

Abstract
The microarray technique requires the organization and analysis of vast amounts of data. These data include information about the samples hybridized, the hybridization images and their extracted data matrices, and information about the physical array, the features and reporter molecules. We present a web-based customizable bioinformatics solution called BioArray Software Environment (BASE) for the management and analysis of all areas of microarray experimentation. All software necessary to run a local server is freely available.
Rationale
Microarrays are emerging as one of the most exciting and promising technologies for biological research and clinical practice [1]. The technology has been utilized in various applications such as the profiling of mRNA [2] and protein levels [3], elucidating protein-DNA interactions [4], assessment of DNA copy number [5], and detection of methylated sequences [6], and today is accessible to even relatively small laboratories. Typically, arrays contain 5,000 to 45,000 reporters, each of which has dozens of biological (for example, gene name, sequence, function) and quality control (QC; for example, sequence verification, purity, number of gel bands) annotations. Each array can be used to analyze up to two biomaterials, each of which can have any number of biological annotations (for example, in vitro treatments, clinical follow-up, mutation status), and in a single hybridization, data spanning tens of megabytes are generated. Whereas microarrays have shed light on many biological processes and disease states, for us [7,8,9,10] and others, a significant bottleneck remains the analysis of hybridization data in the context of biomaterial and reporter annotations. There are a number of separate software systems that individually address some of the needs, such as databases and applications for clustering and visualization of microarray data [11,12,13,14,15,16,17,18], public databases that contain reporter information [19,20,21], commercial laboratory information management systems (LIMS), and various storage methods (such as lab notebooks, clinical charts and public and private databases) for recording biomaterial annotations. However, to our knowledge there are no unified systems capable of organizing all the information surrounding microarray experimentation and which also integrate this information with tools for the analysis of quantified microarray hybridization data.
To address these needs, we developed a system called BioArray Software Environment (BASE) that provides an integrated framework for storing and analyzing microarray information. BASE is a MIAME-supportive [22] customizable database and analysis platform designed to be installed in any microarray laboratory and to serve many users simultaneously via the web. The software was developed on the GNU/Linux operating system (OS) in the PHP language [23], with data being stored in a relational database (MySQL [24]) and communicated to the user through the Apache Webserver [25]. Where needed, the user interface employs Java and JavaScript in addition to plain HTML, and C++ has been used for the more computationally intensive tasks on the server.
In short, the system integrates biomaterial information, raw images and extracted data, and provides a plug-in architecture for data transformation, data viewing and analysis modules. Additionally, for laboratories that fabricate in-house arrays or for groups that wish to track reporter information, the system has array production LIMS features that can be integrated with the data analysis. The structure of BASE was designed to follow the natural workflow of the microarray biologist (Figure 1), and it is compatible with most types of array experiments and data formats (for example, one- or two-channel hybridizations, cDNA/oligos spotted on any substrate, Affymetrix chips, comparative genomic hybridization (CGH) on arrays). With his or her own account and administrated access levels, a user can import data into the database, group array data together into experiments, and in a uniform and streamlined fashion, apply filters and transformations and run analyses. To facilitate online collaboration, users can share almost any object within the database with another user. Data can be exported in a multitude of formats for local analysis and publication. As proof-of-concept of the plug-in architecture, we have implemented in C++ modules featuring locally weighted scatterplot smoothing (LOWESS) [26,27] normalization and multidimensional scaling (MDS) analysis [28].
Figure 1.
