

Abstract
Mesotrypsin is an enigmatic minor human trypsin isoform, which has been recognized for its peculiar resistance to natural trypsin inhibitors such as soybean trypsin inhibitor (SBTI) or human pancreatic secretory trypsin inhibitor (SPINK1). In search of a biological function, two conflicting theories proposed that due to its inhibitor-resistant activity mesotrypsin could prematurely activate or degrade pancreatic zymogens and thus play a pathogenic or protective role in human pancreatitis. In the present study we ruled out both theories by demonstrating that mesotrypsin was grossly defective not only in inhibitor binding, but also in the activation or degradation of pancreatic zymogens. We found that the restricted ability of mesotrypsin to bind inhibitors or to hydrolyze protein substrates was solely due to a single evolutionary mutation, which changed the serine-protease signature glycine-198 residue to arginine. Remarkably, the same mutation endowed mesotrypsin with a novel and unique function: mesotrypsin rapidly hydrolyzed the reactive-site peptide bond of the Kunitz-type trypsin inhibitor SBTI, and irreversibly degraded the Kazal-type temporary inhibitor SPINK1. The observations suggest that the biological function of human mesotrypsin is digestive degradation of trypsin inhibitors. This mechanism can facilitate the digestion of foods rich in natural trypsin inhibitors. Furthermore, the findings raise the possibility that inappropriate activation of mesotrypsinogen in the pancreas might lower protective SPINK1 levels and contribute to the development of human pancreatitis. In this regard, it is noteworthy that the well-known pathological trypsinogen activator cathepsin B exhibited a preference for the activation of mesotrypsinogen of all three human trypsinogen isoforms, suggesting a biochemical mechanism for mesotrypsinogen activation in pancreatic acinar cells.
The human pancreas secretes three isoforms of trypsinogen, which are encoded by the PRSS (protease, serine) genes PRSS1 (OMIM #276000), PRSS2 (OMIM #601564) and PRSS3. On the basis of their electrophoretic mobility, they are commonly referred to as cationic trypsinogen, anionic trypsinogen and mesotrypsinogen. The two major isoforms, cationic and anionic trypsinogen, constitute the bulk of secreted trypsinogen, whereas levels of mesotrypsinogen were reported between 3 and 10 % of total trypsinogen content in normal pancreatic juice (see Table 1 in ref [1]; Fig 8 in ref [2]; and Table 1 in ref. [3]). Rinderknecht et al. first discovered mesotrypsin in 1978 [4] as a new inhibitor-resistant protease found in human pancreatic tissue and fluid, and a systematic characterization was published in 1984 [2]. A cDNA coding for mesotrypsinogen was cloned from human pancreas in 1997 [5], and the crystal structure of mesotrypsin complexed with benzamidine was solved in 2002 [6]. An alternatively spliced form of mesotrypsinogen in which the signal-peptide is replaced with a novel sequence encoded by an alternative exon 1 is expressed in the human brain [7]. Although usually referred to as “brain trypsinogen”, there is no evidence for the activation of this novel chimeric molecule, which might have a function unrelated to proteolytic activity [8].
Table 1.
Kinetic parameters of human trypsins on the synthetic substrate N-CBZ-Gly-Pro-Arg-p-nitroanilide at 22 °C. PRSS1, human cationic trypsin; PRSS2, anionic trypsin; PRSS3, mesotrypsin; R198G, mesotrypsin mutant Arg198→Gly. Data for PRSS1 and PRSS2 were taken from reference [13].
| KM (μM) | kcat (s−1) | kcat/KM (M−1 s−1) | |
|---|---|---|---|
| PRSS1 | 15 ± 1 | 50 ± 1 | 3.3 * 106 |
| PRSS2 | 11 ± 1 | 41 ± 1 | 3.7 * 106 |
| PRSS3 | 22 ± 2 | 148 ± 4 | 6.7 * 106 |
| R198G | 10 ± 2 | 37 ± 1 | 3.7 * 106 |
Figure 8.
Activation of mesotrypsinogen by human trypsins and cathepsin B. Panel A. Mesotrypsinogen (2 μM) was activated with human cationic trypsin (PRSS1), anionic trypsin (PRSS2), mesotrypsin (PRSS3) or enterokinase (200 ng/mL) at 37 °C in 0.1 M Tris-HCl (pH 8.0), 1 mM Ca2+ and 2 mg/mL bovine serum albumin. Panel B. Human cationic trypsinogen (PRSS1), anionic trypsinogen (PRSS2) and mesotrypsinogen (PRSS3) were activated at 2 μM concentration with human cathepsin B (90 μg/mL) at 37 °C in 0.1 M Na-acetate buffer (pH 4.0) in the presence of 1 mM dithiothreitol, 2 mg/mL bovine serum albumin, 1 mM K-EDTA, and 300 μM benzamidine. Aliquots (2.5 μL) were withdrawn at indicated times and trypsin activity was measured on the synthetic substrate N-CBZ-Gly-Pro-Arg-p-nitroanilide. Trypsin activity was expressed as percentage of maximal activity determined by enterokinase activation at pH 8.0, in 20 mM CaCl2 and 2 mg/mL bovine serum albumin.
The most intriguing property of mesotrypsin is its resistance to polypeptide trypsin inhibitors (see Table 5 in ref [2]), such as the Kunitz-type soybean trypsin inhibitor (SBTI) or the Kazal-type pancreatic secretory trypsin inhibitor (SPINK1, serine protease inhibitor, Kazal type 1, OMIM #167790) [2, 5, 6]. Analysis of the recent crystal structure of mesotrypsin provided compelling evidence that the presence of an arginine residue in place of the highly conserved Gly198 (Gly193 in the chymotrypsin numbering system) is responsible for the peculiar inhibitor resistance of mesotrypsin [6]. Arg198 occupies the S2’ subsite and its long side-chain sterically clashes with protein inhibitors and possibly substrates. Furthermore, the charge of the guanidino group contributes to the strong clustering of positive charges around the primary specificity pocket of mesotrypsin. However, no direct experimental evidence has ever been presented for the proposed role of Arg198.
Despite the high-resolution crystal structure, the biological function of mesotrypsin has remained mysterious. In two clearly conflicting theories, it was proposed that premature activation of mesotrypsin in the pancreas might cause or protect against pancreatitis, as the inhibitor-resistant trypsin activity can freely activate or degrade other pancreatic zymogens [2, 5]. It was also suggested that mesotrypsin might have been “abandoned by the process of evolution”, and has no important role in digestion or pancreatic physiology [2, 8]. In the present study we identified a unique and specific role for mesotrypsin in the degradation of trypsin inhibitors. Furthermore, we found that this distinctive enzymatic activity was endowed by the evolutionary selection of Arg198. Finally, we showed that the lysosomal cysteine protease cathepsin B activated mesotrypsinogen at a higher rate, relative to the activation of human cationic and anionic trypsinogens. The observations not only indicate a physiological role for mesotrypsin, but also suggest that premature activation of mesotrypsinogen could contribute to the pathogenesis of human pancreatitis by reducing the protective levels of SPINK1.
EXPERIMENTAL PROCEDURES
Materials
N-CBZ-Gly-Pro-Arg-p-nitroanilide was purchased from Sigma. Suc-Ala-Ala-Pro-Phe-p-nitroanilide was from Bachem (King of Prussia, PA), ultrapure bovine enterokinase was from Biozyme Laboratories (San Diego, CA), and reagent grade bovine serum albumin was from Biocell Laboratories (Rancho Dominguez, CA). Bovine chymotrypsinogen A, TLCK-treated bovine chymotrypsin and TPCK-treated bovine trypsin was obtained from Worthington Biochemical Corp. (Lakewood, NJ). The concentration of bovine trypsin was determined by active-site titration with p-nitrophenyl-p'-guanidinobenzoate (Sigma) as described in [9]. Soybean (Glycine max) trypsin inhibitor (Kunitz type) was from Fluka (#93619), and was further purified on an affinity column containing immobilized S200A mutant human cationic trypsin. Human SPINK1 was expressed in Saccharomyces cerevisiae and purified on the S200A affinity column. Inhibitor concentrations were determined by titration with bovine trypsin. Human pro-elastase II (ELA2A) was expressed in Escherichia coli and purified by ecotin-affinity chromatography. Details of expression for SPINK1 and ELA2A will be reported elsewhere. Ecotin, human cationic trypsinogen and human anionic trypsinogen were expressed and purified as reported previously [10–13]. Human recombinant cathepsin B was a generous gift from Paul M. Steed (Research Department, Novartis Pharmaceuticals, Summit, New Jersey). Before use, cathepsin B was activated with 1 mM dithiothreitol (final concentration) for 30 min on ice.
