

Abstract
The higher-order structure of G protein-coupled receptors (GPCRs) in membranes may involve dimerization and formation of even larger oligomeric complexes. Here, we have investigated the organization of the prototypical GPCR rhodopsin in its native membrane by electron and atomic force microscopy (AFM). Disc membranes from mice were isolated and observed by AFM at room temperature. In all experimental conditions, rhodopsin forms structural dimers organized in paracrystalline arrays. A semi-empirical molecular model for the rhodopsin paracrystal is presented validating our previously reported results. Finally, we compare our model with other currently available models describing the supramolecular structure of GPCRs in the membrane.
Keywords: Atomic force microscopy, G protein-coupled receptor, Membrane protein, Photoreceptor cell, Rhodopsin, Transmission electron microscopy
1. Introduction
The G protein-coupled receptor (GPCR) superfamily consists of three mammalian subfamilies (named A—C) that activate specific subtypes of trimeric G proteins [1,2]. Only the crystal structure of rhodopsin, a member of subfamily A, has been elucidated [3]. The rhodopsin structure serves as a template for the molecular architecture of GPCRs, since the transmembrane segments of these receptors are highly homologous [1,4] including the least conserved members of subfamily C [5]. Other components of the G protein-signaling machinery have conserved structures as well: the high-resolution structures of G proteins (Gtα,Gi1α, and Gsα) and proteins that interact with GPCRs, i.e. arrestins (visual arrestin, β-arrestin 1 and β-arrestin 2), show only small structural variance (reviewed in [6]). For example, the crystal structures of the non-visual arrestins, β-arrestin 2 and β-arrestin 1, differ from each other by an α-carbon root-mean-square deviation of 1.2 Å and 1.8 Å, respectively. These observations suggest that the mechanistic model of the G protein activation by GPCRs and the arrestin-mediated desensitization process must be conserved as well.
How GPCRs operate is one of the most fundamental questions in the field of transmembrane signal transduction. A growing body of pharmacological, biochemical and biophysical data strongly suggests that these receptors form functional homo- and heterodimers as well as higher-order oligomers [7-16]. Among the three subtypes of GPCRs, the issue of dimerization for type C is clearly set. Type C GPCRs form dimers by covalent linkage of disulfide bonds at their extracellular N-terminal domains, e.g. the metabotropic glutamate receptor [17], or by strong non-covalent, intermolecular interactions [18], e.g. two obligatory γ-aminobutyric acid (GABA) receptor subtypes [19-22]. For family A, mutant GPCR-G protein fusions where either G protein or the receptor had been specifically deactivated demonstrate that this class of receptors operates through transactivation [23]. Results from the disulfide-trapping approach provide a new data set for a molecular model of the interfacial surfaces in the complex [24,25]. The contact sites for homodimeric dopamine D2 receptors involve helices IV [25,26]. In other disulfide-trapping studies of the C5a receptor, individual cysteines placed in either intracellular (IC) loop IC-1, IC-2, or the C-tail could mediate specific crosslinking. These results favor a symmetric dimer involving an interface between helix I (H-I) and H-II or H-IV [24]; however, no single dimer model explains all the observed crosslinks suggesting that C5a receptors form higher-order oligomers (e.g. clusters of dimers). Bioluminescence and fluorescence resonance energy transfer techniques have revealed the existence of adenosine A2A (A2AR)—dopamine D2 receptor (D2R) heterodimers in living cells. As interaction sites between D2R and A2AR, helix H-V and/or H-VI and the N-terminal portion of the intracellular loop IC-3 from D2R were found in the vicinity of H-IV and the C-terminal portion of the C-tail from A2AR [27]. CXCR2 forms constitutive oligomers when expressed in HEK cells and in native neurons. The crucial region involved in CXCR2 oligomerization may involve H-III [28]. The GXXXG motif in H-I of the α-factor receptor is similar to the transmembrane dimerization domain of glycophorin A and appears to mediate the oligomerization of the α-factor receptor and other GPCRs [29]. The significantly larger size of G proteins relative to the cytoplasmic surface of GPCRs supports the idea that the activating platform for G proteins is the dimeric form of the receptor [30,31]. In fact, dimeric leukotriene B4 receptor and trimeric G protein form a pentameric complex [32]. Oligomerization of GPCRs may not only be essential for binding and activation of G proteins, binding of receptor kinases and arrestins, but may also be involved in internalization processes [33,34]. Recent in vivo studies clearly demonstrated that the yeast α-factor receptor, the CCR5 receptor and the C5a receptor oligomerize during biogenesis [29,35,36].
For decades the concept of how rhodopsin functions in the disc membrane of retinal rod outer segments (ROS) has been dominated by the hypothesis that rhodopsin rapidly diffuses as a monomeric unit in the fluid membranes which are mostly composed of highly unsaturated phospholipids to encounter the membrane-associated G protein transducin [37]. This view was supported by biophysical measurements of rhodopsin diffusion and rotation in disc membranes, as well as low-resolution neutron diffraction, mostly carried out in amphibian photoreceptors [38-41]. In most of these studies, photoactivation of rhodopsin did not affect the results. From this concept a uniform distribution of the proteins in disc membranes would be expected. However, recent studies demonstrate the existence of detergent-resistant membrane (DRM) microdomains in ROS and therefore a non-uniform distribution of lipid and protein. After light-bleaching of ROS, a large fraction of transducin, the RGS9-1—Gb5L complex, and the p44 isoform of arrestin translocate to DRM rafts [42,43]. In addition, the cholesterol-binding protein caveolin-1 and the Gtα subunit of transducin were also shown to co-localize in such membrane rafts [44]. Thus, alternative interpretations of the early biophysical results [37-41] may be appropriate.