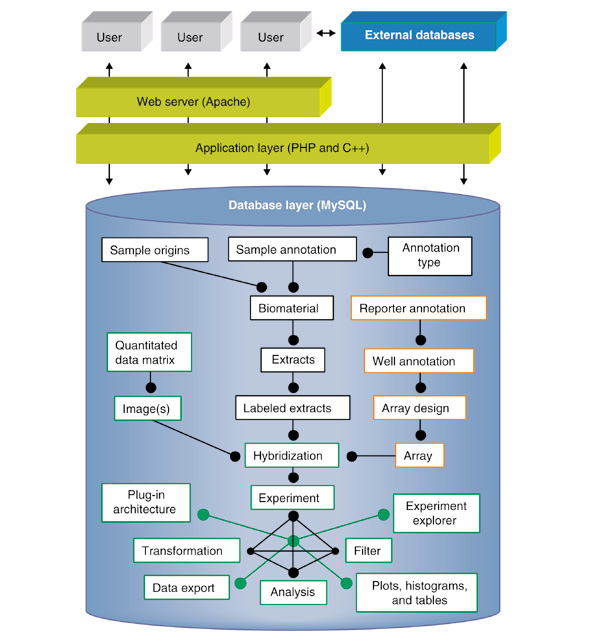
Simplified schematic overview of software structure. Arrows represent the flow of information. Closed circle connectors represent logical relationships between database classes. Database classes outlined by black boxes relate to biomaterials; array production LIMS items are highlighted by orange boxes; and data-analysis features are within green boxes. A detailed database schema is available from our website [34] and with this paper (see Downloading files).
Biomaterials
Full annotation of the samples hybridized enables complex and powerful inquiries. In addition to sample source hierarchies, in vivo or in vitro sample treatments and extraction and labeling protocols, many other types of annotations can be useful to record and correlate with hybridization data. For instance, the genotype, mutation profile, patient data or status of particular proteins as indicated by immunohistochemistry maybe applied to aid in evaluating analysis results. Therefore, we have designed an annotation and tracking system for biomaterials that is user-customizable via a web interface and is integrated with the data analysis (Figure 1). Source organism and cell-type taxonomies can be created, and new annotation types (integer, number, enumerations or free text) can be defined and linked to any sample. Users can enter new samples, annotate them, and create subsequent sample extractions and labelings, storing information such as quantity, quality, events, and protocols at each step.
Array production LIMS
Many laboratories use robot printers to spot their own microarrays on substrates such as derivatized glass or nylon membranes. The printed reporter molecules, such as PCR products, can have dozens of biological and QC annotations, and are often produced through multi-step procedures. For instance, bacterial clones may be received in 96-well microtiter plates and travel through half a dozen or more plates before they are finally spotted. At various stages, products may be run on gels to determine band size and number, or verified by resequencing. Accurate tracking of this information and integration with hybridization results can be useful. For this reason, we have implemented a generic array production LIMS as an optional feature that can be integrated with data analysis (Figures 1, 2b). New plate types can be defined, reporters and their biological annotations imported, and parent-daughter plate relationships tracked. Currently BASE supports 96- and 384-well plate formats, and merging of 96-well plates onto a 384-well plate can be carried out. Moreover, the system is compatible with any reporter type, although we have developed specialized features for human IMAGE clones [29].
Figure 2.

Browsing data with Experiment Explorer. Within BASE, the (a) Experiment Explorer displays data across all hybridizations in an experiment and relates it to the biological sample annotations, and optionally to the back-end array production LIMS. Reporter information is hyperlinked to external databases such as those at NCBI [20]. By clicking on a reporter position, the (b) Reporter Information page displays all raw and transformed data for the reporter as well as its production history from the array LIMS.
A user with the appropriate administrative privileges can define a physical array design by specifying print plates and importing a print map file (created, for example, by the BioRobotics MicroGrid II software). Then, each print run or batch of arrays produced can be managed and the fabrication conditions and protocols recorded, as well as the quantity and identification (barcode or other unique ID) for each physical array. Lastly, a formatted list of array features for use by image processors (such as Axon GenePix) can be downloaded. For laboratories that do not spot cDNA clones, but instead use commercial chips or spot other types of reporter molecules, the array LIMS feature may still be useful to store reporter annotations locally.