Expression and purification of mesotrypsinogen
The gene encoding mesotrypsinogen was PCR-amplified from the IMAGE clone #2659811 (GenBank #AW182356, purchased from Incyte Genomics Reagents & Services, St. Louis, Missouri) and ligated into the expression plasmid pTrap [14] behind the alkaline phosphatase promoter and signal-sequence. Mutation R198G was introduced via oligonucleotide-directed site-specific mutagenesis, using the overlap-extension PCR mutagenesis method. Mesotrypsinogen was expressed in E. coli SM138 [F−, araD139, Δ(lac)U169, relA, rpsL, phoR], as described previously [14, 15]. Typically, 2.4 L cultures of SM138/pTrap in Luria-Bertani medium with 100 μg/mL ampicillin were grown to saturation overnight and periplasmic fractions were isolated by osmotic shock. Tris-HCl (pH 8.0) and NaCl were added to a final concentration of 20 mM and 0.2 M, respectively, and the approximately 260 mL periplasm was applied directly to an ecotin affinity column [10]. The column was washed with 20 mM Tris-HCl (pH 8.0)/0.2 M NaCl, and the zymogen was eluted with 50 mM HCl. The propeptide sequence of the mature secreted trypsinogen expressed from the pTrap plasmid was Ile-Gln-Ala-Phe-Pro-Val-(Asp)4-Lys.
In an attempt to increase yield, the mesotrypsinogen gene was transferred to the pTrap-T7 expression plasmid, which was originally constructed for the high-level expression of human cationic trypsinogen [11, 12]. The pTrap-T7 plasmid harboring the mesotrypsinogen gene was transformed into the E. coli Rosetta(DE3) strain (Novagen), which is a BL21(DE3) derivative strain carrying a chromosomal copy of T7 RNA polymerase under the control of the lacZ promoter. 50 mL cultures were grown in Luria-Bertani medium with 100 μg/mL ampicillin and 34 μg/mL chloramphenicol to an OD600 nm of 0.5, induced with 1 mM isopropyl 1-thio-β-galactopyranoside, and grown for an additional 5 h. Cells were harvested by centrifugation and inclusion bodies were isolated by sonication and centrifugation. In vitro refolding of mesotrypsinogen from the inclusion bodies was accomplished as described previously [11, 12, 16]. Finally, re-folded zymogen was purified on an ecotin affinity column [10]. The propeptide sequence of recombinant mesotrypsinogen expressed from the pTrap-T7 plasmid was Met-Val-Pro-Phe-(Asp)4-Lys. Concentrations of mesotrypsinogen solutions were calculated from the ultraviolet absorbance at 280 nm, using a theoretical extinction coefficient of 40,570 M−1 cm−1.
Protease activity assays
Trypsin and chymotrypsin activity was determined using the synthetic chromogenic substrates N-CBZ-Gly-Pro-Arg-p-nitroanilide (0.1–0.14 mM final concentration) and Suc-Ala-Ala-Pro-Phe-p-nitroanilide (0.15 mM final concentration), respectively. The release of the yellow p-nitroanilide was followed at 405 nm in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2, at 22 °C using a Spectramax Plus 384 microplate reader (Molecular Devices). Elastase was assayed with DQ-elastin fluorescent substrate (Molecular Probes, EnzCheck elastase assay kit) according to the manufacturer’s instructions using a SpectraMax Gemini XS fluorescent microplate reader (Molecular Devices).
Inhibitor assays
Tight-binding inhibition of trypsin by SBTI or SPINK1 was measured by incubating 15–50 nM trypsin with given concentrations of the inhibitor in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 1.5 mg/mL bovine serum albumin for 10 min at room temperature. Residual activity was then determined with N-CBZ-Gly-Pro-Arg-p-nitroanilide as described above. With the exception of mesotrypsin, no significant dissociation of the inhibitor-trypsin complex was detectable during the 1 min assay time.
Inhibitor degradation
SBTI or SPINK1 were incubated with mesotrypsin or the indicated protease in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 1 mg/mL bovine serum albumin at 37 °C. Aliquots were withdrawn at indicated times, mixed with bovine trypsin at a concentration slightly above the initial inhibitor concentration and incubated at room temperature for 1 min before the residual trypsin activity was measured with N-CBZ-Gly-Pro-Arg-p-nitroanilide. Because association of cleaved (“modified”) SBTI to bovine trypsin is considerably slower than association of the intact (“virgin”) SBTI, the SBTI activity detected after 1 min incubation with bovine trypsin is a specific measure of the virgin SBTI concentration [17].
Gel electrophoresis and densitometric analyses
Trypsinogen and SBTI samples were precipitated with trichloroacetic acid (10 % final concentration), the precipitate was dissolved in Laemmli sample buffer containing 100 mM dithiothreitol (final concentration) and samples were heat-denatured at 95 °C for 5 min. Electrophoretic separation was performed on 13 % SDS-PAGE mini gels in standard Tris-glycine buffer. SPINK1 samples were precipitated with 20 % trichloroacetic acid (final concentration), the precipitate was dissolved in sample buffer containing 200 mM Tris-HCl (pH 6.8), 20 % glycerol, 2 % SDS, 0.04 % Coomassie Blue G-250 and 100 mM dithiothreitol (final concentrations). Samples were heat-denatured at 95 °C for 5 min and electrophoretic separation was performed on 16 % SDS-PAGE mini gels in Tris-tricine buffer. Gels were stained with Coomassie Brilliant Blue R for 30 min, and destained with 30 % methanol, 10 % acetic acid overnight. Densitometric quantitation of bands was carried out as described in [18].
RESULTS
Expression of mesotrypsinogen, activation with enterokinase and catalytic properties of mesotrypsin
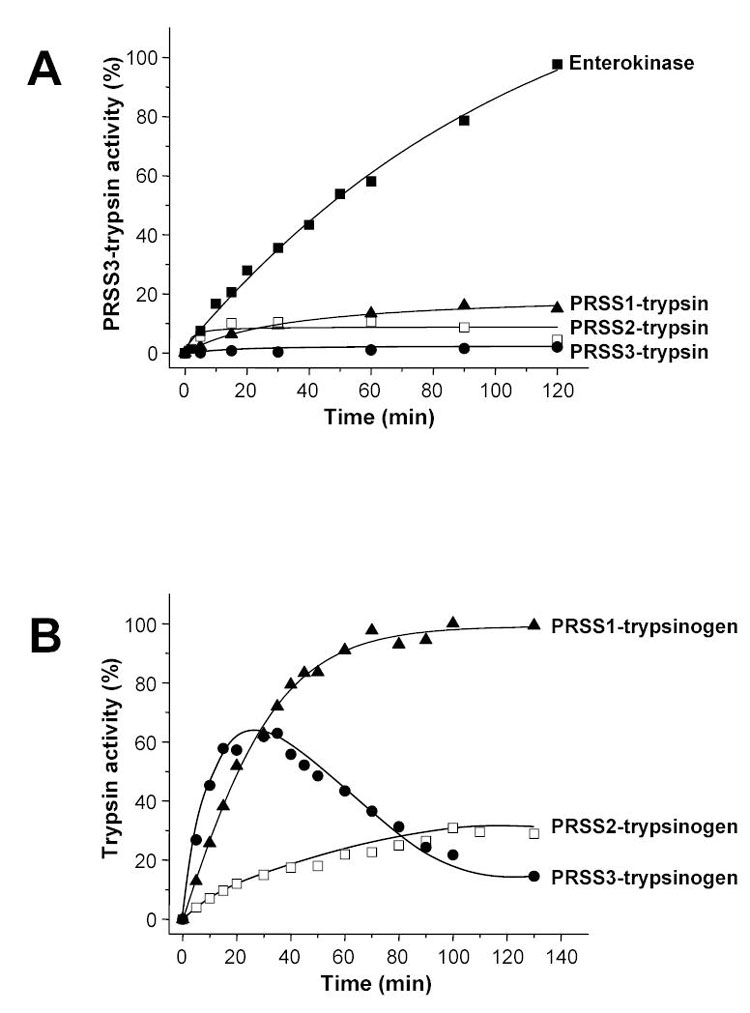
Recombinant human mesotrypsinogen was expressed in two routinely used Escherichia coli expression systems. In the SM138/pTrap expression host/plasmid system trypsinogen is constitutively secreted to the periplasmic space where it acquires its native fold. In the Rosetta(DE3)/pTrap-T7 expression system trypsinogen is produced in a denatured form as inclusion bodies, which are solubilized and re-natured in vitro. Details of expression and purification are given under Experimental Procedures. Importantly, recombinant mesotrypsinogen preparations purified from the two bacterial expression systems were indistinguishable in their functional properties studied here. Mesotrypsinogen was completely activated by bovine enterokinase, albeit at a slower rate than anionic or cationic trypsinogen (Fig 1A and B). Catalytic parameters of activated mesotrypsin were determined with the chromogenic substrate N-CBZ-Gly-Pro-Arg-p-nitroanilide, and mesotrypsin exhibited an approximately 3-fold higher turnover number (kcat) with a comparable Km value relative to cationic or anionic trypsin (Table 1).