We have visualized rhodopsin molecules in their native environment by atomic force microscopy (AFM). These AFM experiments were performed under physiological conditions, i.e. in buffer solution, at room temperature and under normal pressure, without the application of any modification to rhodopsin that may affect its native oligomeric state. We demonstrated that mouse rhodopsin forms dimers and oligomers in native disc membranes [45] and proposed a semi-empirical model for its molecular assembly [46]. The size and geometry allowed the perfect fit of G protein and arrestin on the platform provided by the rhodopsin dimer [46]. The observed crystalline packing was independent of the support, including mica, carbon film and another disc membrane. Furthermore, data obtained by quick-freezing electron microscopy (EM) on the structure of photoreceptor membranes from Drosophila compound eyes [47] support our results from AFM. In this EM study, particles formed regular rows about 10 nm in width on the cytoplasmic surfaces, demonstrating the regular arrangement of rhodopsin molecules in invertebrate photoreceptive membranes. The highly ordered alignment of rhodopsin was also demonstrated for photoreceptors from cephalopod invertebrates by Saibil and Hewat [48]. For vertebrate photoreceptors, recent studies by freeze-etching EM provide evidence of similar crystalline domains in bovine ROS plasma membranes [49].
Rhodopsin has the propensity to form higher-order structures, i.e. two-dimensional (2D) crystals (see [50] for the most recent study). Schertler and colleagues produced 2D crystals of frog rhodopsin by extraction of ROS with Tween detergents [51]. In the p2 crystal form, molecules were unidirectionally oriented and formed contacts between their H-I helices, and between H-IV and H-V along the long unit cell axis [51]. Interestingly, the measured lattice parameters for this p2 crystal form were almost identical to those reported previously for paracrystals of mouse rhodopsin in native disc membranes [45,46]. Both crystals (from mouse and frog) contain a dimer in their unit cell and have the molecules oriented unidirectionally in the membrane, thus as expected for rhodopsin in a physiological configuration. Squid rhodopsin, digested with endoproteinase Glu C, also forms highly ordered 2D crystals consisting of alternating rhodopsin dimer rows exposing the cytoplasmic or the intradiscular surface [52]. In contrast to frog rhodopsin [51], dimers along such rows form symmetrical intradimeric contacts between H-IV, and interdimeric interactions involving the docking of H-8 onto the intracellular loop IC-2 or IC-3 [53]. Based on these interactions and the recent disulfide crosslinking and truncation mutation experiments with the dopamine D2 receptor [25,26], a second model for the dimeric and oligomeric state of a group A GPCR was established [25,54].
The observed packing arrangement of rhodopsin and opsin in native disc membranes [45,46] might be artificially induced by the segregation of protein and lipid at low temperatures[55] (see also [56]). To examine this possibility and to ensure that our previous observations were not a temperature-induced artifact, we have prepared native disc membranes at room temperature throughout, and recorded electron micrographs and AFM topographs. Dimerization and higher-order organization of rhodopsin was again observed in these native membranes. Finally, we generated a computational model of a rhodopsin dimer based on the findings by Guo et al. [25] and Lee et al. [26] who propose the intradimeric interface of two D2Rs to be formed by helices IV. The dimer model was further expanded to an oligomer model (model IV) as proposed by Lee et al. [54] and using structural data from squid rhodopsin [52,53]. We have previously proposed a model (model IV-V) for the higher-order structure of rhodopsin in the native membrane. Model IV-V involves intradimeric contacts through helices H-IV and H-V, whereas contacts mainly between helices I and II, and the cytoplasmic loop connecting helices H-V and H-VI facilitate the formation of rhodopsin dimer rows. In contrast to model IV-V where the distances of the rhodopsin molecules in the paracrystal agree with the data obtained from transmission electron microscopy (TEM) and AFM, model IV produces distances that are too large compared to our experimental results. The work presented here further validates our model IV-V as the arrangement of rhodopsin and possibly other GPCRs in the native membrane.
2. Materials and methods
2.1. Animals
All animal experiments employed procedures approved by the University of Washington Animal Care Committee and conformed to recommendations of The American Veterinary Medical Association Panel on Euthanasia. Wild-type C57BL/6 mice were obtained from the Jackson Laboratory. All animals (4-8 weeks old) were maintained in complete darkness for more than 60 min before they were killed. The eyes were removed and the retinas isolated under complete darkness with the aid of night vision goggles (LAMBDA 9 UV/VIS/NIR filter (transmittance > 560 nm), ITT Industries, VA, USA).
2.2. Isolation of rod outer segments
Twelve mouse retinas were placed in a tube with 120 μl of 8% OptiPrep (Nycomed, Oslo, Norway) in Ringer's buffer (130 mM NaCl, 3.6 mM KCl, 2.4 mM MgCl2 1.2 mM CaCl2,10 mM HEPES-NaOH (pH 7.4), and 0.02 mM EDTA) and vortexed at maximum speed for 1 min. The samples were centrifuged at 200×g for 1 min, and the supernatant containing the ROS was gently removed. The pellet was dissolved in 120 μl of 8% OptiPrep, vortexed and centrifuged again. The vortexing and sedimentation sequence was repeated six times. The collected ROS supernatant ( ∼1.5 ml) was combined and overlaid on a solution of 10-30% continuous gradient of OptiPrep in Ringer's buffer. The samples were centrifuged for 50 min at 26 500×g. ROS were harvested as a second band (about two thirds from the top), diluted three times with Ringer's solution, and centrifuged for 3 min at 500×g to remove the cell nuclei. The supernatant containing ROS was transferred to a new tube and centrifuged for 30 min at 26 500×g. The pelleted material contains pure, osmotically intact ROS. The described isolation of ROS was performed at temperatures between 20 and 25°C.
2.3. Isolation of disc membranes
ROS were disrupted osmotically in 2 ml of 2 mM Tris—HCl (pH 7.4) for 15 h, and the discs were isolated employing a 15-40% continuous gradient of OptiPrep in Ringer's buffer. The sample was centrifuged for 50 min at 26 500×g, and the discs were collected from a faint band located about two thirds from the top of the gradient. The intact discs were harvested, diluted three times with Ringer's solution, and pelleted for 30 min at 26 500×g. The described isolation of disc membranes was performed at temperatures between 20 and 25°C.