Data analysis
A variety of feature-analysis software packages are used to extract quantification matrices from hybridization images, and output is typically in the form of tab-delimited text files. Mining a logical set of such data files, which may have over 1 million data points each, requires a robust informatics system. Moreover, powerful analytical and visualization tools that take advantage of biomaterial and reporter annotations are needed. To this end we have designed a flexible and expandable platform for analyzing microarray data (Figure 1). Within BASE, a user may create associations of labeled extracts, scanned raw images (optional), quantification matrices, and arrays (if the production LIMS is being used) to define individual hybridizations. As a single hybridization can be scanned in more than one scanner and/or under different settings, and each image can be analyzed by different image-processing software with various parameters, these types of relationships and information can also be recorded. Tab-delimited data output from any image processor can be imported into the database using an interactive import wizard, and frequently used formats can be identified automatically. The association of one or two labeled extracts to an imported data matrix appears as a unique data-set object. Within the user's personal workspace, sets of array data objects can be grouped into experiments and annotated. Data sets may be grouped in parallel experiments and thus can be analyzed to test various experimental hypotheses under disparate contexts, and through the object-sharing feature, by more than one user simultaneously.
The analysis of microarray data is a rapidly evolving area of bioinformatics. We have integrated a flexible framework with a plug-in architecture that enables the easy integration of innovative modules that transform, or analyze and visualize microarray data. This architecture consists of three parts: a data standard and format (currently our 'BASEfile' format; MAGE-ML [30] is being considered) for transferring biomaterial, reporter and hybridization data to and from application modules that run on the server, a job handler for execution of application modules and saving results back into the database, and a web interface for the administration and installation of new plug-in modules. A plug-in module may be any type of executable program or script that runs on Linux. We have developed and included three plug-ins: Normalizer performs within-slide global mean or median ratio based normalization; Lowess performs within-slide intensity-dependent LOWESS [26] normalization (Figure 3b); and the MDS module computes a distance metric between samples on the basis of their gene expression, and reduces the high-dimensional space into two- or three-dimensional coordinates [28]. The 3D Data Viewer (Figure 3d) allows the user to visualize and rotate MDS results in relation to biomaterial annotations, and to export figures for publication.
Figure 3.
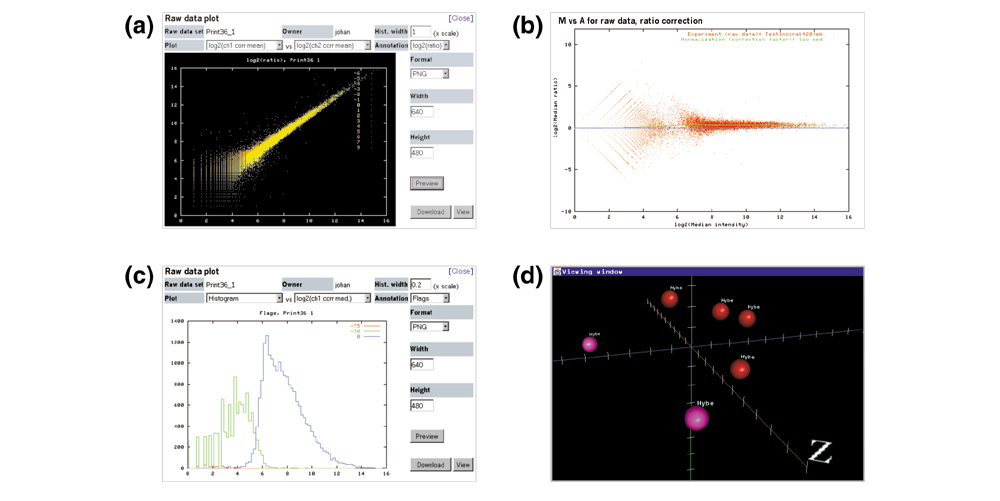
Example data visualization tools within BASE. Images can be resized and exported in several formats. (a) Interactive scatterplot displaying the log2 (ch1 background corrected mean intensity) versus log2 (ch2 corrected mean intensity) pseudocolored according to log2 (ratio). (b) M-A plot [35] of raw data displaying the intensity-dependent LOWESS [26] fitted normalization curve in green. (c) Interactive histogram of log2 (ch1 corrected median intensity) viewed in relation to spot flag annotations from image analysis software. (d) 3D Data Viewer displaying a rotatable and scalable MDS [28] result, in which samples (shown here as spheres) can be visualized in relation to their biomaterial annotations by changing their shape, color, and texture, or by adding floating text.