Figure 1.
Activation of wild-type and R198G mutant mesotrypsinogen (PRSS3) with bovine enterokinase. Panel A. Trypsinogen samples at 2 μM final concentration were incubated with 200 ng/mL enterokinase (final concentration) at 37 °C, in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 2 mg/mL bovine serum albumin in a final volume of 100 μL. Aliquots of 2.5 μL were withdrawn from reaction mixtures at indicated times and trypsin activity was determined with N-CBZ-Gly-Pro-Arg-p-nitroanilide. Trypsin activity was expressed as percentage of potential maximal activity. For comparison, activation of human cationic trypsinogen (PRSS1) and anionic trypsinogen (PRSS2) are also shown. Panels B and C. Reactions incubated without bovine serum albumin were terminated at indicated times by precipitation with 10 % trichloroacetic acid (final concentration), electrophoresed on SDS-polyacrylamide gels (13 %) under reducing conditions and stained with Coomassie blue.
Inhibitor resistance of mesotrypsin is caused by Arg198
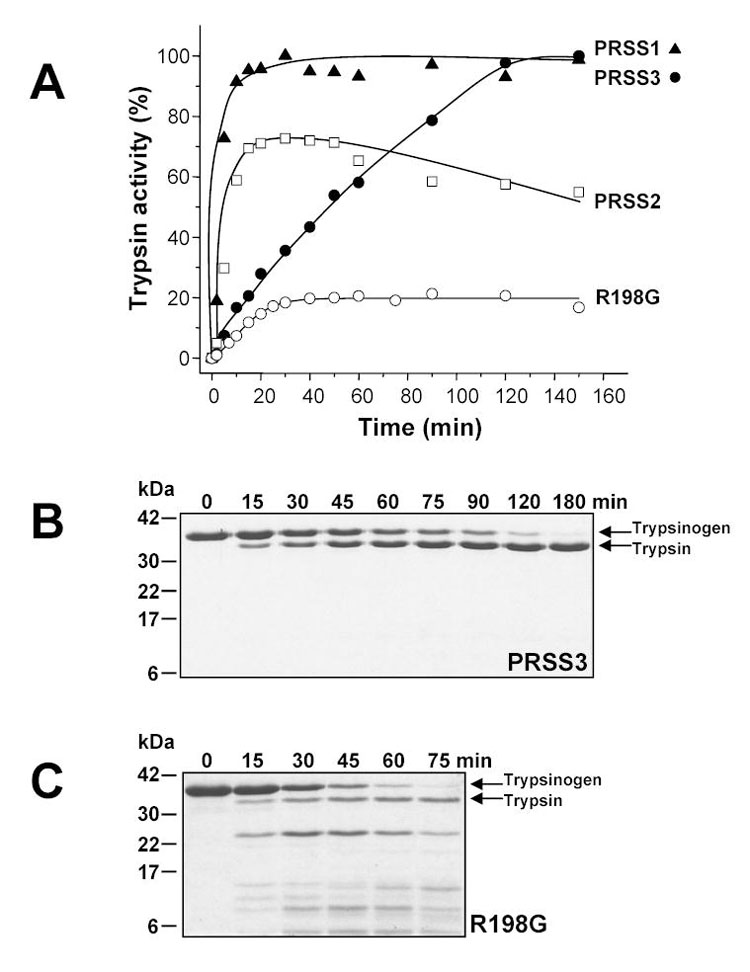
On the basis of sequence alignments [5] and a crystal structure [6], it has been suggested that mesotrypsin is resistant to proteinaceous trypsin inhibitors because of the presence of the Arg198 side chain, which sterically impairs inhibitor binding to the enzyme. However, this notion has never been tested experimentally so far. We have expressed and purified the R198G mesotrypsin mutant, in which the characteristic Gly198 residue -- universally found in chymotrypsin-like serine proteases -- has been restored. Surprisingly, activation of mesotrypsinogen mutant R198G with enterokinase under physiological conditions (pH 8.0, 1 mM Ca2+, 37 °C) yielded only 20 % of the expected activity (Fig 1A). As shown on the gel in Fig 1C, the loss of activity was due to massive degradation of the R198G-zymogen during the activation process. When the reaction was performed in the presence of SPINK1, which inhibited trypsin but not enterokinase, R198G-mesotrypsinogen was quantitatively converted to trypsin, indicating that the degradation was mediated by trypsin and not enterokinase (not shown). Activation in the presence of high Ca2+ concentrations (50 mM) at room temperature (22 °C) increased the trypsin yield, and eventually pure and active R198G-mesotrypsin preparation could be obtained by separating the degradation products on a benzamidine affinity column. Catalytic parameters of R198G-mesotrypsin on N-CBZ-Gly-Pro-Arg-p-nitroanilide were essentially identical to those of cationic or anionic trypsin (Table 1). Strikingly, R198G-mesotrypsin fully regained its sensitivity to protein trypsin inhibitors, and formed tight inhibitory complexes with SBTI or SPINK1 (Fig 2). As expected, wild-type mesotrypsin was resistant to these inhibitors. The experiments confirmed that the unique inhibitor resistance of mesotrypsin was the result of a single evolutionary amino-acid change, which replaced the small conserved Gly198 residue with a bulky Arg. Notably, in addition to rendering mesotrypsin resistant to inhibitors, the evolutionary selection of the potentially trypsin-sensitive Arg198 side-chain also stabilized mesotrypsin(ogen) against autocatalytic degradation. This apparent paradox is resolved if we assume that Arg198 blocks access to mesotrypsin’s active site not only for protein inhibitors, but also for protein substrates, and thus renders mesotrypsin relatively inactive towards its own trypsin-sensitive sites. This notion will be further explored by the subsequent experiments in this paper.
Figure 2.
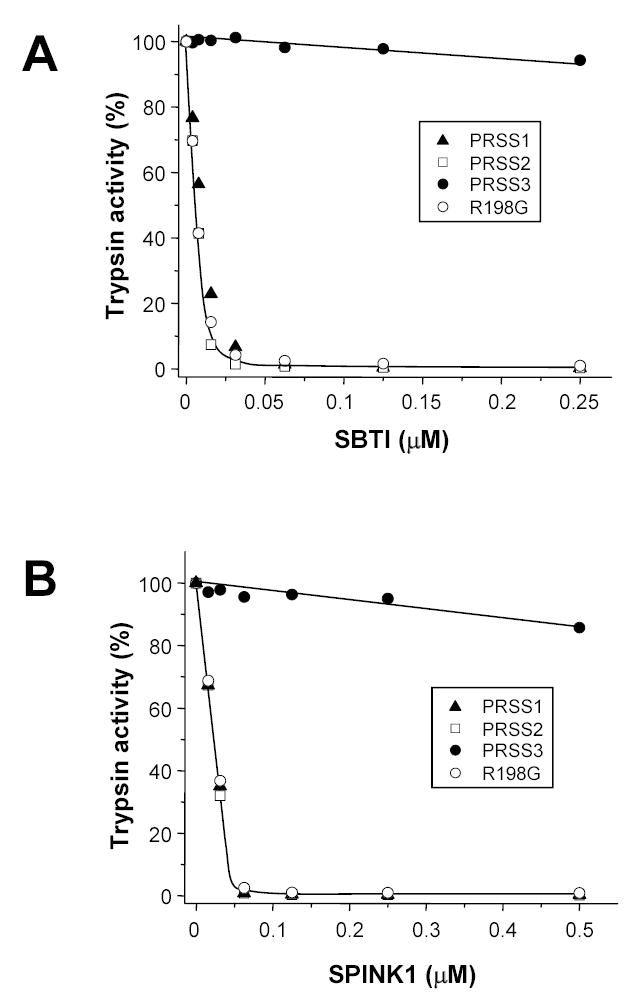
Inhibition of human cationic trypsin (PRSS1), anionic trypsin (PRSS2), wild-type and R198G mesotrypsin (PRSS3) with soybean trypsin inhibitor (SBTI, panel A) and human pancreatic secretory trypsin inhibitor (SPINK1, panel B). Trypsins were incubated with the indicated concentrations of inhibitors at 22 °C in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 1.5 mg/mL bovine serum albumin for 10 min, in a volume of 100 μL. Residual trypsin activity was then determined with N-CBZ-Gly-Pro-Arg-p-nitroanilide as described in Experimental Procedures. Trypsin concentrations used were 25 nM (panel A) and 50 nM (panel B), with the exception of wild-type mesotrypsin, which was used in 15 nM (panel A) and 25 nM (panel B) concentrations.