2.4. TEM of ROS
ROS were fixed in 2.5% glutaraldehyde/1% OsO4/0.13 M sodium phosphate buffer (pH 7.4), for 1 h, washed three times using TEM rinsing buffer (0.13 M NaH2PO4, 0.05% MgCl2 (pH 7.4)) and collected by centrifugation at 16 000×g for 3 min. ROS pellets were suspended in molten 5% phosphate-buffered low-temperature-gelling agarose solution, collected by centrifugation at 16 000×g for 3 min and cooled. The ROS pellets were secondarily fixed with 1% OsO4 in 0.1 M sodium phosphate buffer (pH 7.4), dehydrated with ethanol, and embedded in Eponate12 Resin (Ted Pella, CA, USA). Ultrathin sections (70 nm) were cut and stained with uranyl acetate and lead citrate solution. Electron micrographs were recorded with a Philips CM-10 TEM.
2.5. TEM and image processing of negatively stained disc membranes
Isolated discs were adsorbed to glow-discharged carbon support films mounted on EM grids and negatively stained with 0.5% uranyl acetate. Electron micrographs of disc membranes were recorded with a Hitachi H-7000 electron microscope operated at 100 kV. Single power spectra of electron micrographs and averages of several power spectra were calculated with the SEMPER image processing system [57].
2.6. Sample preparation for tapping and contact mode AFM
For the tapping mode AFM experiment in air (see Fig. 1), 70 nm ultrathin sections of ROS were deposited on freshly cleaved mica and dried (see Section 2.4). For the contact mode AFM experiment in liquid, washed disc membranes were adsorbed to freshly cleaved mica in 2 mM Tris—HCl (pH 7.4) for 15-20 min and washed with 20 mM Tris—HCl (pH 7.8), 150 mM KCl, 25 mM MgCl2 (imaging buffer).
Fig. 1.
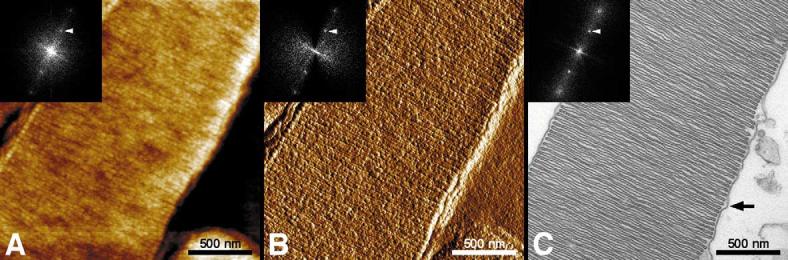
Morphology of isolated ultrathin sectioned ROS. AFM height (A) and amplitude image (B) recorded by tapping mode in air, and TEM micrograph (C). Disc membranes in a rod are stacked and surrounded by plasma membrane (arrow). The calculated power spectrum of the corresponding image is shown as inset. Vertical brightness ranges: 10 nm (A) and 1 nm (B).
2.7. Tapping and contact mode AFM
Tapping and contact mode AFM measurements were performed with a Nanoscope IV multimode AFM (Veeco/Digital Instruments, Santa Barbara, CA, USA) equipped with a 690 nm wavelength laser diode and an E-scanner (scan range up to 13 μm). For tapping mode AFM in air rectangular silicon cantilevers of 160 μm length and a nominal resonance frequency of 300 kHz (OMCL-AC160TS; Olympus, Tokyo, Japan) were used. These were excited slightly below their free resonance frequency. Two frames of 512 by 512 pixels were recorded simultaneously showing height and amplitude signals at scan frequencies below 2 Hz. For contact mode AFM in liquid oxidesharpened silicon nitride probes on V-shaped micro-cantilevers of 100 and 200 μm length, and nominal spring constants of 0.06 and0.32 N/m (NP-S; Veeco/Digital Instruments) were used. Two frames of 512 by 512 pixels (scan frequency 4.7 Hz) in trace and retrace direction were recorded simultaneously showing height and deflection signals. AFM images in contact mode were acquired at minimal loading forces.
2.8. Modeling
The presented model IV-V of rhodopsin in its oligomer state is based on the 1N3M Protein Data Bank entry (see [46] for details on the modeling of 1N3M). Model IV was built as described [46] considering the biochemical and structural information of [25,26,52-54]. In both models, phospholipids (dipalmitoyl phosphatidylcholine) were inserted between rhodopsin monomers, and optimized by molecular dynamics followed by energy minimization with rhodopsin monomers frozen to their initial positions. Next, the complex of six rhodopsin monomers was Ljected to several steps of short molecular dynamics followed by energy minimization to remove disallowed contacts. Favorable interactions were created during 10 ps molecular dynamics. No water was considered. The distances between the rhodopsin monomers in the paracrystal of model IV-V remained unchanged after addition of phospholipids and model optimization.
3. Results and discussion
3.1. AFM and TEM of ultrathin sectioned ROS
AFM allows imaging of biomolecules in buffer solutions and it also provides images of superb clarity exhibiting a vertical resolution of ± 2 °A. When calibrated with the TEM, lateral dimensions measured in AFM topographs are accurate to ± 2 Å as well (see [58] for a recent review on AFM). Here, we demonstrate another application of the AFM. Thin sections of isolated ROS fixed and embedded in Eponate12 resin were imaged by tapping mode AFM in air (Fig. 1A,B) and TEM (Fig. 1C). In all three, the AFM height and amplitude images as well as the TEM micrograph, wellorganized disc membranes were apparent with a lamellar spacing of about 35 nm. This value was measured from the calculated power spectra of the corresponding image (see Fig. 1, insets). In Fig. 1C, arrow, the plasma membrane enveloping the discs can clearly be discerned.
3.2. TEM and AFM of disc membranes isolated at room temperature
In our previous studies, ROS disc membranes were isolated at temperatures between 0 and 5°C [45,46]. To investigate whether low temperatures cause segregation of lipid from rhodopsin, and therefore disruption of the native organization of rhodopsin, disc membranes from mice were isolated at room temperature.
