35 ml) was equilibrated with 150 mm NaCl, 20 mm Tris, pH 8.0, and calibrated with mixtures of radioactive SUVs and GLTP. Protein/vesicle mixtures (1–1.5 ml) were loaded, and 1-ml fractions were collected at a flow rate of 0.5 ml/min while washing the column with buffer. The vesicles (∼2 × 106 Da) (31) eluted near the void volume (fractions 11–14). The fractions (20–25), corresponding to free soluble protein, were analyzed by Bradford assay and SDS-PAGE. The radioactivity was measured by liquid scintillation counting. Control experiments showed that optimal sensitivity for detection of glycolipid complexation by GLTP was observed when the molar amount of GLTP was in excess relative to glycolipid.
Synthesis of [3H]LacCer—The [3H]N-hydroxysuccinimide ester of palmitic acid (4 μCi) was dried under nitrogen and combined with d-lactosyl-β1-1-d-erythrosphingosine and then dissolved in tetrahydrofuran and N-ethyldiisopropylamine. The mixture was gently bath sonicated. The reaction tube was heated at 50–52 °C. The reaction progress was checked periodically over 43 h by TLC using the solvent CHCl3: MeOH:H2O (50:21:3, v/v/v). N-Hydroxysuccinimide-palmitate and 18:1 LacCer were used as standards and detected on the silica gel plate by spraying with primulin reagent and viewing under long wave ultraviolet light. The silica gel bands were scrapped into counting vials. H2O(1 ml) and Ecolume scintillation mixture (10 ml) were added to each vial to measure radioactivity by scintillation counting. To recover pure [3H]LacCer, a Waters solid phase extraction Sep-Pak cartridge (100 mg; WAT023595) was equilibrated with 10 ml of chloroform. The reaction mixture was dried under nitrogen, resuspended in chloroform (100 μl), and loaded on the column. To release N-hydroxysuccinimide fatty acyl derivatives and related reaction side products, the column was eluted with 3.5 ml of CHCl3 (+1% MeOH). Then, 5 ml of acetone:MeOH (9:1, v/v) was added to elute glycolipid. All column fractions were dried under nitrogen and dissolved in 1 ml of CHCl3:MeOH (2:1, v/v). The purity of recovered [3H]16:0 LacCer was checked by TLC and fluorography using EN3HANCE spray (PerkinElmer Life Sciences).
RESULTS
Our initial goal was to evaluate the effect of point mutation at different sites within the liganding region of GLTP by analyzing the time-dependent response of glycolipid complexation. Previously, SEC on Sephadex G-75 had been used to separate porcine GLTP from phospholipid vesicles containing glycolipid and to show that GLTP can ligand glycolipid to form a soluble protein/lipid complex (4). However, the long separation times and poor protein recoveries reported with Sephadex G-75 were deemed impractical for effectively analyzing the glycolipid liganding capabilities of mutant GLTPs in time-dependent fashion. We investigated whether GLTP could acquire glycolipid immobilized on glass as hydrated films. We reasoned that this strategy would allow rapid sampling of GLTP in solution without the need for additional manipulations to achieve separation. Also, we recently found glass-immobilized glycolipid films to be useful for loading GLTP with glycolipid prior to crystallization for structural analysis.3
Fig. 1A shows the time-dependent accumulation of [3H]GalCer into aqueous solutions in the presence and absence of human rGLTP. The GalCer was presented as an immobilized film adsorbed to the walls of a glass vial and was composed of identical trace amounts of [3H]GalCer (2 pmol) mixed with different amounts of nonradioactive GalCer (0, 7, 20, 60 nmol). As expected, a competition effect was observed between radioactive and nonradioactive GalCer, causing an apparent decrease in the acquisition rate of [3H]GalCer by GLTP as the amount of nonradioactive GalCer increased, causing dilution of the radioactive GalCer. Assessment of the competition effect between radioactive and nonradioactive GalCer enabled optimization of assay conditions for monitoring the acquisition of glycolipid by GLTP. As alluded to under “Experimental Procedures,” incubation of GLTP with a large excess of glycolipid ligand suppressed the detection sensitivity of glycolipid complexation by GLTP, probably because of the compromised availability of immobilized glycolipid substrate (55). Thus, we generally monitored complex formation using only a slight excess of glycolipid ligand relative to GLTP (e.g. 2:1) and over an experimentally convenient time interval (1–1.5 h), to avoid protein instability problems observed with long incubations for some mutant GLTPs. It is noteworthy, however, that extended incubation of wtGTLP with GalCer films results in a 1:1 GLTP/glycolipid complex (Fig. 1C).