Mesotrypsin cannot activate pancreatic zymogens
Next, we tested the hypothesis that because of its inhibitor-resistance mesotrypsin can activate pancreatic zymogens unopposed by SPINK1, and this mechanism might play a role in the development of human pancreatitis [2, 5]. This theory was already contradicted by sporadic observations indicating that mesotrypsin was defective in activating bovine chymotrypsinogen [2] or human cationic and anionic trypsinogen [16]. As shown in Fig 3, these previous findings were fully confirmed by our experiments in which mesotrypsin was used to activate human anionic and cationic trypsinogen. Both zymogens autoactivated spontaneously to trypsin as a function of time, and addition of cationic trypsin, anionic trypsin or R198G-mesotrypsin markedly enhanced this process. In contrast, inclusion of mesotrypsin had no appreciable effect on trypsinogen activation, and mesotrypsinogen itself exhibited no autoactivation either. Wild-type mesotrypsin did not activate bovine chymotrypsinogen A, whereas R198G-mesotrypsin was essentially as active as cationic or anionic trypsin. When rates of chymotrypsinogen activation were compared quantitatively, mesotrypsin proved to be 500–1000-fold less efficient than cationic trypsin (not shown). Finally, the activation experiments were extended to human pro-elastase 2 (ELA2A), which was efficiently activated by human cationic trypsin, and even better by R198G-mesotrypsin, but was totally resistant to wild-type mesotrypsin (Fig 3). Importantly, under the conditions used (pH 8.0, 1 mM Ca2+), mesotrypsin did not degrade any of the zymogens tested to a detectable extent. Taken together, these experiments convincingly rule out a possible role for mesotrypsin in promoting intra-pancreatic zymogen activation. Furthermore, our notion that the presence of Arg198 renders mesotrypsin catalytically impaired toward protein substrates gained clear experimental support. In terms of cleaving the activation peptide bonds of pancreatic zymogens, mesotrypsin appears to be 2–3 orders of magnitude less active than the major trypsin isoforms, and this defect can be fully repaired by the restoration of Gly198.
Figure 3.
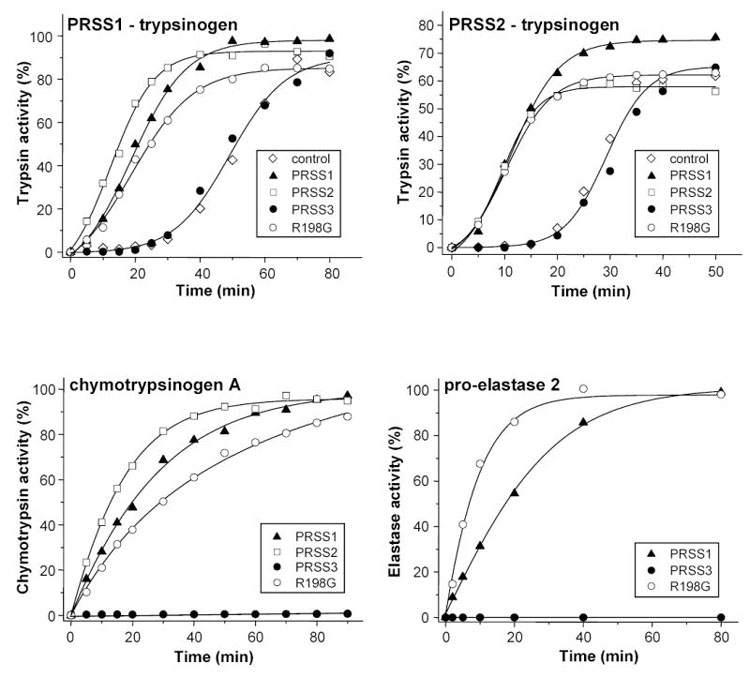
Activation of pancreatic zymogens with wild-type and R198G mutant mesotrypsin (PRSS3). Human cationic trypsinogen (PRSS1-trypsinogen, 2 μM), human anionic trypsinogen (PRSS2-trypsinogen, 2 μM), bovine chymotrypsinogen A (9 μM) and human pro-elastase 2 (1 μM, final concentrations) were incubated at 37 °C, in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 2 mg/mL bovine serum albumin. To test trypsinogen activation, incubations were performed in the absence of added trypsin (control) or in the presence of 120 nM (final concentration) cationic trypsin (PRSS1), anionic trypsin (PRSS2), wild-type mesotrypsin (PRSS3) or R198G mesotrypsin mutant. Bovine chymotrypsinogen A and human pro-elastase 2 were activated with 32 nM and 75 nM trypsins, respectively. Although not shown, in other experiments mesotrypsin at 125 nM concentration activated approximately 5 % of bovine chymotrypsinogen in 90 min, but had no measurable activating effect on human pro-elastase 2. Protease activities were determined as described in Experimental Procedures, and expressed as percent of maximal activity.
Role of mesotrypsin in trypsinogen degradation
Previous work by Rinderknecht et al. indicated that mesotrypsin can degrade bovine trypsinogen and possibly other pancreatic zymogens [2]. This observation spawned the theory, that inappropriate trypsinogen activation in the pancreas is curbed not only by SPINK1, which mops up active trypsin, but also by the SPINK1-resistant mesotrypsin, which eliminates trypsinogen, and thus limits further escalation of the activation cascade [2, 5]. To test this model, we examined the activity of mesotrypsin on the degradation of human cationic and anionic trypsinogen. For these experiments we used activation-resistant mutant trypsinogens, in which Lys23 in the activation site was replaced with Gln (K23Q). K23Q-trypsinogen is an ideal model substrate to study trypsinogen degradation without interference from trypsinogen activation [13, 18]. Figure 4 shows that in the absence of Ca2+ (in 1 mM EDTA, pH 8.0, 37 °C) mesotrypsin slowly cleaved the Arg122-Val123 peptide bond in cationic K23Q-trypsinogen. We demonstrated previously that this peptide bond is the most sensitive trypsinolytic site in cationic trypsinogen [18]. In contrast to a widely held belief, cleavage at this site does not result in any further degradation or inactivation, but yields a double-chain trypsin(ogen) species, which is functionally equivalent to its single-chain parent enzyme. Furthermore, cleavage never proceeds to completion, but due to trypsin-mediated re-synthesis of the Arg122-Val123 peptide bond an equilibrium is established between the single-chain and the double-chain species. Mesotrypsin-mediated digestion of the Arg122-Val123 peptide bond in 1 mM EDTA resulted in the expected hydrolysis equilibrium with approximately 18 % single-chain and 82 % double-chain trypsinogen present (Fig 4A and C). Addition of 1 mM Ca2+ significantly decreased the rate of cleavage, and at 240 min only 20 % double-chain trypsinogen was observed (Fig 4B and C). This was not a true equilibrium yet, because previous studies indicated that in the presence of Ca2+ the equilibrium mixture should contain 40 % double-chain and 60 % single-chain trypsinogen [18]. The results clearly demonstrate that mesotrypsin is capable of cleaving the Arg122-Val123 peptide bond. However, comparison of mesotrypsin (Fig 4) and cationic trypsin (cf. Fig 4 in reference [18]) in their ability to cleave cationic K23Q-trypsinogen revealed that mesotrypsin was at least 500-fold less active. Other than cleaving at Arg122-Val123, mesotrypsin did not degrade single-chain or double-chain cationic K23Q-trypsinogen to any extent.
Figure 4.