Isolated disc membranes were adsorbed on carbon-coated EM grids, negatively stained and inspected by TEM. Discs had a circular shape with diameters of 0.8—1.5 μm. Fig. 2A shows the morphology of an intact disc adsorbed on carbon film. Power spectra (Fig. 2B) calculated from areas on the disc membrane, e.g. broken circle (1), revealed a diffuse ring at ∼(4.5 nm)-1 indicating paracrystallinity of rhodopsin in the native membrane. As a negative control, power spectra from regions on the carbon film, e.g. broken circle (2), were calculated, but no diffraction was apparent.
Fig. 2.
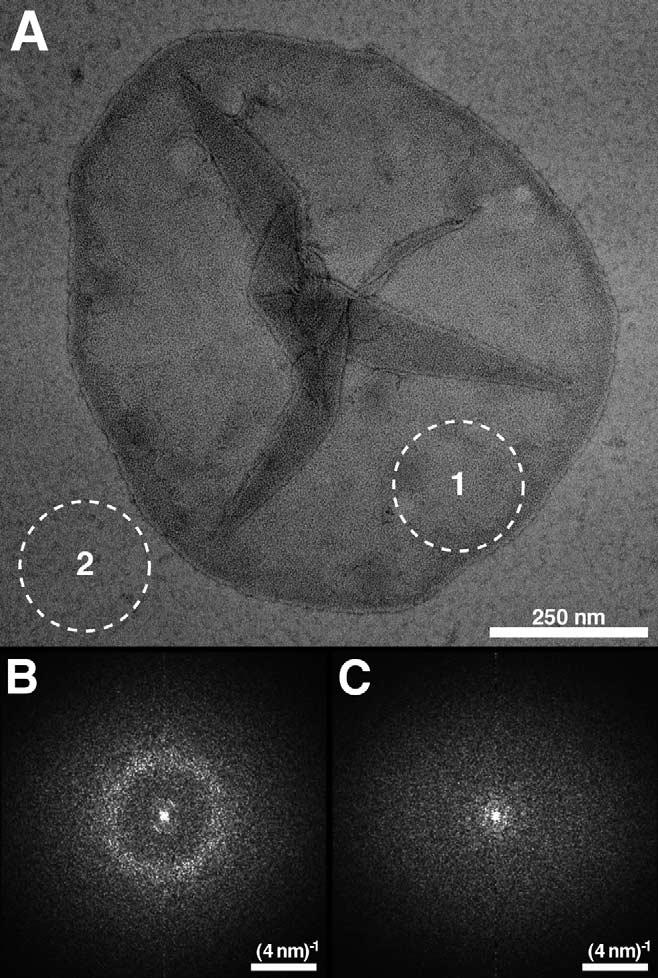
EM of negatively stained native disc membranes adsorbed on carbon film. A: Morphology of a native disc membrane from mouse isolated at room temperature and never exposed to low temperatures. B: Average of six power spectra calculated from regions on the displayed disc membrane, e.g. area marked by the broken circle (1). Powder diffraction is evident indicating paracrystallinity of the disc. C: Average of six power spectra calculated from regions on the carbon film, e.g. area marked by the broken circle (2). No powder diffraction is evident.
Native disc membranes were adsorbed to mica and imaged by AFM in buffer solution. Fig. 3A displays an overview topograph of an open, spread-flattened disc recorded at low magnification. Four different surface types are discerned: the rhodopsin surface (types 1 and 2), lipid (type 3) and mica (type 4). From height profiles such as that shown in Fig. 3B, heights of 3.68 ± 0.14 nm and 7.96 ± 0.24 nm above the mica surface were measured for domains of bare lipid (Fig. 3A, type 3) and of densely packed rhodopsin molecules (Fig. 3A, types 1 and 2), respectively. The thickness of the latter indicated that the disc membrane in Fig. 3A was single-layered, opened by the osmotic treatment (see Section 2). As observed previously [46], two different types of rhodopsin domains were identified: large uniform paracrystals (Fig. 3A, type 1) and rafts of smaller rhodopsin paracrystals separated by lipid (Fig. 3A, type 2). Topographs recorded at higher magnification (Fig. 3C, inset) unveiled rows (arrow) of rhodopsin dimers (arrowheads) forming the paracrystal identifying rhodopsin dimers as building blocks. From such crystalline areas the lattice parameters were determined: a = 8.2 ± 0.2 nm, b = 3.9 ± 0.1 nm, γ = 84 ± 2° (n = 7), in good agreement with previous measurements [45,46].
Fig. 3.
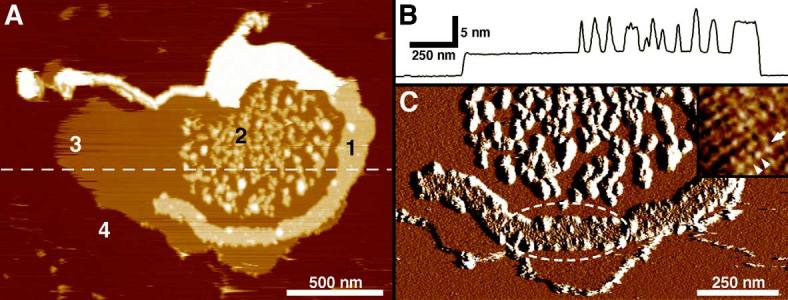
AFM of a native disc membrane adsorbed on mica and imaged in buffer solution. A: Overview height image of an open, spread-flattened single-layered disc membrane isolated at room temperature and never exposed to low temperatures. Four different surface types are discerned: the rhodopsin packed areas (types 1 and 2), lipid (type 3) and mica (type 4). B: Height profile along the broken line in A. C: Deflection image of A at higher magnification: rhodopsin paracrystals are evident (broken ellipse). Inset in C: Deflection image of a rhodopsin paracrystal at higher resolution. One row (arrow) of rhodopsin dimers (arrowheads) forming the paracrystal is indicated. Frame size of the inset in C: 56 nm. Vertical brightness ranges: 19 nm (A), 0.5 nm (C) and 0.3 nm (C, inset).