Furthermore, to allow for any combination and series of data filtering, transformation and number-crunching steps, we created a data-analysis interface that is organized hierarchically. An unmodified data set can be filtered and sent to a plug-in module; subsequently the output can be filtered and transformed again, and so on to create transformed data and resultant subsets. In this way, the original unmodified data set can be filtered under different settings and sent to alternative modules, to create many branches under the same experiment. All parameters and settings are stored at each step for later reference, and the entire analysis history can be seen as a textual dendrogram.
Data can be visualized at several stages of analysis. Unmodified and transformed data sets can be plotted interactively as scatter plots (Figure 3a,3b), displayed in histograms (Figure 3c), or viewed as tables. Entire experiments can be displayed in various overview plots in the context of how they are annotated, and figures and tables can be exported for publication. From any data-analysis step the experiment can be imported into a data-visualization interface that we have included called Experiment Explorer (Figure 2a), in which the data can be browsed and viewed, reporter by reporter, in the context of sample and reporter annotations. Data can also be exported for custom analyses (for example, for algorithms that are very expensive of computer power and time) and local development of new analysis methods, and in various defined formats for use in external analysis programs such as Cluster [12] and J-Express [14].
Requirements and availability
All additional software required such as the OS, database, webserver, and languages are freely available from their developers. With some modification, BASE can be made to run under other OS and database environments. BASE has already been successfully installed on the Solaris operating system, and with some modification can be made to run under other database and OS environments such as Windows, Macintosh OS X, and other Unix varieties. The hardware requirements are quite modest (a PC with 100 gigabyte hard disk can manage over 3,000 hybridizations, each with 30,000 features and several analysis steps), making BASE a realistic alternative even for users with a limited budget. As user requirements increase, additional servers and storage space can be added to a BASE installation.
The future of BASE
There are many data-transformation and analysis algorithms that we would like to integrate within BASE as plug-in modules (see [31] for a review), and features we anticipate will be desirable in the future and intend to support (for example, MAGE-ML [30] export for data deposition in public repositories such as ArrayExpress [17] and GEO [32], and hybridizations using three or more channels). By providing an open-source platform to build on and by continuously developing new plug-in applications ourselves, we hope to stimulate researchers to use the system. We encourage academic and commercial contribution, and hope that end-users will not only customize BASE to suit their own needs, but also share their experiences, source code and new plug-in modules with the community of BASE users.
Downloading files
All BASE source code is publicly available to academic and commercial sites under the GNU General Public License [33] and may be downloaded from our website, along with an operating manual [34]. The manual and a diagram of the software are available with this article.
Supplementary Material
Software operating manual
A diagram of the software
Acknowledgments
Acknowledgements
This work was in part supported by the Knut and Alice Wallenberg Foundation through the SWEGENE consortium and by the Swedish Cancer Society. We thank the numerous beta test laboratories for their feedback and suggestions. We are grateful to Mario Gianota for creating the 3D Data Viewer, and Björn Samuelsson for his work on the normalizers.
Disclaimer: Whilst every effort is made by the authors, editors and publishers to ensure that the software performs as described, they accept no liability whatsoever for the consequences of downloading or using it.