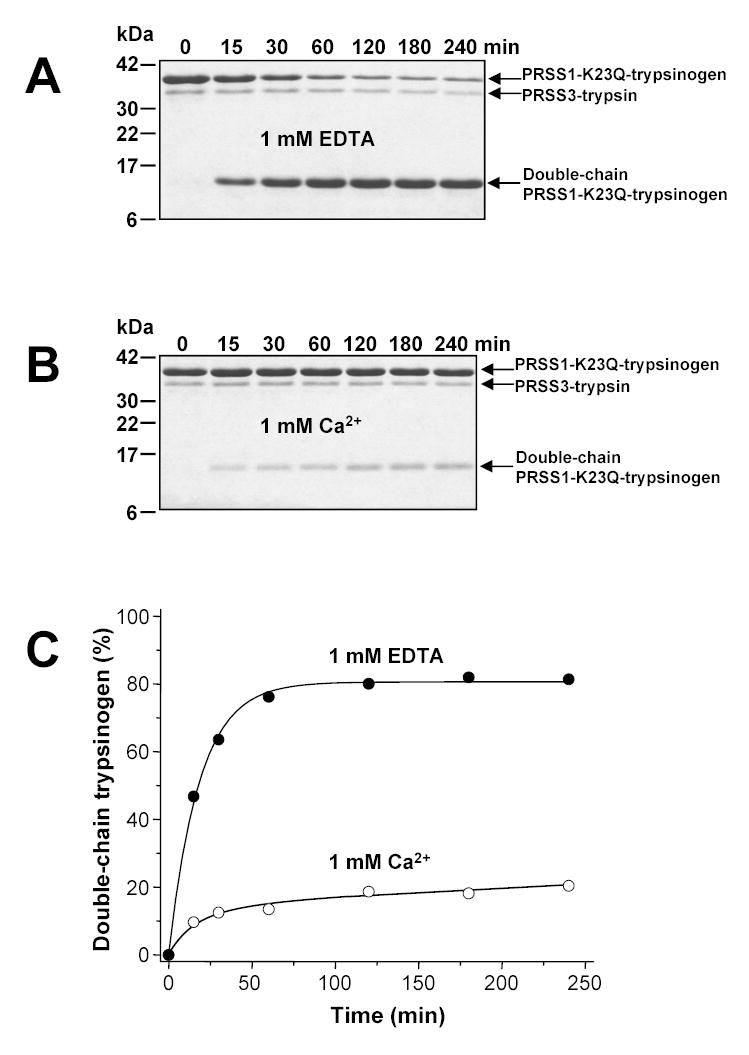
Cleavage of the Arg122-Val123 peptide bond in human cationic trypsinogen (PRSS1) by mesotrypsin (PRSS3). Cationic K23Q-trypsinogen (2 μM final concentration) was digested with mesotrypsin (200 nM final concentration) at 37 °C in 0.1 M Tris-HCl (pH 8.0) containing 1 mM EDTA (panel A) or 1 mM Ca2+ (panel B). Samples were precipitated with trichloroacetic acid (10 % final concentration) at the indicated times and analyzed by SDS-PAGE and Coomassie-blue staining. Panel C. Densitometric quantitation of the double-chain cationic trypsinogen forms.
In contrast to cationic K23Q-trypsinogen, digestion of the proteolytically less stable anionic K23Q-trypsinogen by mesotrypsin resulted in complete zymogen degradation, with a t1/2 of approximately 50 min (Fig 5A and C). Under similar conditions (pH 8.0, 1 mM EDTA, 37 °C, 1:10 trypsin-to-zymogen ratio), cationic trypsin degraded anionic K23Q-trypsinogen 22-fold more rapidly, with a half-life of 2.25 min (cf. Fig 3 in reference [13]). Addition of 1 mM Ca2+ almost completely stabilized anionic K23Q-trypsinogen and only approximately 25 % mesotrypsin-mediated degradation was detected over the 240 min time course studied (Fig 5B and C). Taken together, the results demonstrate that the two major human trypsinogen isoforms are poor substrates for mesotrypsin, and dispute a protective role for mesotrypsin-mediated zymogen degradation in pancreatic physiology. Furthermore, the slow but measurable degradation of anionic trypsinogen by mesotrypsin also indicates that the loss of affinity towards protein substrates is not always several orders of magnitude, and a “specific” mesotrypsin substrate might exist, which can avoid the guarding side-chain of Arg198.
Figure 5.
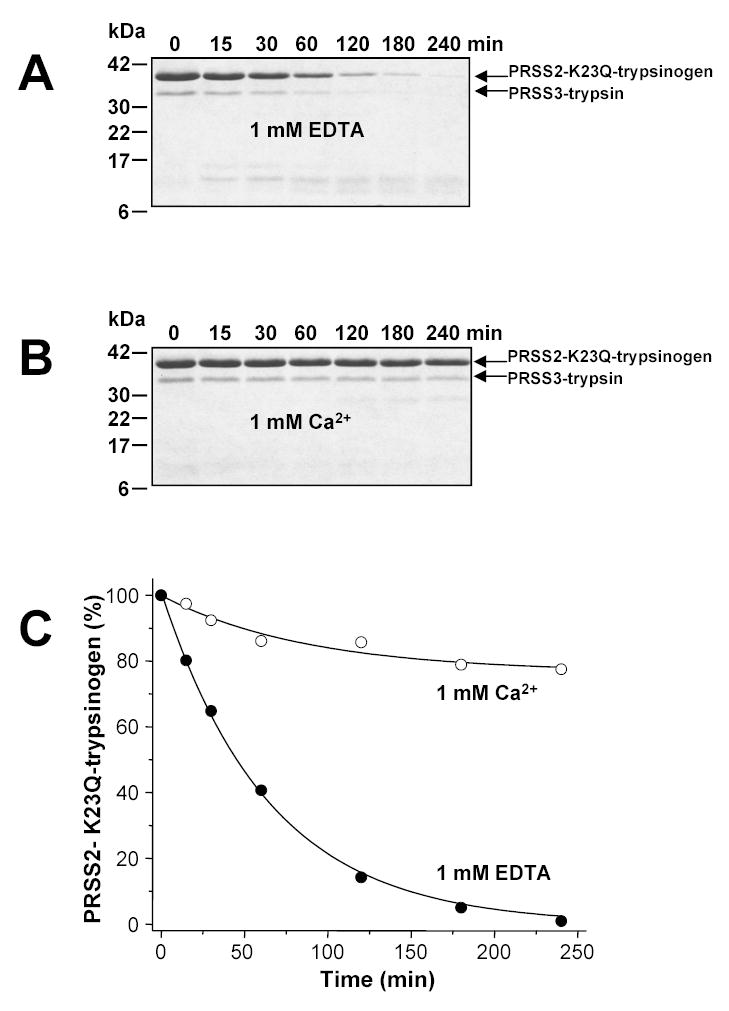
Degradation of human anionic trypsinogen (PRSS2) by mesotrypsin (PRSS3). Anionic K23Q-trypsinogen (2 μM final concentration) was digested with mesotrypsin (200 nM final concentration) at 37 °C in 0.1 M Tris-HCl (pH 8.0) containing 1 mM EDTA (panel A) or in 1 mM Ca2+ (panel B). Samples were precipitated with trichloroacetic acid (10 % final concentration) at the indicated times and analyzed by SDS-PAGE and Coomassie-blue staining. Panel C. Densitometric quantitation of the intact anionic K23Q-trypsinogen band.
Mesotrypsin is weakly inhibited by SPINK1 or SBTI
We hypothesized that although mesotrypsin degrades human anionic trypsinogen very slowly, this process might still be significant under conditions when all other trypsins are inhibited by SPINK1 and only mesotrypsin is active. To demonstrate the feasibility of such a scenario, we digested anionic K23Q-trypsinogen with mesotrypsin in the presence of SPINK1. To our surprise, SPINK1 inhibited mesotrypsin activity even in submicromolar concentrations, and no degradation of anionic trypsinogen was detectable (not shown). These data seemed to contradict the observation in Fig 2 that SPINK1 does not bind to mesotrypsin. However, the experimental setup used in Fig 2 only tests for tight-binding inhibition, which withstands dissociation during the assay. Relatively weak inhibitory complexes would quickly dissociate when the sample was diluted into the substrate-containing assay buffer, and no inhibition would be apparent. Thus, the assay used in Fig 2 is not appropriate for testing weak inhibition. To characterize the relatively weaker inhibition of mesotrypsin by SPINK1, catalytic parameters (Km, kcat) of mesotrypsin were measured on N-CBZ-Gly-Pro-Arg-p-nitroanilide in the presence of increasing concentrations of inhibitor. From [inhibitor] versus Km plots a Ki value of 1.5 μM was estimated (not shown). Similar experiments with SBTI yielded a Ki value of 0.42 μM (not shown), which was identical to the Ki value determined by Katona et al. using progress curve analysis [6]. Interestingly, while SBTI acted in a purely competitive fashion, SPINK1 in the higher concentration range also exhibited non-competitive inhibition of mesotrypsin, for reasons that are not readily apparent. In any event, the results clearly indicate that mesotrypsin retained low but significant affinity toward trypsin inhibitors, and challenge the notion that mesotrypsin can act uncontrolled in the presence of SPINK1.