3.3. Molecular models of rhodopsin arrangements in disc membranes
Knowledge of the organization of functional GPCR dimers in native membranes is essential to understand how interacting proteins dock on the surface of these receptors. Our initial model of rhodopsin in its oligomeric state [46] (Protein Data Bank, identifier 1N3M) was further refined. Phospholipids were inserted and optimized by molecular dynamics followed by energy minimization (see Section 2). Distances between rhodopsin monomers were unaffected by the addition of phospholipids. The final model (model IV-V) for the rhodopsin dimer and oligomer is shown in Fig. 4 and all amino acid residues involved in contacts are listed in Table 1. The distances in the model are in agreement with the experimental data from TEM and AFM.
Fig. 4.
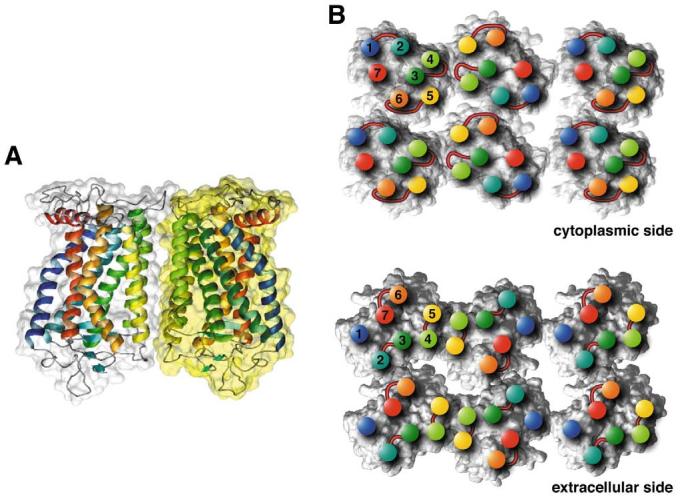
Model for the packing arrangement of rhodopsin molecules within the paracrystalline arrays of native disc membranes (based on [45,46]). A: Side view of a rhodopsin dimer, the building block of the paracrystal. Contacts between monomers are formed by the transmembrane helices IV (yellow-green) and V (yellow). Most of the interacting residues are located on the cytoplasmic loop between helices H-III and H-IV, and on the C-terminal region. Other interaction sites are located within the membrane. Both areas contain hydrogen bonds as well as interactions of hydrophobic nature. B: Cytoplasmic side (top) and extracellular side (bottom) of rhodopsin oligomers. Positions of helix ends are marked by colored discs and the corresponding helix numbers. Extracellular and cytoplasmic loops are drawn schematically at the corresponding locations. Contacts between dimers are created entirely by the intracellular loop between H-V and H-VI from one monomer with the loop between H-I and H-II and the C-terminal residues from the partner monomer. Only half of a second row of rhodopsins is shown. The contact between double rows is created mainly by hydrophobic residues from H-I close to the extracellular side. The lipid molecules initially included in the model for molecular dynamics are not displayed.
Table 1.
Contacts between amino acid residues in the oligomeric assembly of rhodopsin according to model IV-V
| Interaction type | Residues and their location on helices (H) | Location of the bond |
|---|---|---|
| Hydrogen bonds between rhodopsin molecules in the dimer | Met-143 loop H-III-H-IV—Asn-1514.40 H-IV | cytoplasmic |
| Ser-144 loop H-III-H-IV—Pro-347 C-terminal region | ||
| Ser-144 loop H-III-H-IV—Ala-348 C-terminal region | ||
| Asn-145 loop H-III-H-IV—Ala-348 C-terminal region | ||
| Arg-147 loop H-III-H-IV—Asn-145 loop H-III-H-IV | ||
| Asn-151 4.40 H-IV—Asn-145 loop H-III-H-IV | ||
| Pro-347 C-terminal region—Asn-145 loop H-III-H-IV | ||
| Ala-348 C-terminal region—Asn-145 loop H-III-H-IV | ||
| Trp-175 loop H-IV-H-V—Ser-202 5.37 H-V | transmembrane | |
| Asn-199 loop H-IV-H-V—Ser-202 5.37 H-V | ||
| Ser-202 5.37 H-V—Asn-199 loop H-IV-H-V | ||
| Hydrophobic interactions between rhodopsin molecules in the dimer | Phe-146 loop H-III-H-IV—Phe-146 loop H-III-H-IV | cytoplasmic |
| Phe-146 loop H-III-H-IV—His-152 loop H-IV | ||
| Phe-148 loop H-III-H-IV—Phe-146 loop H-III-H-IV | ||
| His-152 H-IV 4.41—Phe-146 loop H-III-H-IV | ||
| Trp-175 loop H-IV-H-V—Trp-175 loop H-IV-H-V | transmembrane | |
| Tyr-206 5.41 H-V—Tyr-206 5.41 H-V | ||
| Met-155 4.44 H-IV—Met-155 4.44 H-IV | ||
| Met-1634.52 H-IV-Met-1634.52 H-IV | ||
| Hydrogen bonds between dimers of rhodopsins | Lys-231 loop H-V-H-VI—Tyr-742.41 H-II | cytoplasmic |
| Lys-231 loop H-V-H-VI—Lys-66 loop H-I-H-II | ||
| Lys-231 loop H-V-H-VI—Gln-344 C-terminal region | ||
| Ala-235 loop H-V-H-VI—Lys-339 C-terminal region | ||
| Gln-238 loop H-V-H-VI—Thr-340 C-terminal region | ||
| Gln-238 loop H-V-H-VI—Ser-338 C-terminal region | ||
| carbohydrates—carbohydrates | extracellular | |
| Ionic bonds between double rows of rhodopsin dimers | Asp-330 C-terminal region—Lys-325 C-terminal region | cytoplasmic |
| (or Lys-235 loop H-V-H-VI—Gln-237 loop H-V-H-VI) | ||
| Hydrogen bonds between double rows of rhodopsin dimers | Trp-351.30 H-I—Glu-33 N-terminal region | extracellular |
| Hydrophobic interactions between double rows of rhodopsin dimers | Leu-328 C-terminal region—Leu-328 C-terminal region | cytoplasmic |
| Trp-351.30 H-I—Trp-351.30 H-I | transmembrane | |
| Met-391.34 H-I—Met-391.34 H-I |
An alternative dimer model was formulated based on the results from disulfide crosslinking [25] and truncation mutation [26] with the dopamine D2 receptor that unveiled H-IV as the dimer interface (see Fig. 5). In the former study [25], the site of crosslinking in D2R was identified as Cys-1684.58 (Ala-1694.58 in rhodopsin) at the extracellular end of H-IV. Throughout this paper, we indexed amino acid residues based on the bovine opsin sequence and the universal indexing (as superscript) of GPCRs [59,60]. We have built a computer model (model IV) for a rhodopsin paracrystal containing the D2R dimer configuration and the protein—protein interactions of the physiologically oriented dimers in the squid rhodopsin 2D crystal [52—54]. As for model IV-V, model IV was optimized by molecular dynamics followed by energy minimization (see Section 2). Molecular dynamics was fundamental to obtain the contacts between unidirectionally oriented rhodopsin dimer rows since this information was not available from the squid rhodopsin 2D crystal [52,53]. The interdimeric contacts in model IV are achieved by interaction of H-8 with the ends of helices III and V. The intradimer distance between rhodopsin monomers is 35 Å and between dimers 45 Å (Fig. 5). For comparison, the intradimer distance in model IV-V and that between rhodopsin dimers in rows is 38 Å for both. In model IV, the distance between double rows, e.g. horizontal distance from one yellow to the next yellow monomer in Fig. 5, is ∼77 Å,compared to 84 Å in model IV-V. The distance of 77 Å (model IV) is inconsistent with the distance of 84 Å obtained from TEM and AFM. The interaction between double rows in model IV is achieved by the flexible and elongated cytoplasmic loop IC-3 between the transmembrane helices H-V and H-VI (Fig. 5).