Fig. 1.
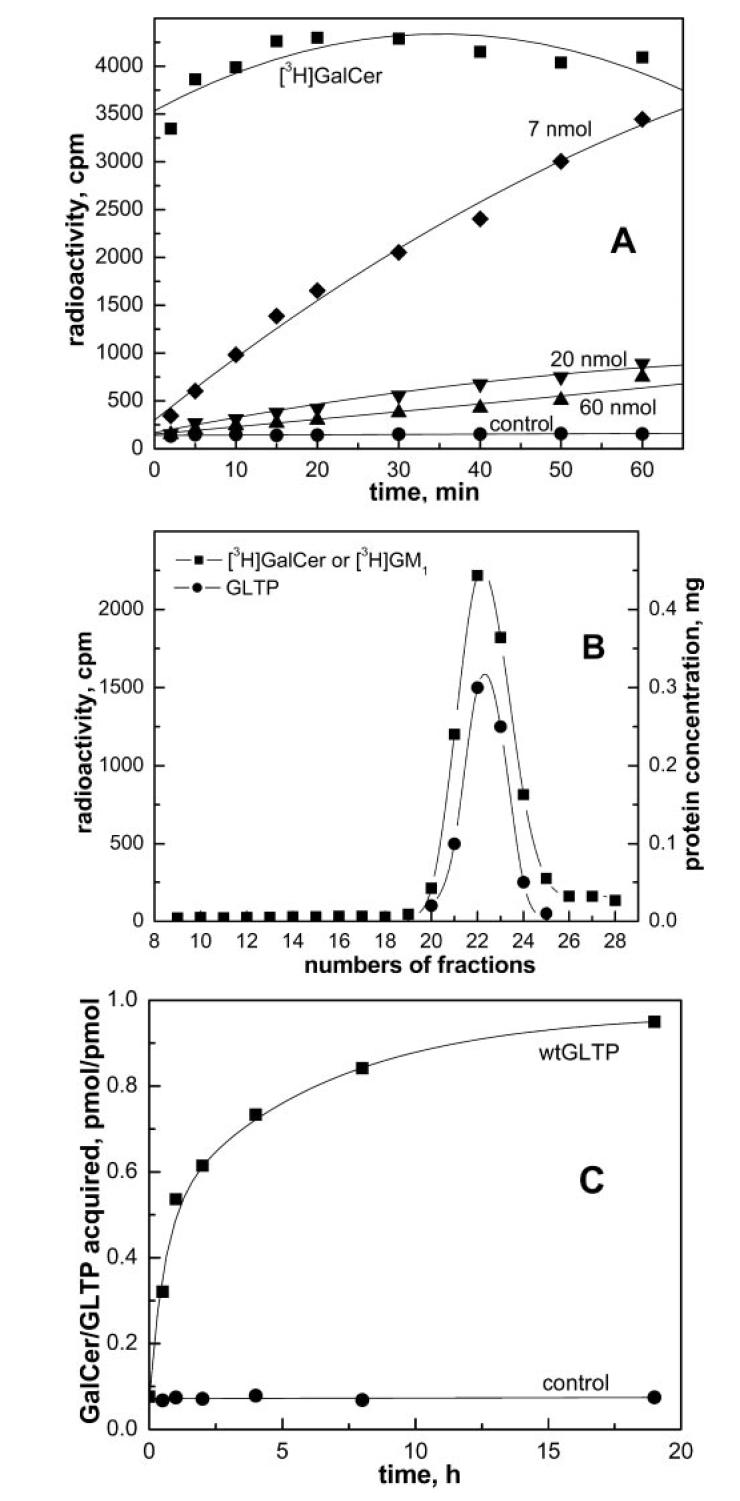
Formation of GLTP-glycolipid complexes using immobilized glycolipid films. A, time-dependent acquisition of GalCer by wtGLTP from glass-adsorbed, immobilized glycolipid films. GLTP (0.5 mg; ∼20 nmol) was incubated at room temperature with glass-adsorbed glycolipid films prepared from [3H]GalCer (∼2 pmol) and various amounts of unlabeled GalCer (0 (■), 7 (◆), 20 (▼), or 60 nmol (▲)). Also shown are control incubations without GLTP (●). B, association of glycolipid with soluble GLTP. GLTP/[3H]GalCer complex was produced by incubation with immobilized GalCer film as described above. GLTP/[3H]GM1 complex was formed by incubation with aqueous dispersions of GM1 (5 μm) followed by elution through Ni-NTA resin as described in the legend for Fig. 5 (panel A). Analysis of either GLTP/glycolipid complex by through a Sephacryl S-300 column shows coincident protein (●) and glycolipid (■) elution profiles. C, extended incubation of wt-GTLP with glass immobilized glycolipid film. wtGLTP (200 μg, ∼8 nmol) was incubated at room temperature with immobilized glycolipid film (16 nmol unlabeled GalCer + [3H]GalCer, ∼30 nCi). (■) indicates [3H]GalCer acquisition by wtGLTP. ● represents control incubation with buffer containing no GLTP.
To confirm that the accumulated [3H]GalCer in solution was because of glycolipid complexation with GLTP rather than detachment of glycolipid aggregates from the glass surface, two control experiments were performed. First, immobilized [3H]GalCer was incubated under identical experimental conditions with buffer containing no protein. An insignificant amount of radioactive glycolipid was found in solution (Fig. 1A, [unk]). Second, after incubation with immobilized films of [3H]GalCer, the solution containing GLTP was analyzed by SEC using Sephacryl S-300. The elution profile of radioactive [3H]GalCer coincided with that of the soluble protein (fractions 20–25, Fig. 1B). There was no evidence of glycolipid aggregates (molecular masses > 500 kDa) eluting prior to GLTP. Thus, formation of a stable, soluble GLTP/glycolipid complex was deemed the most likely explanation for the time-dependent accumulation of [3H]GalCer in solution.
Acquisition of GalCer by GTLP Point Mutants—Fig. 2 illustrates the three-dimensional structure of the GLTP/LacCer complex as revealed by x-ray diffraction (13). The labeled amino acids correspond to residues within the glycolipid liganding site that were point-mutated to assess their role in glycolipid complex formation. Each mutation affected different aspects of the sugar headgroup interaction or the accommodation of the ceramide hydrocarbon chains. Specifically, the point mutations disrupted stacking between the indole and sugar rings (Trp-96); hydrogen bonding of the sugar C2 carbon (Asp-48) and the sugar C3 carbon (Asn-52); gating action by the Phe-148 phenyl group that moves to permit entry of C9–C18 carbons of the sphingoid base chain; hydrophobic contacts involving the C10–C12 carbons of the sphingoid base chain (Leu-136); and channel accommodation of the terminal methyl group of the acyl chain (Phe-183).
Fig. 2.
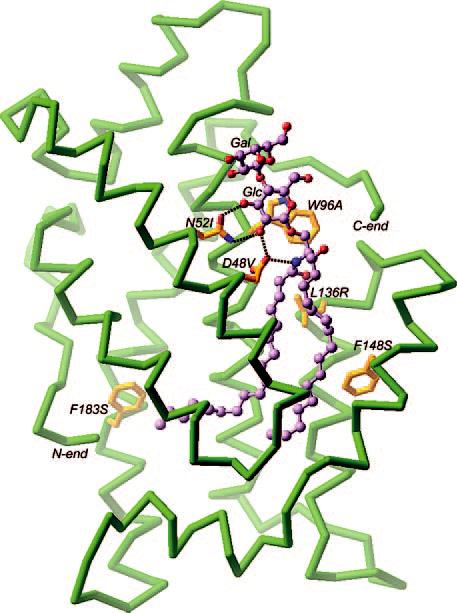
Locations of point mutations in the glycolipid liganding site of human GLTP. Analyses of the crystal structures of apo-GLTP (1.65Å) and of LacCer-GLTP (1.95Å) have recently been presented elsewhere (13). Depicted are amino acid side chains (gold) involved in the liganding of LacCer (lavender) and located in either the sugar headgroup recognition center (W96A, N52I, D48V) or the ceramide-accommodating hydrophobic channel (L136R, F148S, F183S). The GLTP backbone appears in lime green. Hydrogen bonds between side chains and glycolipid are indicated by dotted black lines.