Mesotrypsin rapidly cleaves the reactive-site peptide bond of SBTI
Seminal work from the Laskowski laboratory demonstrated that in the complexes of proteases and canonical protease inhibitors, the reactive-site peptide bond of the inhibitor gets slowly cleaved, resulting in an equilibrium mixture of double-chain “modified” inhibitor and single-chain “virgin” inhibitor [19 and references therein]. We hypothesized that due to their low but still significant affinity, mesotrypsin recognizes protein trypsin inhibitors as substrates and may rapidly hydrolyze their reactive-site peptide bonds. A precedent for this notion was found by Estell and Laskowski [20, 21], who demonstrated that trypsin-1 from the starfish Dermasterias imbricata (leather star) cleaved the reactive sites of SBTI and bovine pancreatic trypsin inhibitor at an increased rate. In the experiment shown in Fig 6A, we incubated 500 nM SBTI with 10 nM mesotrypsin (final concentrations) and at indicated times aliquots were taken and the virgin (intact) SBTI concentration was determined using bovine trypsin (see Experimental Procedures for a description of the assay). Remarkably, a rapid decrease in the concentration of virgin SBTI was observed, and the reaction reached a plateau in about 20 min, with approximately 40 % of the original virgin inhibitor remaining. Analysis of the digestion reactions on reducing SDS-PAGE revealed that mesotrypsin cleaved SBTI at a single site (Fig 6B), resulting in the appearance of two new bands on the gels. N-terminal sequencing of the major fragment yielded a sequence of Ile-Arg-Phe-Ile-Ala, confirming that the cleaved peptide bond was the Arg88-Ile89 reactive site of SBTI (numbering starts with Met1). Densitometric quantitation of the virgin SBTI band indicated that approximately 40–44 % virgin (intact) and 56–60 % modified (cleaved) inhibitor was present in the equilibrium mixture, which was in good agreement with the results of the functional assay in Fig 6A. At the same pH but under somewhat different assay conditions (0.5 M KCl, 21 °C) the Laskowski laboratory reported a hydrolysis equilibrium containing 78 % modified SBTI after digestion with Dermasterias imbricata trypsin-1 or bovine trypsin [20]. In control experiments 50 nM cationic or anionic trypsin was incubated with 500 nM SBTI (final concentrations), and after the initial 50 nM decrease due to complex formation between trypsin and SBTI, no further change in the concentration of free virgin SBTI was detectable for 70 hours.
Figure 6.
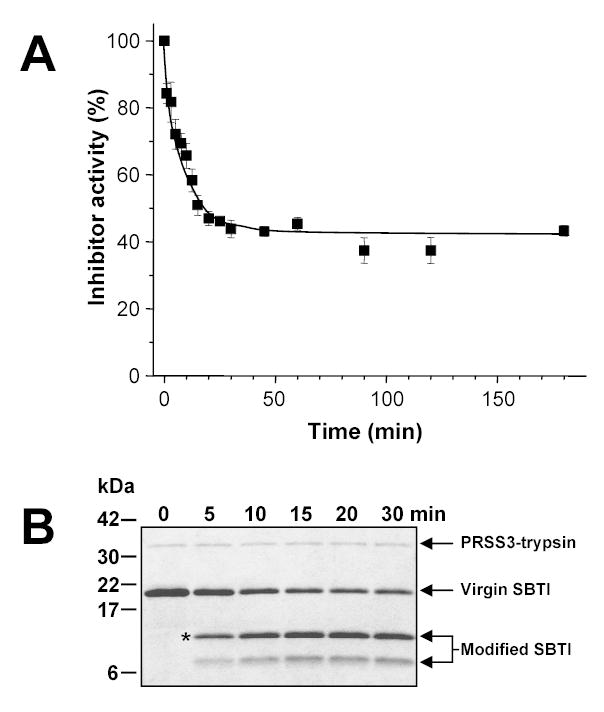
Hydrolysis of the reactive site of SBTI by mesotrypsin. Panel A. SBTI (500 nM) was incubated with 10 nM mesotrypsin (final concentrations) at 37 °C in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 1 mg/mL bovine serum albumin. Cleavage of the reactive site was followed by measuring the decrease in the association rate of modified SBTI with bovine trypsin, as described in Experimental Procedures. Panel B. Alternatively, samples were precipitated with 10 % trichloroacetic acid (final concentration) and analyzed by reducing SDS-PAGE and Coomassie-blue staining. The asterisk indicates the band subjected to N-terminal protein sequencing.
Mesotrypsin degrades human SPINK1
In contrast to SBTI, which forms stable complexes with trypsin, SPINK1 is a so-called “temporary inhibitor”, because trypsin-SPINK1 complexes irreversibly dissociate over time [22]. First, reversible digestion of the Lys41-Ile42 reactive site peptide bond occurs, which is followed by the irreversibly inactivating cleavage of the Arg67-Gln68 bond [23–25]. In addition, peptide bonds Arg28-Glu29, Arg65-Lys66 and Lys75-Ser76 are also subject to tryptic attack [23–25]. In our experiments, temporary inhibition followed a relatively rapid time-course (t1/2 2–4 hours) when human cationic or anionic trypsin was in some excess to SPINK1, however, at lower trypsin-to-SPINK1 ratios the reaction proceeded dramatically slower. Thus, when cationic or anionic trypsin (50 nM, final concentration) was reacted with SPINK1 (500 nM, final concentration) at a 1:10 ratio, no trypsin activity was detectable up to 90 hours, and free SPINK1 levels did not change measurably either (not shown). In sharp contrast, incubation of 50 nM mesotrypsin with 500 nM SPINK1 (final concentrations) resulted in a much more rapid decrease of active SPINK1 concentration (Fig 7A), which eventually resulted in complete elimination of SPINK1 activity. In control experiments, incubation of 500 nM bovine chymotrypsin or human elastase 2 with 500 nM SPINK1 (final concentrations) did not result in any appreciable SPINK1 degradation up to 20 h, indicating that mesotrypsin can be regarded as a specific degrading enzyme for SPINK1.
Figure 7.
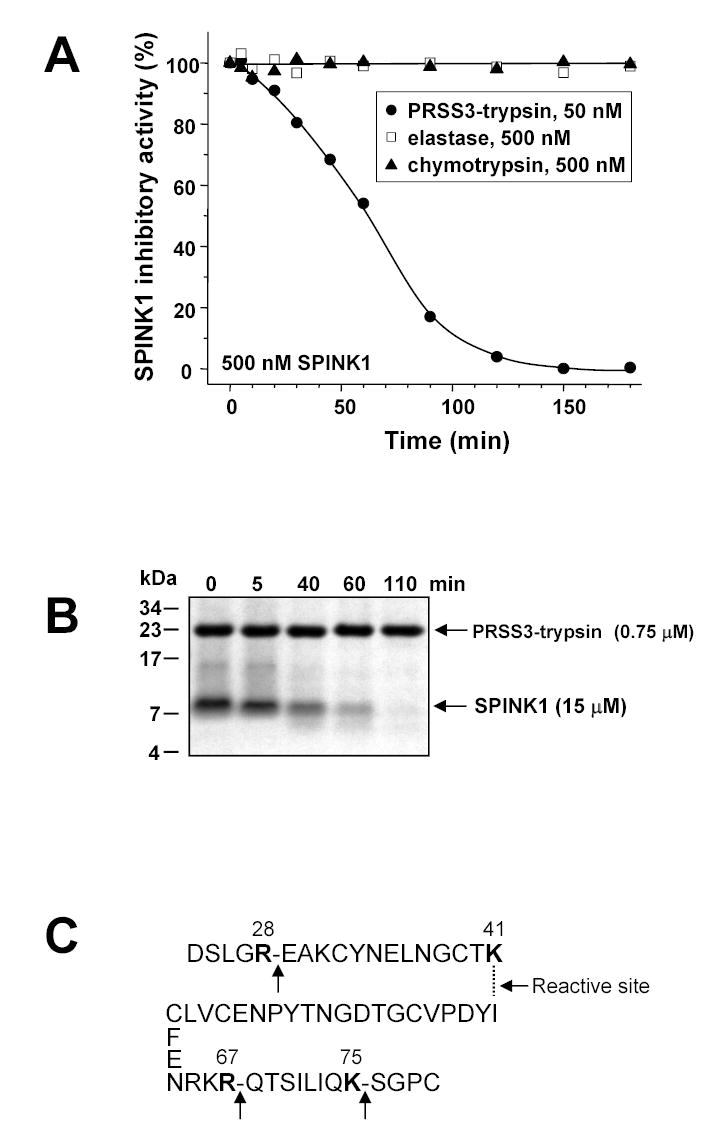
Degradation of human SPINK1 by mesotrypsin. Panel A. SPINK1 (500 nM) was incubated with 50 nM mesotrypsin, 500 nM bovine chymotrypsin or 500 nM human elastase 2 (final concentrations) at 37 °C in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 1 mg/mL bovine serum albumin. Residual inhibitory activity was measured with bovine trypsin, as described in Experimental Procedures. Panel B. SPINK1 (15 μM) was incubated with 0.75 μM mesotrypsin (final concentrations) at 37 °C in 0.1 M Tris-HCl (pH 8.0) and 1 mM CaCl2. Samples were precipitated with 20 % trichloroacetic acid (final concentration) at indicated times; separated on 16 % tricine-SDS gels under reducing conditions and visualized by Coomassie blue staining. Note that SPINK1 stains very poorly, and the relative intensities of the mesotrypsin and SPINK1 bands do not reflect the actual ratio of their concentrations in the reaction mixtures. Panel C. Sites of mesotrypsin cleavage in SPINK1, as determined from N-terminal sequencing of a sample of the digestion mixture at 40 min in Fig 7B.