Fig. 5.
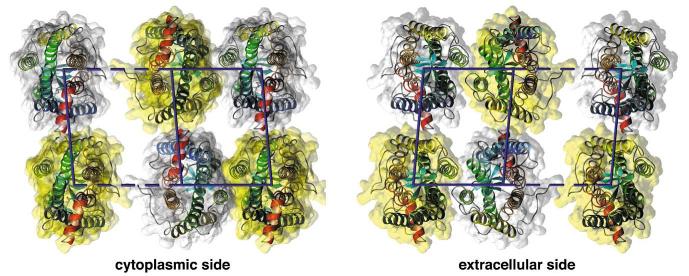
Alternative H-IV arrangement of rhodopsin in the membranes (based on [25,26,52—54]). Two views are displayed: cytoplasmic (left) and extracellular side (right). Intradimeric contacts are formed mainly by H-IV. The distance between two monomers in the dimer is 35 Å (horizontal, solid line), between dimers along the rows (oblique, solid line) 45 Å, and between double rows (horizontally, e.g. yellow to yellow monomer distance) maximally 77 Å. Contact between dimers is only possible at the cytoplasmic side between H-8 and the ends of helices III and V. Contacts between double rows are formed by IC-3, the cytoplasmic loop between H-V and H-VI. The lipid molecules initially included in the model for molecular dynamics are not displayed
Although the arrangement of GPCRs could be type-specific and extrapolation of the data from one receptor to another may not be justified [61], the interface involved in the interaction of partner proteins, G proteins, receptor kinases, and arrestins is likely to be preserved within this superfamily of transmembrane receptors.
3.4. Rhodopsin: dimer and a single photon response
The vertebrate retina registers absorption of a single photon of visible light [62]. This observation has been confermed by single cell recordings [63]. The absorption of light that leads to conformational change of photoactivated rhodopsin needs to be significantly amplified to evoke a change in the ‘dark current’; in photoreceptor cells that is ultimately coupled to changes in the neurotransmitter release from the photoreceptors to adjacent bipolar cells. In the first amplification step, photoactivated rhodopsin catalyzes nucleotide exchange on the α-subunit of G protein from GDP to GTP, a step that is repeated multiple times [64]. These observations and the fact that rhodopsin exists as a dimer according to our AFM experiments raise several questions that call for future experiments. Is one rhodopsin in the dimer sufficient for efficient activation? What is the role of the individual rhodopsin molecules in the activation process and interaction with the G protein transducin? How will the activation of transducin change when there is a single or two photoactivated rhodopsin molecules in the dimer? Most importantly, the atomic structure of the rhodopsin—transducin and rhodopsin—arrestin complexes should be determined.
Although there are no direct answers to these questions, we can provide some possible interpretation based on the work on the dimeric GABAB receptor. This GPCR of subfamily C [4,65,66] forms a heterodimer consisting of two rhodopsin-like subunits (GB1 and GB2) [18,20—22]. Despite a significant homology between these subunits, only the N-terminal domain of GB1 binds GABA. Mutant receptor complexes with GB2 N-termini on both GB1 and GB2 subunits did not respond to GABA, whereas receptors with two GB1 N-termini showed increased basal activity and responded to GABA with inhibition, rather than activation of GIRK channels via Gi [19,67]. The authors suggested that this inhibition is a consequence of GABA binding to the N-terminal region of GB2 subunit, when the N-terminal domain of the GB2 subunit was replaced with the N-terminal region of the GB1 subunit. Moreover, receptors with reciprocal exchange of N-terminal domains between the subunits displayed wild-type phenotype. GB2 intracellular segments are solely responsible for specific coupling of GABAB receptors to their physiologic effectors, Gi and G protein-activated K+ channels [67]. Therefore it is reasonable to speculate that one molecule of rhodopsin in the dimer is needed for the activation, while the second one provides a partial platform to dock subunits of transducin. It is unclear if activation of both rhodopsin molecules in the dimer is equal to the activation of only one molecule. If the activity of the doubly activated dimer is similar or lower than that of singly bleached rhodopsin in the dimer, such a property would contribute to light activation and disproportional desensitization of the visual system (non-linear relationship between the light intensity and the electrophysiological response). With this clearly testable model it will be possible to answer these emerging questions.