The acquisition of [3H]GalCer by the point mutant GLTPs from glass-immobilized, glycolipid films is shown in Fig. 3A. Careful examination of the data shows that loading is not complete by 1 h and is a slow process that continues for many hours (Fig. 1C) (except for completely inactive mutants). Point mutation F183S, which affects the bottom of the hydrophobic tunnel, only slightly impaired acquisition of [3H]GalCer as compared with wtGLTP. In contrast, mutation F148S, which is located near the tunnel entrance, and recognition center mutant N52I, which affects hydrogen bonds to the sugar headgroup, had reduced adsorption of GalCer (∼50 – 60%). Recognition center mutant D48V, which eliminates hydrogen bonds to the sugar headgroup and to the amide nitrogen, showed substantially diminished GalCer acquisition (∼25%) as compared with wtGLTP. Point mutant W96A, which eliminated the stacking interaction with the sugar ring, and L136R, which blocked the entrance of the hydrophobic tunnel, showed almost no capacity to acquire GalCer (∼1%) as compared with wt-GTLP and resembled the no protein control curve (Fig. 3A). Similar patterns of relative glycolipid acquisition were observed among the point mutants, including N52I and D48V, after incubation with vesicles containing [3H]GalCer for 1 h and separation by Sephacryl S-300 gel filtration chromatography (not shown).
Fig. 3.

Acquisition and release of glycolipid by GLTP point mutants. A, acquisition of [3H]GalCer from immobilized glycolipid films by the wtGLTP and its mutants. wtGLTP (■) and mutants, F183S (▲), F148S (▼), N52I ([unk]), D48V (◆), and W96A and L136R ([unk]) were incubated (200 μg each, ∼8 nmol) with glass-adsorbed, immobilized glycolipid films (16 nmol unlabeled GalCer + [3H]GalCer, ∼30 nCi) at room temperature. Aliquots were sampled at different time points. Control incubation in the absence of protein is indicated by (●). B, release of [3H]GalCer from isolated wtGTLP/GalCer complex following incubation with POPC acceptor vesicles. The [3H]GalCer/GLTP complex was produced by incubation of wtGTLP (4 nmol) with immobilized glycolipid film (5 nmol of GalCer + [3H]GalCer, 100 nCi) for 2 h at room temperature (see “Experimental Procedures”). Recovered GLTP-GalCer complex (1 (▲),2(■), or 5(▼) μg) was incubated with 100 mole% POPC vesicles (50 mm,10 μl) at 37 °C and separated on DEAE-minicolumns at different time intervals. Calculation of the vesicle-to-protein molar ratio, as described under “Discussion,” revealed a 5:1 ratio. ◆ indicates glycolipid release to 4× POPC acceptors from GLTP-GalCer complex (2 μg), representing a vesicle-to-protein molar ratio of 20:1. The dotted line (◻) shows glycolipid release to POPC acceptors from GLTP-GalCer complex (2 μg), representing a vesicle-to-protein molar ratio of 5:1, in the presence of added glycolipid-free GLTP (1.5 μg). Control experiments were performed (●) in which [3H]GalCer/GLTP complex was incubated without acceptor vesicles. C, release of [3H]GalCer from isolated point mutant GLTP/[3H]GalCer complexes following incubation with POPC acceptor vesicles. Experimental conditions for complex production and release of glycolipid to acceptor vesicles were the same as for panel B. Each point mutant complex (2 μg) was incubated with POPC vesicles at molar vesicle-to-protein ratios of 5:1. The glycolipid/protein molar ratios (pmol GalCer/pmol protein) after acquisition incubations (time 0 of the release assays) were: wtGTLP (0.45), F183S (0.42), F148S and N52I (0.31), and D48V (0.14). The symbols for various mutants are the same as for panel A. D, intervesicular transfer activity for wtGLTP and GLTP point mutants. Donor vesicles comprised of POPC:DPPA:GalCer (88:10:2 mol/mol) and including [3H]GalCer were preincubated with protein (1 μg) for 5 min at 37 °C. The reaction was initiated by addition of acceptor POPC SUVs (5× molar vesicle excess as compared with protein). The reaction buffer was 150 mm NaCl, 20 mm Tris-Cl (pH 8.0). At different times, aliquots were eluted through DEAE minicolumns to recover the POPC acceptor vesicles for liquid scintillation counting.
Release of GalCer from the Complex to Acceptor Vesicles—A key issue regarding liganding of glycolipid by GLTP and by point mutants is the functionality of the resulting complex. Among the questions considered were the following. Can the soluble complex formed by GLTP/glycolipid transfer glycolipid to acceptor vesicles, and can point mutants be distinguished on the basis of their capacities to release the bound glycolipid in the presence of acceptor membranes? In other words, do certain mutants maintain a wtGTLP-like capacity to complex glycolipid, while being defective in their ability to release the bound ligand when incubated with membrane? To address such issues, we monitored the membrane-dependent release of [3H]GalCer from the GLTP/glycolipid complex produced by incubation with immobilized glycolipid films. The transfer of glycolipid from the GLTP/GalCer complex to acceptor POPC SUVs was then monitored by rapidly separating the vesicles from protein-lipid complex using DEAE minicolumns.
Fig. 3B shows that both the rate and the extent of glycolipid release from the wtGLTP/glycolipid complex to the acceptor POPC SUVs depended on the amount of protein complex. When GLTP (1, 2, or 5 μg) containing complex was incubated with acceptor vesicles, the molar transfer of GalCer increased with higher protein amounts. However, the percentage of total GalCer release remained similar (∼30%). For instance, GLTP (2 μg) loaded with 31 pmol of GalCer released only one-third of the glycolipid (∼10 pmol) during a 1-h incubation with POPC vesicles.
Fig. 3B also shows that the observed release of glycolipid from the GLTP/glycolipid complex required the presence of POPC vesicles. It is noteworthy that a 4-fold increase in acceptor vesicle concentration resulted in only slightly faster glycolipid release from the GLTP/glycolipid complex and in only a slightly higher equilibrium level of glycolipid release. This finding suggested that release of glycolipid from the GLTP/glycolipid complex was not limited by the availability of acceptor vesicles. In the presence of excess POPC acceptor membrane, the equilibrium distribution of [3H]GalCer is expected to reflect the relative affinities of GLTP and the acceptor membrane for the glycolipid because the ligand is not covalently attached to the protein. Interestingly, the glycolipid showed stronger affinity for GLTP than POPC vesicles, with ∼75% remaining associated with GLTP. However, because the complex produced by incubation with immobilized glycolipid films also contained “glycolipid-free” protein, there was a possibility that the observed [3H]GalCer release to the POPC acceptor SUVs might be affected by the glycolipid-free protein. To evaluate this possibility, additional experiments were performed. Glycolipid-free GLTP (1.5 μg) was added to assay mixtures containing partially loaded GLTP (2 μg) and POPC SUVs (Fig. 3B, dotted line). However, no significant change in the glycolipid release profile to the acceptors was observed. Thus, the presence of glycolipid-free protein mixed with the complex did not affect the release of glycolipid by GLTP.