To visualize the degradation of SPINK1, an experiment was performed using high concentrations of inhibitor (15 μM) and 20-fold less mesotrypsin (0.75 μM). Under these conditions, the reaction proceeded faster, and more than 50 % of SPINK1 activity was abolished in 40 min (not shown). Samples were analyzed under reducing conditions on SDS-tricine polyacrylamide gels, where gradual disappearance of the SPINK1 band was apparent (Fig 7B). Only a faint and transiently visible cleavage product was detected, indicating that SPINK1 suffered more extensive proteolysis beyond the cleavage of the reactive-site peptide bond. To confirm this notion, a sample of the digestion mixture taken at 40 min was applied to a ProSorb PVDF cartridge (Applied Biosystems) and subjected to Edman-degradation. In addition to the native N terminus, two major new N termini were identified, which indicated cleavages at peptide bonds Lys41-Ile42 (the reactive site) and Lys75-Ser76. Two additional N termini were found in smaller yield, which revealed cleavages at Arg28-Glu29 and Arg67-Gln68. Although we could not establish the exact order of cleavages from these data, the results clearly confirmed that one of the major targets of mesotrypsin was the reactive site of SPINK1, and the other tryptic cleavages corresponded to those previously described. Therefore, we conclude that mesotrypsin-mediated SPINK1 degradation followed the established mechanism of “temporary inhibition”, but at a markedly higher rate.
Cathepsin B is a potential pathological activator of mesotrypsinogen
The observations described above directly suggest the idea that premature activation of mesotrypsinogen in the pancreas can degrade protective SPINK1 and eventually cause pancreatitis. Since we found that mesotrypsin cannot autoactivate, we were left with the dilemma of identifying a possible activating enzyme for mesotrypsinogen in the pancreas. In this regard, the two major trypsin isoforms appeared to be good candidates. However, when mesotrypsinogen was activated with cationic trypsin, anionic trypsin or enterokinase under physiologically optimal conditions (37 °C, pH 8.0, 1 mM Ca2+), human trypsins generated less than 20 % of the potentially maximal mesotrypsin activity in 2 hours (Fig 8A). Gel analysis indicated that the mesotrypsinogen band was slowly converted to trypsin, and after 2 hours approximately half of the mesotrypsinogen remained unactivated. In addition, a considerable fraction of mesotrypsinogen was degraded, despite the presence of 1 mM Ca2+ (not shown). In the absence of Ca2+, mesotrypsinogen was highly susceptible to degradation by cationic or anionic trypsin, and half-lives of 2.3 min and 0.5 min were measured, respectively. The t1/2 value of degradation by cationic trypsin was almost identical to the previously determined half-life of anionic K23Q-trypsinogen in the presence of cationic trypsin (2.25 min, see Fig 3 in ref [13]). Thus, mesotrypsinogen seems to be similar in its proteolytic stability to anionic trypsinogen, while cationic trypsinogen is at least 20-fold more stable (cf. Fig 3 in ref [13]). In conclusion, the results indicate that cationic and anionic trypsin are more likely to play a role in mesotrypsinogen degradation than activation.
The lysosomal cysteine protease cathepsin B was shown to play a critical role in various experimental pancreatitis models as the intracellular catalyst of pathological trypsinogen activation [26, 27 and references therein]. Cathepsin B is also found in the secretory pathway of the human pancreas, where it is co-localized with trypsinogen [27]. In vitro, cathepsin B is a potent activator of cationic trypsinogen, with a pH optimum of 4.0 [16, 27, 28]. When activation of the three human trypsinogen isoforms was compared at pH 4.0, an interesting picture emerged (Fig 8B). Rapid activation of mesotrypsinogen was apparent, which was followed by slow degradation. Compared to cationic and anionic trypsinogen, the initial rate of mesotrypsinogen activation was approximately 2-fold and 6-fold higher, respectively. Therefore, under certain conditions, cathepsin B can rather selectively activate mesotrypsinogen, and potentially initiate the mesotrypsin-mediated degradation of SPINK1.
DISCUSSION
The experiments presented in this study provide three important observations: (1) Mesotrypsin can rapidly hydrolyze the reactive-site peptide bond of Kunitz-type trypsin inhibitors and degrade Kazal-type temporary inhibitors. (2) This unique activity is caused by the evolutionary selection of Arg198, which also rendered mesotrypsin resistant to protein inhibitors and limited its capacity to cleave protein substrates. (3) Cathepsin B can preferentially activate mesotrypsinogen of the three human trypsinogen isoforms, under certain conditions. These observations clearly define a physiological purpose for mesotrypsin and also suggest a potential pathological role, in which prematurely activated mesotrypsin can degrade protective SPINK1 and cause pancreatitis.
The results strongly argue that human mesotrypsin plays a unique and highly specialized role in the degradation of trypsin inhibitors. Obviously, such a function would be advantageous in the digestion of foods rich in naturally occurring trypsin inhibitors, and would provide a rationale for the evolution of this trypsin isoform. Such a specialized function can also explain the relatively low levels of mesotrypsin secretion, as opposed to the largely non-specific digestive enzymes. Conceivably, mesotrypsin exerts its effect in concert with other digestive proteases, which might attack the partially digested inhibitors. A known example is carboxypeptidase B, which can cleave off the newly generated C-terminal Lys or Arg residues after hydrolysis of the reactive-site peptide bonds and thus completely inactivate trypsin inhibitors (see also discussion in [19]). We hypothesized that mesotrypsin might be also responsible for the elimination of endogenous SPINK1 secreted to the duodenum, however, model experiments indicated that at physiological concentrations SPINK1 was bound by cationic or anionic trypsin and the presence of mesotrypsin did not facilitate the irreversible dissociation of SPINK1 (data not shown). Thus, elimination of unwanted SPINK1 seems to occur through the “temporary inhibition” mechanism by major trypsin isoforms. From the experimental data it appears that mesotrypsin-mediated degradation of inhibitors becomes significant at high inhibitor concentrations, while in the absence of inhibitors mesotrypsin is probably degraded by the true digestive trypsins.
Undoubtedly, the most exciting implication of the observations presented here is that inappropriate activation of human mesotrypsinogen in the pancreas might degrade protective SPINK1 levels and cause pancreatitis. The role of SPINK1 in protecting against pancreatitis is clearly supported by the association of SPINK1 mutations with certain forms of chronic pancreatitis [e.g. 29–31]. Should mesotrypsinogen get converted to mesotrypsin prematurely in the pancreas, the ensuing degradation of SPINK1 might represent an immediate risk factor for the development of pancreatitis. Such a model of disease onset would require a relatively specific activator for mesotrypsinogen in the pancreatic acinar cells. Remarkably, in our in vitro experiments cathepsin B robustly activated mesotrypsinogen, with a measurable preference over cationic or anionic trypsinogen. Thus, the biochemical basis clearly exists for a putative mesotrypsin-induced pancreatitis model, and future research will confirm or rule out the medical significance of such a mechanism. In this regard, identification of pancreatitis-associated mutations that stabilize mesotrypsin or loss-of-function mutations that protect against pancreatitis could provide particularly strong evidence.