Acknowledgements:
We would like to thank Drs.Kevin Ridge, Jonathan Javitch and Thomas Baranski for comments on the manuscript. This research was supported in part by U.S. Public Health Service Grants EY01730 and EY08061 from the National Eye Institute, National Institutes of Health, Bethesda, MD, an unrestricted grant from Research to Prevent Blindness, Inc.(RPB), New York, NY to the Department of Ophthalmology at the University of Washington, grants from the E.K. Bishop Foundation, from the Foundation Fighting Blindness, and from the Jim and Jane Lea Research Fund. A.E. acknowledges support by the Swiss National Research Foundation, the M.E. Muüller Foundation, the Swiss National Center of Competence in Research (NCCR) ‘Structural Biology’ and the NCCR ‘Nanoscale Science’.
Abbreviations:
- AFM
atomic force microscope/microscopy
- GPCR
G protein-coupled receptor
- H-I
helix I
- IC
intracellular
- ROS
rod outer segment(s)
- TEM
transmission electron microscopy
References
- Ballesteros J, Palczewski K. Curr. Opin. Drug Discov. Dev. 2001;4:561–574. [PMC free article] [PubMed] [Google Scholar]
- Gether U. Endocr. Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- Palczewski K, et al. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Mirzadegan T, Benko G, Filipek S, Palczewski K. Biochemistry. 2003;42:2759–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G, Nadin BM, Brasier DJ, Glauner KS, Shah RD, Heinemann SF, Karim F, Gereau RW. J. Biol. Chem. 2003;278:30294–30301. doi: 10.1074/jbc.M303258200. [DOI] [PubMed] [Google Scholar]
- Ridge KD, Abdulaev NG, Sousa M, Palczewski K. Trends Biochem. Sci. 2003;28:479–487. doi: 10.1016/S0968-0004(03)00172-5. [DOI] [PubMed] [Google Scholar]
- Angers S, Bouvier M. Trends Pharmacol. Sci. 2000;21:326–326. doi: 10.1016/s0165-6147(00)01518-2. [DOI] [PubMed] [Google Scholar]
- Angers S, Salahpour A, Bouvier M. Annu. Rev. Pharmacol. Toxicol. 2002;42:409–435. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- Bai M. Cell Signal. 2004;16:175–186. doi: 10.1016/s0898-6568(03)00128-1. [DOI] [PubMed] [Google Scholar]
- Bouvier M. Nat. Rev. Neurosci. 2001;2:274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- Milligan G. J. Cell Sci. 2001;114:1265–1271. doi: 10.1242/jcs.114.7.1265. [DOI] [PubMed] [Google Scholar]
- Milligan G, Ramsay D, Pascal G, Carrillo JJ. Life Sci. 2003;74:181–188. doi: 10.1016/j.lfs.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Nat. Rev. Mol. Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- George SR, O'Dowd BF, Lee SP. Nat. Rev. Drug Discov. 2002;1:808–820. doi: 10.1038/nrd913. [DOI] [PubMed] [Google Scholar]
- Van Brocklyn JR, Behbahani B, Lee NH. Biochim. Biophys. Acta. 2002;1582:89–93. doi: 10.1016/s1388-1981(02)00141-5. [DOI] [PubMed] [Google Scholar]
- Rios CD, Jordan BA, Gomes I, Devi LA. Pharmacol. Ther. 2001;92:71–87. doi: 10.1016/s0163-7258(01)00160-7. [DOI] [PubMed] [Google Scholar]
- Kunishima N, et al. Nature. 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- Kuner R, Kohr G, Grunewald S, Eisenhardt G, Bach A, Kornau HC. Science. 1999;283:74–77. doi: 10.1126/science.283.5398.74. [DOI] [PubMed] [Google Scholar]
- Margeta-Mitrovic M, Jan YN, Jan LY. Proc. Natl. Acad. Sci. USA. 2001;98:14643–14648. doi: 10.1073/pnas.251554798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupmann K, et al. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- Jones KA, et al. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- White JH, et al. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- Carrillo JJ, Pediani J, Milligan G. J. Biol. Chem. 2003;278:42578–42587. doi: 10.1074/jbc.M306165200. [DOI] [PubMed] [Google Scholar]
- Klco JM, Lassere TB, Baranski TJ. J. Biol. Chem. 2003;278:35345–35353. doi: 10.1074/jbc.M305606200. [DOI] [PubMed] [Google Scholar]
- Guo W, Shi L, Javitch JA. J. Biol. Chem. 2003;278:4385–4388. doi: 10.1074/jbc.C200679200. [DOI] [PubMed] [Google Scholar]
- Lee SP, O'Dowd BF, Rajaram RD, Nguyen T, George SR. Biochemistry. 2003;42:11023–11031. doi: 10.1021/bi0345539. [DOI] [PubMed] [Google Scholar]
- Canals M, et al. J. Biol. Chem. 2003;278:46741–46749. doi: 10.1074/jbc.M306451200. [DOI] [PubMed] [Google Scholar]
- Trettel F, Di Bartolomeo S, Lauro C, Catalano M, Ciotti TM, Limatola C. J. Biol. Chem. 2003;278:40980–40988. doi: 10.1074/jbc.M306815200. [DOI] [PubMed] [Google Scholar]
- Overton MC, Chinault SL, Blumer KJ. J. Biol. Chem. 2003;278:49369–49377. doi: 10.1074/jbc.M308654200. [DOI] [PubMed] [Google Scholar]
- Hamm HE. Proc. Natl. Acad. Sci. USA. 2001;98:4819–4821. doi: 10.1073/pnas.011099798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimoto R, Kisselev OG, Makara GM, Marshall GR. Biophys. J. 2001;81:3285–3293. doi: 10.1016/S0006-3495(01)75962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baneres JL, Parello J. J. Mol. Biol. 2003;329:815–829. doi: 10.1016/s0022-2836(03)00439-x. [DOI] [PubMed] [Google Scholar]