Release of GalCer from GLTP Point Mutant Complexes to Acceptor Vesicles—The release of glycolipid from isolated point mutant GLTP/GalCer complexes to acceptor POPC SUVs was investigated under identical conditions as wtGTLP (Fig. 3C). As with wtGTLP, the initial rates of glycolipid release to POPC acceptor vesicles were much faster than their acquisition rates from immobilized films of pure glycolipid. This outcome was not surprising considering the inherent differences between immobilized lipid films and SUVs, i.e. surface curvature, and the known effects that surface curvature and the lipid packing state have on GLTP transfer activity (3, 5, 10). Among mutant GLTP/glycolipid complexes, the glycolipid release to POPC acceptor vesicles followed a pattern that was similar to the rank order of their glycolipid uptake pattern (Fig. 3, C versus A). For instance, glycolipid release by point mutants F183S and F148S also attained values that were ∼80 and ∼50%, respectively, of that observed for wtGTLP (Fig. 3C). Although these findings are not surprising because of the expected thermodynamic reversibility of ligand acquisition and release, interesting deviations were observed in the response of point mutants N52I (∼35%) and D48V (0%), which showed lower than expected release values as compared with wtGTLP. The implications of the altered glycolipid release by point mutants N52I and D48V are elaborated below and under “Discussion.”
Intermembrane Transfer Kinetics of GLTP Point Mutants— Our next goal was to evaluate the acquisition and release of glycolipid by wtGTLP and related point mutants within the context of the intermembrane transfer process of glycolipid. Fig. 3D shows the kinetic time course of [3H]GalCer transfer from donor to acceptor vesicles for each point mutant and for wtGTLP. Recognition center mutants D48V and N52I were significantly inactivated (residual activities of ∼12 and ∼7%, respectively), and mutant W96A was almost completely inactive (∼1% activity). Hydrophobic tunnel mutants F183S and F148S retained considerable activity (residual activity of ∼70 and 40%, respectively), but L136R was severely inactivated (∼1% activity). Comparison of the time-dependent intervesicular glycolipid transfer activities of the GLTP point mutants (Fig. 3D) with their patterns of glycolipid acquisition (Fig. 3A) and membrane-stimulated release (Fig. 3C) provides insights into the functional consequences of the defects. A basic tenet that emerges is the requirement for GLTP to form a soluble complex with glycolipid to catalyze the intermembrane transfer of glycolipid, consistent with a carrier mode of GLTP action. This tenet is supported by the finding that none of the mutants could catalyze the intermembrane transfer of glycolipid without forming a soluble GLTP/glycolipid complex. In contrast, point mutant N52I was severely impaired in its ability to transfer glycolipid between vesicles, although it could acquire and release glycolipid moderately well (Fig. 3, A and D). Although a similar situation appeared to exist for point mutant D48V, the more restricted ability to acquire glycolipid (∼25% as compared with that of wtGTLP) made unequivocal quantitation of glycolipid release more difficult to establish.
Complexation of LacCer and Ganglioside GM1 by GLTP— Mammalian GLTPs efficiently transfer GalCer, GlcCer, and LacCer, but the selection specificity among more complex glycolipids, including gangliosides, is less clear (1, 2, 5, 6, 11, 12, 29, 32). To determine whether wtGTLP can ligand LacCer and GM1, we initially attempted to use the immobilized glycolipid film approach that had been successful with GalCer. However, difficulties were encountered because both LacCer and GM1 did not remain tightly adsorbed to the glass surface after hydration and tended to desorb into buffer alone (data not shown). To circumvent the problem, a separation approach based on Ni-NTA affinity chromatography was developed. We reasoned that, after incubation of His·GLTP with SUVs containing glycolipid, the mixture could be separated on Ni-NTA resin because His·GLTP would bind strongly to the resin, but the vesicles would not. The strategy was initially tested by incubating His·GLTP (1 mg) with SUVs comprised of POPC:GalCer (99:1 mol/mol) for 1 h at room temperature with moderate shaking. After incubation, Ni-NTA resin was added to the GLTP/SUVs mixture to bind the His fusion protein to the resin. Washing of the resin to remove vesicles was continued until background levels of radioactivity were reached (Fig. 4A, fractions 1–10). Bound His fusion GLTP, which contained significant amounts of [3H]GalCer, was released by elution with buffer containing 200 mm and 1 m imidazole (Fig. 4A, fractions 11–15). The elution profiles for GLTP and radioactive [3H]GalCer coincided. Control experiments with 100 mol % POPC SUVs showed that no [14C]POPC co-eluted with soluble protein, confirming that rGLTP selectively extracted GalCer from SUVs (data not shown). The [3H]GalCer/GLTP complex, recovered from the Ni-NTA resin, was evaluated to address the possibility that vesicles containing residual [3H]GalCer were responsible for the glycolipid counts that co-eluted with the soluble protein. Analysis by SEC using Sephacryl S-300 revealed no radioactivity in fractions corresponding to the elution position of SUVs containing residual radioactive glycolipid (Fig. 1B, fractions 10–15). All radioactive [3H]GalCer co-eluted with protein (Fig. 1B, fractions 20–25). We concluded that the radioactive glycolipid recovered in the 0.2 and 1 m imidazole washes represents glycolipid liganded to GLTP to form a soluble GLTP/glycolipid complex. The Ni-NTA approach was further validated by using it to assess the liganding capacity of hydrophobic channel mutant F148S and sugar headgroup recognition center mutant D48V. Fig. 4B shows that F148S accumulated GalCer much better than D48V, consistent with the liganding data obtained using the immobilized GalCer films (Fig. 3A). We then used the Ni-NTA approach to evaluate the ability of wtGTLP to acquire LacCer from POPC vesicles. [3H]LacCer was found co-eluting with imidazole-released wt-GTLP, suggesting the formation of a GLTP/LacCer complex (data not shown). Parallel SEC experiments using Sephacryl S-300 to separate mixtures of protein and SUVs containing [3H]LacCer provided complementary evidence for formation of the GLTP/LacCer complex. Fig. 5 shows that, in addition to the excess residual [3H]LacCer remaining with the POPC SUV peak (fractions 11–15), substantial [3H]LacCer co-eluted with the soluble wtGLTP peak (fractions 20–26).
Fig. 4.