Is mesotrypsin unique to humans? Rinderknecht et al. reported that no inhibitor-resistant trypsin activity was found in pancreatic extracts from dog, cow, pig, rat, mouse or hamster or in pancreatic juice from dog or hamster [2]. Nonetheless, examination of the numerous rat trypsinogen genes found in databanks revealed that trypsinogen V contains a bulky tyrosine residue at position 198. While there is no evidence that functional protein is expressed from the rat trypsinogen V mRNA, this protein would be expected to exhibit mesotrypsin-like inhibitor-resistance and inhibitor-degrading properties. Interestingly, the corresponding mouse gene is probably a pseudogene with a 9 amino-acid deletion in the N-terminal half of trypsinogen. No other sequenced trypsinogen gene carries the mesotrypsin signature mutation. Trypsin-1 from Dermasterias imbricata [20, 21] is clearly a mesotrypsin-like enzyme, however, the protein or DNA sequence has not been determined for this trypsin yet. With the ongoing sequencing of several vertebrate genomes, the discovery of new mesotrypsin orthologs can be expected in the near future. Finally, evolutionary mutations in trypsin at positions other than 198 can also result in mesotrypsin-like properties. In this regard, rat trypsinogen IV was described as partially inhibitor resistant, presumably due to the presence of a negatively charged aspartic acid, which replaced the conserved neutral glutamine residue found at position 197 (mesotrypsinogen numbering) in other mammalian trypsinogens [32].
Acknowledgments
Thanks are due to Lan Guan for the densitometric evaluations, and Edit Szepessy for help with the cloning and expression of human pro-elastase 2. We are indebted to Miklós Tóth (Department of Medical Chemistry, Semmelweis University, Budapest, Hungary) for critical reading of the manuscript and valuable discussions. Protein sequencing was carried out by David W. McCourt (Midwest Analytical, Inc., St. Louis, MO).
Footnotes
This work was supported by NIH grant DK58088 to M. S.-T.
References
- 1.Rinderknecht H, Renner IG, Carmack C. Gut. 1979;20:886–891. doi: 10.1136/gut.20.10.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rinderknecht H, Renner IG, Abramson SB, Carmack C. Gastroenterology. 1984;86:681–692. [PubMed] [Google Scholar]
- 3.Rinderknecht H, Stace NH, Renner IG. Dig Dis Sci. 1985;30:65–71. doi: 10.1007/BF01318373. [DOI] [PubMed] [Google Scholar]
- 4.Rinderknecht H, Renner IG, Carmack C, Friedman R, Koyama P. Clin Res. 1978;26 :112A. [Google Scholar]
- 5.Nyaruhucha CNM, Kito M, Fukuoka SI. J Biol Chem. 1997;272:10573–10578. doi: 10.1074/jbc.272.16.10573. [DOI] [PubMed] [Google Scholar]
- 6.Katona G, Berglund GI, Hajdu J, Gráf L, Szilágyi L. J Mol Biol. 2002;315:1209–1218. doi: 10.1006/jmbi.2001.5305. [DOI] [PubMed] [Google Scholar]
- 7.Wiegand U, Corbach S, Minn A, Kang J, Müller-Hill B. Gene. 1993;136 :167–75. doi: 10.1016/0378-1119(93)90460-k. [DOI] [PubMed] [Google Scholar]
- 8.Chen, J.-M. and Ferec, C. (2003) In Cooper, D. N. (ed.), Nature Encyclopedia of the Human Genome Nature Publishing Group, London, pp. 645–650.
- 9.Chase T, Jr, Shaw E. Biochem Biophys Res Comm. 1967;29:508–514. doi: 10.1016/0006-291x(67)90513-x. [DOI] [PubMed] [Google Scholar]
- 10.Lengyel Z, Pál G, Sahin-Tóth M. Prot Expr Purif. 1998;12 :291–294. doi: 10.1006/prep.1997.0837. [DOI] [PubMed] [Google Scholar]
- 11.Sahin-Tóth M. J Biol Chem. 2000;275 :22750–22755. doi: 10.1074/jbc.M002943200. [DOI] [PubMed] [Google Scholar]
- 12.Sahin-Tóth M, Tóth M. Biochem Biophys Res Commun. 2000;278 :286–289. doi: 10.1006/bbrc.2000.3797. [DOI] [PubMed] [Google Scholar]
- 13.Kukor Z, Tóth M, Sahin-Tóth M. Eur J Biochem. 2003;270 :2047–2058. doi: 10.1046/j.1432-1033.2003.03581.x. [DOI] [PubMed] [Google Scholar]
- 14.Gráf L, Craik CS, Patthy A, Roczniak S, Fletterick RJ, Rutter WJ. Biochemistry. 1987;26 :2616–2623. doi: 10.1021/bi00383a031. [DOI] [PubMed] [Google Scholar]
- 15.Sahin-Tóth M. J Biol Chem. 1999;274 :29699–29704. doi: 10.1074/jbc.274.42.29699. [DOI] [PubMed] [Google Scholar]
- 16.Szilágyi L, Kénesi E, Katona G, Kaslik G, Juhász G, Gráf L. J Biol Chem. 2001;276:24574–24580. doi: 10.1074/jbc.M011374200. [DOI] [PubMed] [Google Scholar]
- 17.Luthy JA, Praissman M, Finkenstadt WR, Laskowski M., Jr J Biol Chem. 1973;248:1760–1771. [PubMed] [Google Scholar]
- 18.Kukor Z, Tóth M, Pál G, Sahin-Tóth M. J Biol Chem. 2002;277 :6111–6117. doi: 10.1074/jbc.M110959200. [DOI] [PubMed] [Google Scholar]
- 19.Laskowski M, Jr, Qasim MA. Biochim Biophys Acta. 2000;1477 :324–337. doi: 10.1016/s0167-4838(99)00284-8. [DOI] [PubMed] [Google Scholar]
- 20.Estell DA, Laskowski M., Jr Biochemistry. 1980;19:124–131. doi: 10.1021/bi00542a019. [DOI] [PubMed] [Google Scholar]
- 21.Estell DA, Wilson KA, Laskowski M., Jr Biochemistry. 1980;19:131–137. doi: 10.1021/bi00542a020. [DOI] [PubMed] [Google Scholar]
- 22.Laskowski M, Wu FC. J Biol Chem. 1953;204 :797–806. [PubMed] [Google Scholar]
- 23.Schneider SL, Stasiuk L, Laskowski M., Sr J Biol Chem. 1973;248 :7207–7214. [PubMed] [Google Scholar]
- 24.Schneider SL, Laskowski M., Sr J Biol Chem. 1974;249 :2009–2015. [PubMed] [Google Scholar]
- 25.Kikuchi N, Nagata K, Shin M, Mitsushima K, Teraoka H, Yoshida N. J Biochem (Tokyo) 1989;106:1059–1063. doi: 10.1093/oxfordjournals.jbchem.a122964. [DOI] [PubMed] [Google Scholar]
- 26.Halangk W, Lerch MM, Brandt-Nedelev B, Roth W, Ruthenbuerger M, Reinheckel T, Domschke W, Lippert H, Peters C, Deussing J. J Clin Invest. 2000;106:773–781. doi: 10.1172/JCI9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kukor Z, Mayerle J, Krüger B, Tóth M, Steed PM, Halangk W, Lerch MM, Sahin-Tóth M. J Biol Chem. 2002;277 :21389–21396. doi: 10.1074/jbc.M200878200. [DOI] [PubMed] [Google Scholar]
- 28.Figarella C, Miszczuk-Jamnska B, Barrett A. Biol Chem Hoppe-Seyler. 1988;369(Suppl):293–298. [PubMed] [Google Scholar]
- 29.Witt H, Luck W, Hennies HC, Clasen M, Kage A, Las U, Landt O, Becker M. Nat Genet. 2000;25 :213–216. doi: 10.1038/76088. [DOI] [PubMed] [Google Scholar]
- 30.Pfützer RH, Barmada MM, Brunskill AP, Finch R, Hart PS, Neoptolemos J, Furey WF, Whitcomb DC. Gastroenterology. 2000;119:615–623. doi: 10.1053/gast.2000.18017. [DOI] [PubMed] [Google Scholar]
- 31.Le Maréchal C, Chen JM, Le Gall C, Plessis G, Chipponi J, Chuzhanova NA, Raguénès, O, Férec C. Hum Mut. 2003 doi: 10.1002/humu.9212. in press. [DOI] [PubMed] [Google Scholar]
- 32.Fukuoka SI, Nyaruhucha CM. Biochim Biophys Acta. 2002;1588 :106–112. doi: 10.1016/s0925-4439(02)00153-9. [DOI] [PubMed] [Google Scholar]