References
- Schulze A, Downward J. Navigating gene expression using microarrays - a technology review. Nat Cell Biol. 2001;3:E190–E195. doi: 10.1038/35087138. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Haab BB, Dunham MJ, Brown PO. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2001;2:research0004.1–0004.13. doi: 10.1186/gb-2001-2-2-research0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, Brown PO. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Williams CF, Jeffrey SS, Botstein D, Brown PO. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet. 1999;23:41–46. doi: 10.1038/12640. [DOI] [PubMed] [Google Scholar]
- Yan PS, Chen CM, Shi H, Rahmatpanah F, Wei SH, Caldwell CW, Huang TH. Dissecting complex epigenetic alterations in breast cancer using CpG island microarrays. Cancer Res. 2001;61:8375–8380. [PubMed] [Google Scholar]
- Hedenfalk I, Duggan D, Chen Y, Radmacher M, Bittner M, Simon R, Meltzer P, Gusterson B, Esteller M, Kallioniemi OP, et al. Gene-expression profiles in hereditary breast cancer. N Engl J Med. 2001;344:539–548. doi: 10.1056/NEJM200102223440801. [DOI] [PubMed] [Google Scholar]
- Khan J, Bittner ML, Saal LH, Teichmann U, Azorsa DO, Gooden GC, Pavan WJ, Trent JM, Meltzer PS. cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc Natl Acad Sci USA. 1999;96:13264–13269. doi: 10.1073/pnas.96.23.13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu CR, Peterson C, Meltzer PS. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7:673–679. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruvberger S, Ringner M, Chen Y, Panavally S, Saal LH, Borg A, Ferno M, Peterson C, Meltzer PS. Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res. 2001;61:5979–5984. [PubMed] [Google Scholar]
- Ermolaeva O, Rastogi M, Pruitt KD, Schuler GD, Bittner ML, Chen Y, Simon R, Meltzer P, Trent JM, Boguski MS. Data management and analysis for gene expression arrays. Nat Genet. 1998;20:19–23. doi: 10.1038/1670. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander ES, Golub TR. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dysvik B, Jonassen I. J-Express: exploring gene expression data using Java. Bioinformatics. 2001;17:369–370. doi: 10.1093/bioinformatics/17.4.369. [DOI] [PubMed] [Google Scholar]
- Sherlock G, Hernandez-Boussard T, Kasarskis A, Binkley G, Matese JC, Dwight SS, Kaloper M, Weng S, Jin H, Ball CA, et al. The Stanford Microarray Database. Nucleic Acids Res. 2001;29:152–155. doi: 10.1093/nar/29.1.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18:207–208. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- ArrayExpress http://www.ebi.ac.uk/microarray/ArrayExpress/arrayexpress.html
- Expression Profiler http://ep.ebi.ac.uk/EP/
- Tsai J, Sultana R, Lee Y, Pertea G, Karamycheva S, Antonescu V, Cho J, Parvizi B, Cheung F, Quackenbush J. RESOURCERER: a database for annotating and linking microarray resources within and across species. Genome Biol. 2001;2:software0002.1–0002.4. doi: 10.1186/gb-2001-2-11-software0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov
- Affymetrix NetAffx http://www.affymetrix.com/analysis/index.affx
- Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, et al. Minimum information about a microarray experiment (MIAME) - toward standards for microarray data. Nat Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- PHP: Hypertext preprocessor http://www.php.net
- MySQL http://www.mysql.com
- Apache HTTP Server Project http://httpd.apache.org
- Cleveland WS, Devlin SJ. Locally weighted regression: an approach to regression analysis by local fitting. J Am Stat Assoc. 1988;83:596–610. [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J, Simon R, Bittner M, Chen Y, Leighton SB, Pohida T, Smith PD, Jiang Y, Gooden GC, Trent JM, Meltzer PS. Gene expression profiling of alveolar rhabdomyosarcoma with cDNA microarrays. Cancer Res. 1998;58:5009–5013. [PubMed] [Google Scholar]
- Lennon G, Auffray C, Polymeropoulos M, Soares MB. The I.M.A.G.E. Consortium: an integrated molecular analysis of genomes and their expression. Genomics. 1996;33:151–152. doi: 10.1006/geno.1996.0177. [DOI] [PubMed] [Google Scholar]
- Microarray and gene expression - MAGE http://www.mged.org/Workgroups/MAGE/mage.html
- Quackenbush J. Computational analysis of microarray data. Nat Rev Genet. 2001;2:418–427. doi: 10.1038/35076576. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GNU general public license http://www.gnu.org/licenses/licenses.html#GPL
- BioArray Software Environment http://base.thep.lu.se
- Yang YH, Dudoit S, Luu P, Speed TP. Normalization for cDNA Microarray Data. Department of Statistics, UC Berkeley Technical Report (Preprint) 2001;number 589 http://www.stat.berkeley.edu/tech-reports/index.html [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Software operating manual
A diagram of the software