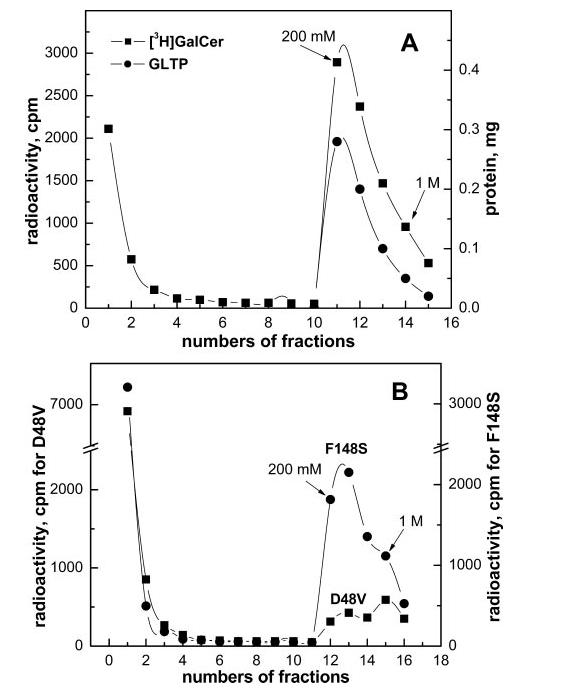
Ni-NTA affinity separation of His fusion GLTP from SUVs containing glycolipid. His fusion wtGLTP (1 mg) was incubated for 1 h with SUVs (0.5 ml) comprised of POPC:GalCer (99:1 mol/mol; 5 nmol total [3H]GalCer) and loaded on Ni-NTA resin. Fractions 1–10 corresponded to SUVs (containing glycolipid) that eluted by washing with buffer. Fractions 11–15 corresponded to the protein that eluted by washing with imidazole buffer (200 mm and 1 m). A similar elution profile was observed when GM dispersions were incubated with wtGTLP and then separated using Ni-NTA affinity resin (A). B, use of the Ni-NTA affinity separation approach with His fusion GLTP point mutants. GLTP mutants (1 mg), F148S (●) or D48V (■), were incubated for 1 h at room temperature with SUVs comprised of POPC:GalCer (99:1 mol/mol) (5 nmol total GalCer) and [3H]GalCer. Separation was achieved by elution through the Ni-NTA resin as described for panel A.
Fig. 5.
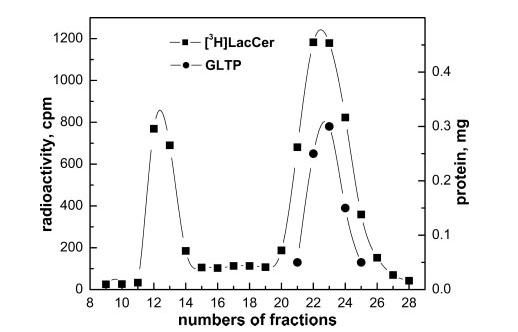
Liganding of LacCer by GLTP. GLTP (1 mg) was incubated for 1 h with SUVs (0.5 ml) comprised of POPC:LacCer (99:1 mol/mol; 5 nmol total [3H]LacCer) and then separated on a Sephacryl S-300 column. The lipid vesicles (■) eluted first (fractions 11–14) and were followed by the soluble GLTP/LacCer complex (●) (fractions 20–26).
We also determined whether rGLTP could form a complex with ganglioside GM1. Because GM1 contains a five-sugar, negatively charged headgroup, this glycolipid is at least 1000-fold more soluble than GalCer and disperses as micelles rather than bilayers at higher concentrations in aqueous buffer. To test whether GLTP could acquire aqueously dispersed ganglioside GM1, wtGTLP (1 mg) was incubated with [3H]GM1 (5 μm total concentration). After 1 h, the solution was applied on the Ni-NTA resin. All unbound ganglioside was removed from the resin during washing, and the protein was eluted with imidazole buffer. The results were similar with those shown in Fig. 4. Most of the [3H]GM1 co-eluted with the protein (fractions 10 – 15). In a separate control experiment, wtGTLP was incubated with [3H]GM1 (5 μm) for 1 h and then applied directly to a Sephacryl S-300 SEC column. The result was identical with that in Fig. 1B. The [3H]GM1 elution peak coincided with the protein peak. The result indicated that GLTP is capable of acquiring glycolipid dispersed in nonbilayer forms, such as ganglioside micelles and/or possibly monomers.
DISCUSSION
GLTP utilizes a unique layering of α-helices to form a glycolipid liganding site that consists of a sugar headgroup recognition center and a molded-to-fit hydrophobic cavity for the hydrocarbon chains of ceramide (13). The all α-helix conformation of GLTP, achieved without intramolecular disulfide bridges, contrasts the situation in other lipid-binding and transfer proteins, which generally use motifs dominated by β-sheet, i.e. β-grooves/-concave cups and β-barrels, or helical bundles stabilized by multiple disulfide bridges, i.e. saposin-folds. Such proteins include nonspecific lipid transfer proteins, CD1 proteins, steroidogenic acute regulatory protein-related lipid transfer proteins, fatty acid-binding proteins, lipocalins, plant lipid transfer proteins, and saposins (14–23, 33–37). Both the conformation and the primary sequence of GLTP suggest that this protein represents an emerging new family of proteins that bind/transfer glycolipids. As such, it was of interest to investigate the interrelationships between the liganding and transferring of glycolipid by GLTP.
Although previous studies with porcine and bovine GLTP had established the ability of the protein to selectively accelerate the intermembrane transfer of glycolipids in vitro (1–7), the mechanism of action of the protein has remained unclear. Abe and Sasaki (4) used nondenaturing polyacrylamide gel electrophoresis and Sephadex G-75 SEC to show formation of a soluble GLTP/GalCer complex along with no protein bound to vesicles containing glycolipid and proposed that GLTP functions as a soluble intermediate that carries glycolipids between membranes. However, only 13 mol % of the porcine GLTP molecules acquired GalCer (4, 38, 39). Other attempts to demonstrate formation of soluble GLTP/glycolipid complexes have resulted in conflicting data. Metz and Radin (1) found that cerebroside transfer protein from bovine spleen binds small amounts of GalCer (∼4%) but also observed that the diffusion of the protein was reduced by mixing with liposomes or red cell ghosts that contained glycolipids. They speculated that the protein desorbs from the membrane surface as a protein/lipid complex that then rapidly dissociates in solution before reaching an acceptor membrane. Wong et al. (3) found that a substantial fraction (30 – 40%) of partially purified bovine brain GLTP coelutes with POPC:GalCer vesicles but were unable to detect glycolipid acquisition by the protein. Brown et al. (5) reported weak association of bovine brain GLTP with pyrene-labeled glucosylceramide using fluorescence approaches but found no evidence of protein binding to vesicles containing glycolipid. Sasaki and colleagues (40) showed that porcine brain GLTP acquires pyrene-labeled GalCer from vesicles and forms a complex but found no GLTP/glycolipid complex in the subphase beneath radiolabeled GalCer monolayers (41). In all of these previous studies, GLTP was isolated using differing approaches from animal tissues known to contain various glycosphingolipids. Consequently, at least partial occupation of the glycolipid liganding site would be expected, adding to the difficulties associated with such binding studies.