- Cheng ZJ, Harikumar KG, Holicky EL, Miller LJ. J. Biol. Chem. 2003;278:52972–52979. doi: 10.1074/jbc.M310090200. [DOI] [PubMed] [Google Scholar]
- Stanasila L, Perez JB, Vogel H, Cotecchia S. J. Biol. Chem. 2003;278:40239–40251. doi: 10.1074/jbc.M306085200. [DOI] [PubMed] [Google Scholar]
- Issafras H, Angers S, Bulenger S, Blanpain C, Parmentier M, Labbe-Jullie C, Bouvier M, Marullo S. J. Biol. Chem. 2002;277:34666–34673. doi: 10.1074/jbc.M202386200. [DOI] [PubMed] [Google Scholar]
- Floyd DH, Geva A, Bruinsma SP, Overton MC, Blumer KJ, Baranski TJ. J. Biol. Chem. 2003;278:35354–35361. doi: 10.1074/jbc.M305607200. [DOI] [PubMed] [Google Scholar]
- Liebman PA, Parker KR, Dratz EA. Annu. Rev. Physiol. 1987;49:765–791. doi: 10.1146/annurev.ph.49.030187.004001. [DOI] [PubMed] [Google Scholar]
- Cone RA. Nat. New Biol. 1972;236:39–43. doi: 10.1038/newbio236039a0. [DOI] [PubMed] [Google Scholar]
- Poo M, Cone RA. Nature. 1974;247:438–441. doi: 10.1038/247438a0. [DOI] [PubMed] [Google Scholar]
- Liebman PA, Entine G. Science. 1974;185:457–459. doi: 10.1126/science.185.4149.457. [DOI] [PubMed] [Google Scholar]
- Saibil H, Chabre M, Worcester D. Nature. 1976;262:266–270. doi: 10.1038/262266a0. [DOI] [PubMed] [Google Scholar]
- Seno K, et al. J. Biol. Chem. 2001;276:20813–20816. doi: 10.1074/jbc.C100032200. [DOI] [PubMed] [Google Scholar]
- Nair KS, Balasubramanian N, Slepak VZ. Curr. Biol. 2002;12:421–425. doi: 10.1016/s0960-9822(02)00691-7. [DOI] [PubMed] [Google Scholar]
- Elliott MH, Fliesler SJ, Ghalayini AJ. Biochemistry. 2003;42:7892–7903. doi: 10.1021/bi027162n. [DOI] [PubMed] [Google Scholar]
- Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. Nature. 2003;421:127–128. doi: 10.1038/421127a. [DOI] [PubMed] [Google Scholar]
- Liang Y, Fotiadis D, Filipek S, Saperstein DA, Palczewski K, Engel A. J. Biol. Chem. 2003;278:21655–21662. doi: 10.1074/jbc.M302536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki E, Katayama E, Hirosawa K. J. Electron Microsc. (Tokyo) 1993;42:178–184. [PubMed] [Google Scholar]
- Saibil H, Hewat E. J. Cell Biol. 1987;105:19–28. doi: 10.1083/jcb.105.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura N, Harada Y, Usukura J. J. Electron Microsc. (Tokyo) 2000;49:691–697. doi: 10.1093/oxfordjournals.jmicro.a023860. [DOI] [PubMed] [Google Scholar]
- Krebs A, Edwards PC, Villa C, Li J, Schertler GF. J. Biol. Chem. 2003;278:50217–50225. doi: 10.1074/jbc.M307995200. [DOI] [PubMed] [Google Scholar]
- Schertler GF, Hargrave PA. Proc. Natl. Acad. Sci. USA. 1995;92:11578–11582. doi: 10.1073/pnas.92.25.11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A, Schertler GF, Gowen BE, Saibil HR. J. Struct. Biol. 1996;117:36–44. doi: 10.1006/jsbi.1996.0067. [DOI] [PubMed] [Google Scholar]
- Davies A, Gowen BE, Krebs AM, Schertler GF, Saibil HR. J. Mol. Biol. 2001;314:455–463. doi: 10.1006/jmbi.2001.5167. [DOI] [PubMed] [Google Scholar]
- Lee SP, O'Dowd BF, George SR. Life Sci. 2003;74:173–180. doi: 10.1016/j.lfs.2003.09.028. [DOI] [PubMed] [Google Scholar]
- Chabre M, Cone R, Saibil H. Nature. 2003;426:30–31. doi: 10.1038/426030b. [DOI] [PubMed] [Google Scholar]
- Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. Nature. 2003;426:31. doi: 10.1038/421127a. [DOI] [PubMed] [Google Scholar]
- Saxton WO. J. Struct. Biol. 1996;116:230–236. doi: 10.1006/jsbi.1996.0035. [DOI] [PubMed] [Google Scholar]
- Fotiadis D, Scheuring S, Müller SA, Engel A, Müller DJ. Micron. 2002;33:385–397. doi: 10.1016/s0968-4328(01)00026-9. [DOI] [PubMed] [Google Scholar]
- Visiers I, Ballesteros JA, Weinstein H. Methods Enzymol. 2002;343:329–371. doi: 10.1016/s0076-6879(02)43145-x. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Shi L, Javitch JA. Mol. Pharmacol. 2001;60:1–19. [PubMed] [Google Scholar]
- Soyer OS, Dimmic MW, Neubig RR, Goldstein RA. Biochemistry. 2003;42:14522–14531. doi: 10.1021/bi035097r. [DOI] [PubMed] [Google Scholar]
- Hech S, Shlaer S, Pirenne MH. J. Gen. Physiol. 1942;25:819–840. doi: 10.1085/jgp.25.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylor DA, Lamb TD, Yau KW. J. Physiol. 1979;288:613–634. [PMC free article] [PubMed] [Google Scholar]
- Kwok-Keung Fung B., Stryer L. Proc. Natl. Acad. Sci. USA. 1980;77:2500–2504. doi: 10.1073/pnas.77.5.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilatis DK, et al. Proc. Natl. Acad. Sci. USA. 2003;100:4903–4908. doi: 10.1073/pnas.0230374100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. Mol. Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Margeta-Mitrovic M, Jan YN, Jan LY. Proc. Natl. Acad. Sci. USA. 2001;98:14649–14654. doi: 10.1073/pnas.251554498. [DOI] [PMC free article] [PubMed] [Google Scholar]