In the present study, we used recombinant GLTP cloned from human skin fibroblasts (9, 13). Aside from being 98% homologous to the bovine and porcine GLTPs, our human rGLTP is purified free of glycosphingolipid because E. coli do not synthesize glycosphingolipids. By developing new separation strategies involving immobilized glycolipid films and Ni-NTA agarose, we could achieve sufficiently rapid isolation of GLTP to enable efficient monitoring of glycolipid acquisition and release by GLTP. Interestingly, we observed similar outcomes regardless of whether the glycolipid ligand-to-GLTP molar ratio during loading was 10:1 or 2:1, probably because of the compromised conditions resulting from the ligand being insoluble and associated with surfaces such as glass or phosphatidyl choline vesicles. Nonetheless, we found no significant difference in the relative response pattern of the ligand site point mutants as compared with wtGTLP with respect to GalCer uptake and release. All our data consistently indicated that wtGLTP forms a soluble, stable complex with glycolipids, that release of glycolipid from the wtGTLP/complex is observed in the presence of acceptor membranes, and that these abilities correlate with the intermembrane transfer activity of the protein. Interestingly, however, upon mixing GLTP/glycolipid complex with acceptor vesicles, the glycolipid release was limited to ∼25% of the total complexed glycolipid. Calculation of the acceptor vesicle-to-protein molar ratio revealed a 5:1 ratio, assuming that each vesicle consisted of ∼2,500 lipids (42), and confirmed that excess target membrane surface was available for interaction with GLTP. A 4-fold increase in the acceptor concentration only slightly increased glycolipid release from the GLTP/glycolipid complex. Thus, there was a clear tendency of glycolipid to remain complexed with wtGLTP, even in the presence of excess bilayer vesicles. We are currently exploring whether differing conformations of the glycolipid ligand within the GLTP liganding site might provide a rational explanation for our observations.
Our point mutational disruptions of GLTP targeted key interaction sites within the sugar recognition center and the hydrophobic channel that comprise the single liganding site, enabling analysis of the interrelationship between glycolipid liganding and transfer. The mutations selectively altered the stacking between the sugar and indole rings (W96A); the hydrogen bonding of the sugar C2 hydroxy (D48V) and the sugar C3 hydroxy (N52I); the gating action by the Phe-148 phenyl group that affects entry of the sphingoid base chain (F148S); the hydrophobic contacts in the channel involving the C10–C12 carbons of the sphingoid base chain (L136R); and accommodation of the terminal methyl group of the acyl chain (F183S) in the channel. Functional analyses of glycolipid acquisition and release by the point mutants suggested two fundamentally different types of defects: mutants with impaired ability to form a soluble GLTP/glycolipid complex and mutants with impaired ability to acquire and release glycolipid. Either defect impaired the glycolipid intermembrane transfer activity of GLTP. Because mutants of the first type could unload their glycolipid ligand as efficiently as wtGTLP (∼25–30%) in the presence of membranes, the defect within these point mutants appears to primarily affect the affinity of the liganding interaction with glycolipid and not the membrane-dependent release of glycolipid by the mutants. Examples are point mutants F183S and F148S. In contrast, because the second type of mutants show impaired abilities both to acquire and to release (∼17%) glycolipid ligand, the defect may reflect a diminished capacity to properly adsorb/desorb with the membranes themselves. Examples are point mutant N52I and possibly D48V. Verification of this suggestion will require direct assessment of the physical association between membranes and GLTP (and related point mutants) during the glycolipid acquisition and release processes to establish whether GLTP utilizes an interfacial or noninterfacial mechanism (43) to ligand and release glycolipid. The development of such assessment capabilities for GLTP is currently underway (44). In any case, liganding of glycolipid by GLTP appears to be a necessary and prerequisite step for accomplishing transfer via a carrier mode of action.
The results of the present study showed that the glycolipid ligand need not be dispersed among phosphoglycerides within a fluid-phase bilayer environment to be acquired by GLTP. Liganding of GalCer was observed by GLTP upon incubation with pure GalCer films adsorbed to glass. The GalCer in such pure films can be expected to be tightly packed in a lateral sense because the main thermotropic transition temperature of GalCer occurs at ∼80 °C (45). GLTP also interacted efficiently with ganglioside GM1. This result is noteworthy for two reasons. First, some earlier reports indicated that complex glycolipids are not transferred efficiently by GLTP (1, 2, 6, 11). However, our present data indicated that ganglioside GM1 is acquired by GLTP, supporting the previously reported ability of GLTP to transfer GM1 between vesicles (5, 6, 32). Secondly, the GM1 used in the present study was dispersed directly into aqueous buffer in the absence of phosphoglyceride, showing that GLTP can acquire glycolipid from nonbilayer sources. Because our GM1 aqueous concentration was 5 μm, it appeared likely that both micelles and monomers of GM1 were available to interact with GLTP. Although the critical micelle concentration of GM1 was originally reported to be in the 10-5-10-6 m range (46 – 48), more recent determinations indicate critical micelle concentration values in the 10-7-10-8 m range with highly purified GM1 and with correction for nonspecific adsorption to tube walls (49–54). Although our data raised the possibility that GLTP can directly acquire monomeric GM1 from solution without interacting with GM1 micelles, as has recently been proposed for human cytosolic sialidase action on gangliosides GM1, GD1a, and GM2 (54), the dramatically increased local concentration of GM1 within micelles might serve to enhance the efficiency of uptake upon interaction with GLTP. Regardless of whether GLTP binds GM1 monomers or extracts the ganglioside from the micelles, it is clear that ganglioside GM1 need not be dispersed in phosphoglyceride membranes to be acquired by GLTP.
IMPLICATIONS
In the present study, we used point mutational analysis of the GLTP liganding site, guided by our recent structural determinations, to elaborate a framework for understanding how GLTP employs its novel conformational motif to acquire and release glycolipids during the intermembrane transfer of glycolipids. Although our data are consistent with GLTP functioning as a glycolipid carrier, our observations also raise the possibility that GLTP action in vivo may not be limited to membrane-to-membrane transfer of glycolipids, as has been proposed previously (55). The finding that glycolipids need not be integrated into fluid phase membranes to be acquired by GLTP and the finding that the POPC vesicle-dependent release of glycolipid from GLTP/glycolipid complexes occurs inefficiently suggests that other in vitro roles for GLTP be considered and explored. Other possibilities include a role in the presentation and/or delivery of glycolipids to protein receptors, analogous to the situation existing for glycolipid antigens and endosomal sphingolipid activator proteins (21). Another potential in vivo role might be as a glucosylceramide intracellular sensor to help control intracellular ceramide levels, thereby regulating programmed cell death. In any case, because of the highly conserved and novel folding motif used by mammalian GLTPs to effectively and selectively ligand glycolipids, studies are presently underway to assess how glycolipid liganding affects the thermodynamic and conformational stability of the protein.
Acknowledgments
Acknowledgements—We thank Prof. Taeowan Chung of Yeungnam University for the helpful advice during the cloning and mutagenesis of GLTP, Prof. Anthony J. Windebank for help with DNA sequencing at the Mayo Molecular Biology Core Facility, Dr. Xin-Min Li for synthesizing and purifying the unlabeled N-16:0 LacCer, and Dr. Barbara Malewicz for valuable suggestions during preparation of the manuscript.
Footnotes
This work was supported in part by NIGMS National Institutes of Health Grant 45928 and a grant from the Hormel Foundation. Parts of this study were presented in preliminary form at the 48th Biophysical Society Annual Meeting in Baltimore, MD (Malakhova, M. L., Malewicz, B. M., Pike, H. M., Brown, R. E. (2004) In vitro association of glycolipid transfer protein with glycolipid. Biophys. J. 86, 562a, 2916 – Pos). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- GLTP
- glycolipid transfer protein
- rGLTP
- recombinant GLTP
- wtGLTP
- wild type GLTP
- GalCer
- galactosylceramide
- LacCer
- lactosylceramide
- POPC
- 1-palmitoyl-2-oleyl-phosphatidylcholine
- GM1
- monosialoganglioside
- SUV
- small unilamellar vesicle
- Ni-NTA
- nickel-nitrilotriacetic acid
- SEC
- size exclusion chromatography.
Nucleotide sequences for mammalian GLTP can be accessed in the GenBank™/EBI Databank: human skin fibroblast (AF209704), bovine brain (AF209701, NM016433), porcine brain (AF209702, NM016433), murine JB6 epidermal cells (AF209703). Corresponding amino acids sequences can be accessed in the NCBI Protein Database: human (AAF33210, NP_057517), bovine (AAF33207, P17403, NP_786993), porcine (AAF33208, P17403), murine (AAF33209, Q9JL62).
M. L. Malakhova, L. Malinina, H. M. Pike, A. T. Kanack, D. J. Patel, and R. E. Brown, unpublished observation.
REFERENCES
- Metz RJ, Radin NS. J. Biol. Chem. 1982;257:12901–12907. [PubMed] [Google Scholar]
- Abe A, Yamada K, Sasaki T. Biochem. Biophys. Res. Commun. 1982;104:1386–1393. doi: 10.1016/0006-291x(82)91403-6. [DOI] [PubMed] [Google Scholar]
- Wong M, Brown RE, Barenholz Y, Thompson TE. Biochemistry. 1984;23:6498–6505. doi: 10.1021/bi00321a035. [DOI] [PubMed] [Google Scholar]
- Abe A, Sasaki T. J. Biol. Chem. 1985;260:11231–11239. [PubMed] [Google Scholar]
- Brown RE, Stephenson FA, Markello TC, Barenholz Y, Thompson TE. Chem. Phys. Lipids. 1985;38:79–93. doi: 10.1016/0009-3084(85)90059-3. [DOI] [PubMed] [Google Scholar]
- Gammon CM, Vaswani KK, Ledeen RW. Biochemistry. 1987;26:6239–6243. doi: 10.1021/bi00393a043. [DOI] [PubMed] [Google Scholar]
- Brown RE, Jarvis KL, Hyland KJ. Biochim. Biophys. Acta. 1990;1044:77–83. doi: 10.1016/0005-2760(90)90221-i. [DOI] [PubMed] [Google Scholar]
- Lin X, Mattjus P, Pike HM, Windebank AJ, Brown RE. J. Biol. Chem. 2000;275:5104–5110. doi: 10.1074/jbc.275.7.5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-M, Malakhova ML, Lin X, Pike HM, Chung T, Molotkovsky JG, Brown RE. Biochemistry. 2004;43:10285–10294. doi: 10.1021/bi0495432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao CS, Pike HM, Molotkovsky JG, Brown RE. Biochemistry. 2004;43:13805–13815. doi: 10.1021/bi0492197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Abe A, Sasaki T. J. Biol. Chem. 1985;260:4615–4621. [PubMed] [Google Scholar]
- Yamada K, Abe A, Sasaki T. Biochim. Biophys. Acta. 1986;879:345–349. [PubMed] [Google Scholar]
- Malinina L, Malakhova ML, Brown RE, Patel DJ. Nature. 2004;430:1048–1053. doi: 10.1038/nature02856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz KWA. Biochem. J. 1977;324:353–360. [Google Scholar]
- Choinowski T, Hauster H, Piontek K. Biochemistry. 2000;39:1897–1902. doi: 10.1021/bi992742e. [DOI] [PubMed] [Google Scholar]
- Stolowich NJ, Petrescu AD, Huang H, Martin GG, Scott AI, Schroeder FS. CMLS Cell. Mol. Life Sci. 2002;59:193–212. doi: 10.1007/s00018-002-8416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert Wright C, Zhao Q, Rastinejad F. J. Mol. Biol. 2003;331:951–964. doi: 10.1016/s0022-2836(03)00794-0. [DOI] [PubMed] [Google Scholar]
- Ahn VE, Faull KF, Whitelegge JP, Fluharty AL, Prive GG. Proc. Natl. Acad. Sci. U. S. A. 2003;100:38–43. doi: 10.1073/pnas.0136947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alba E, Weiler S, Tjandra N. Biochemistry. 2003;42:14729–14740. doi: 10.1021/bi0301338. [DOI] [PubMed] [Google Scholar]
- Schuette CG, Pierstorff B, Huettler S, Sandhoff K. Glycobiology. 2001;11:81R–90R. doi: 10.1093/glycob/11.6.81r. [DOI] [PubMed] [Google Scholar]
- Zhou D, Cantu C, Sagiv Y, Schrantz N, Kulkarni AS, Qi X, Mahuran DJ, Morales CR, Grabowski GA, Benlagha K, Savage P, Bendelac A, Teyton L. Science. 2004;303:523–527. doi: 10.1126/science.1092009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadola SD, Zaccai NR, Harlos K, Shepherd D, Castro-Palomino JC, Ritter G, Schmidt RR, Jones EY, Cerundolo V. Nat. Immunol. 2002;3:721–726. doi: 10.1038/ni821. [DOI] [PubMed] [Google Scholar]
- Zajonc DM, Elsliger MA, Teyton L, Wilson IA. Nat. Immunol. 2003;4:808–815. doi: 10.1038/ni948. [DOI] [PubMed] [Google Scholar]
- Godi A, DiCampli AD, Konstantakopoulos A, Di Tullio G, Alessi DR, Kular GS, Daniele T, Marra P, Lucocq JM, De Matteis MA. Nat. Cell Biol. 2004;6:393–404. doi: 10.1038/ncb1119. [DOI] [PubMed] [Google Scholar]
- De Matties MA, Godi A. Biochim. Biophys. Acta. 2004;1666:264–274. doi: 10.1016/j.bbamem.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Broedersen P, Petersen M, Pike HM, Olszak B, Skov S, Odum N, Jorgensen LB, Brown RE, Mundy J. Genes Dev. 2002;16:490–502. doi: 10.1101/gad.218202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattjus P, Turcq B, Pike HM, Molotkovsky JG, Brown RE. Biochemistry. 2003;42:535–542. doi: 10.1021/bi026896x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Thompson TE. Biochemistry. 1987;26:5454–5460. doi: 10.1021/bi00391a036. [DOI] [PubMed] [Google Scholar]
- Brown RE, Hyland KJ. Biochemistry. 1992;31:1062–1069. doi: 10.1021/bi00158a024. [DOI] [PubMed] [Google Scholar]
- Barenholz Y, Gibbes D, Litman BJ, Goll J, Thompson TE, Carlson FD. Biochemistry. 1977;16:2806–2810. doi: 10.1021/bi00631a035. [DOI] [PubMed] [Google Scholar]
- Mattjus P, Pike HM, Molotkovsky JG, Brown RE. Biochemistry. 2000;39:1067–1075. doi: 10.1021/bi991810u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Sugár IP, Thompson TE. Biochemistry. 1985;24:4082–4091. doi: 10.1021/bi00336a042. [DOI] [PubMed] [Google Scholar]
- Soccio RE, Breslow JL. J. Biol. Chem. 2003;278:22183–22186. doi: 10.1074/jbc.R300003200. [DOI] [PubMed] [Google Scholar]
- Roderick SL, Chan WW, Agate DS, Olsen LR, Vetting MW, Rajashankar KR, Cohen DE. Nat. Struct. Biol. 2002;9:507–511. doi: 10.1038/nsb812. [DOI] [PubMed] [Google Scholar]
- Hamilton JA. Prog. Lipid Res. 2004;43:177–199. doi: 10.1016/j.plipres.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Campanaccia V, Nurizzob D, Spinellia S, Valenciaa C, Tegonia M, Cambillaua C. FEBS Lett. 2004;562:183–188. doi: 10.1016/S0014-5793(04)00199-1. [DOI] [PubMed] [Google Scholar]
- Kader J-C. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996;47:627–654. doi: 10.1146/annurev.arplant.47.1.627. [DOI] [PubMed] [Google Scholar]
- Sasaki T. Chem. Phys. Lipids. 1985;38:63–77. doi: 10.1016/0009-3084(85)90058-1. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Abe A, Roerink F. Subcell. Biochem. 1990;16:113–127. doi: 10.1007/978-1-4899-1621-1_5. [DOI] [PubMed] [Google Scholar]
- Abe A, Yamada K, Sakagami T, Sasaki S. Biochim. Biophys. Acta. 1984;778:239–244. doi: 10.1016/0005-2736(84)90364-x. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Demel RA. Biochemistry. 1985;24:1079–1083. doi: 10.1021/bi00326a002. [DOI] [PubMed] [Google Scholar]
- Huang C, Thompson TE. Methods Enzymol. 1974;32:485–489. doi: 10.1016/0076-6879(74)32048-4. [DOI] [PubMed] [Google Scholar]
- Gelb MH, Jung-Hyun M, Jain MK. Biochim. Biophys. Acta. 2000;1488:20–27. doi: 10.1016/s1388-1981(00)00106-2. [DOI] [PubMed] [Google Scholar]
- Rao CS, Chung T, Pike HM, Brown RE. Biophys. J. 2005;89:4017–4028. doi: 10.1529/biophysj.105.070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koynova R, Caffrey M. Biochim. Biophys. Acta. 1995;1255:213–236. doi: 10.1016/0005-2760(94)00202-a. [DOI] [PubMed] [Google Scholar]
- Howard RE, Burton RM. Biochim. Biophys. Acta. 1964;84:435–440. doi: 10.1016/0926-6542(64)90007-1. [DOI] [PubMed] [Google Scholar]
- Yohe HC, Rosenberg A. Chem. Phys. Lipids. 1972;9:279–294. doi: 10.1016/0009-3084(72)90015-1. [DOI] [PubMed] [Google Scholar]
- Rauvala H. Eur. J. Biochem. 1979;97:555–564. doi: 10.1111/j.1432-1033.1979.tb13144.x. [DOI] [PubMed] [Google Scholar]
- Formisano S, Johnson ML, Edelhoch H. Biochemistry. 1979;18:1119–1124. doi: 10.1021/bi00573a028. [DOI] [PubMed] [Google Scholar]
- Corti M, Degiorgio V. Chem. Phys. Lipids. 1980;26:225–238. doi: 10.1016/0009-3084(80)90053-5. [DOI] [PubMed] [Google Scholar]
- Ulrich-Bott B, Wiegand H. J. Lipid Res. 1984;25:1233–1245. [PubMed] [Google Scholar]
- Sonnino S, Cantu L, Corti M, Acquotti D, Venerando B. Chem. Phys. Lipids. 1994;71:21–45. doi: 10.1016/0009-3084(94)02304-2. [DOI] [PubMed] [Google Scholar]
- Cantù L, Corti M, Del Favero E, Raudino A. J. Phys. C. 1997;9:5033–5055. [Google Scholar]
- Tringal C, Papini N, Fusi P, Croci G, Borsani G, Preti A, Tortora P, Tettamanti G, Venerando B, Monti E. J. Biol. Chem. 2004;279:3169–3179. doi: 10.1074/jbc.M308381200. [DOI] [PubMed] [Google Scholar]
- Sasaki T. Experientia. 1990;46:611–616. doi: 10.1007/BF01939700. [DOI] [PubMed] [Google Scholar]
- Tischer W, Kasche V. Trends Biotechnol. 1999;17:326–335. doi: 10.1016/s0167-7799(99)01322-0. [DOI] [PubMed] [Google Scholar]
- Berg OG, Gelb MH, Tsai MD, Jain MK. Chem. Rev. 2001;101:2613–2653. doi: 10.1021/cr990139w. [DOI] [PubMed] [Google Scholar]