

Abstract
Inheritance of germline BRCA1 mutations is associated with a high risk of breast and ovarian cancers. A multitude of cellular functions has been ascribed to BRCA1, including transcription activation and various aspects of DNA repair. So far, indirect evidence has indicated a role for BRCA1 in the repair of double-strand breaks. Recently, an elegant gene targeting design was used to provide definitive evidence that BRCA1 promotes homologous recombination and limits nonhomologous mutagenic repair processes. This reaffirms the role of BRCA1 as caretaker in preserving genomic integrity.
Keywords: Brca1, Brca2, homologous recombination, nonhomologous end-joining, double-strand break, Rad51, RecA, breast cancer
Introduction
Ever since the cloning of the two breast cancer susceptibility genes BRCA1 and BRCA2, intense scientific effort has been invested into elucidating their biologic functions. Both genes encode large proteins (1863 and 3418 amino acids, respectively) that are coordinately expressed during cellular proliferation and differentiation [1]. The BRCA1 and BRCA2 proteins are expressed in most tissues and most abundantly during the S phase of the cell cycle. A plethora of functions have been ascribed to BRCA1 and BRCA2, including transcription activation [2,3,4], chromatin remodelling [5,6], centrosome duplication [7,8], apoptosis [9], and transcription-coupled [10] and homology directed [11,12,13] DNA repair.
Murine models for BRCA1 and BRCA2 mutations establish that both genes are essential for development [14,15,16,17], because early embryonic lethality is a common phenotype. This early lethal phenotype can be partly rescued by introducing a null mutation in the damage checkpoint gene p53 or p21 [18]. This suggests that loss of BRCA1 or BRCA2 may trigger the activation of a cell-cycle checkpoint through the accumulation of DNA damage. In agreement with this, both Brca1-/- and Brca2 -/- homozygous cells have been found to be hypersensitive to γ-irradiation [17,18] and spontaneously exhibit chromosomal abnormalities [11,13].
The first indication that BRCA1 and BRCA2 function in homologous recombination came from coimmunoprecipitation and yeast two-hybrid studies [17,19,20], which demonstrated that both BRCA1 and BRCA2 interacted with Rad51, a mammalian homologue of the Escherichia coli RecA protein that is involved in DNA damage repair. (The association of BRCA1 with Rad51, however, is likely to be indirect and possibly mediated by BRCA2 [21]). Immunostaining further suggests that BRCA1, BRCA2 and Rad51 work together in a protein complex as they coalesce to form discrete nuclear dots during S phase and after DNA damage [21,22]. Moreover, in meiotic cells extensive colocalization of BRCA1, BRCA2 and Rad51 have been demonstrated on synaptonemal complexes [21].
As already mentioned, Rad51 is part of homologous recombination. In E coli, and the budding yeast Saccharomyces cerevisiae, homologous recombination is a major pathway for repairing double-strand breaks (although no homologues of BRCA1 or BRCA2 have yet been identified in these organisms). In mammalian cells, it has been estimated that around 30-50% of DNA breaks are repaired by homologous recombination [23], the remainder being dealt with mainly by a process known as nonhomologous end-joining (NHEJ). Unlike homologous recombination, which repairs through genetic exchange with a homologous chromosome, NHEJ simply involves direct joining of the broken ends. This frequently results in small deletions or insertions at the site of breakage. Whereas NHEJ is a relatively well-understood pathway in mammalian cells [24], little is known about the mechanisms involved in mammalian homologous recombination. Despite all speculations that BRCA1 and BRCA2 participate in Rad51-mediated homologous recombination, direct evidence is lacking. Furthermore, to delineate the players involved in homologous recombination in mammalian cells, it is necessary to have a robust reporter assay system. It is to this challenge that Jasin [25] provided a valuable tool.
BRCA1 controls homologous recombination
A Brca1-deficient murine embryonic stem cell line developed by Gowen et al [14] was employed. The mutant Brca1 gene here was created by replacing the terminal portion of intron 10 and the beginning of exon 11 with a selectable marker gene, resulting in the loss of the full-length Brca1 transcript. Exon 11 is the largest coding exon of Brca1 and encodes approximately 60% of the protein, including the putative Rad51-interacting domain [20]. Murine embryos that harbour the Brca1 mutation described by Gowen et al [14] typically survive to embryonic day 9 or 10.
Moynahan et al [12] devised two assays to test the integrity of homologous recombination in the Brca1-/- embryonic stem cells. First, the rate of targeted integration of transfected DNA was evaluated (Fig. 1). Moynahan et al found that gene-targeting efficiency was 23-fold lower in Brca1-/-cells when the promotorless pim1-Q targeting vector was transfected. This was not due to the lack of a promotor, because when an Rb-O targeting vector that contained a pgk promoter was used, Brca1-/-cells had a 13-fold decrease in gene targeting and a fourfold increase in random integration compared with Brca1 +/- control cells.
Figure 1.

Schematic representations of the (A) pim1-Q and (B) Rb-O targeting vectors. (A) Gene targeting strategy at the pim1 locus. Expression of the promotorless hygromycin resistance relies on the endogenous pim1 promotor once it is integrated by homologous recombination. Transfected cells are selected in hygromycin. (B) Gene targeting strategy at the Rb locus. The Rb-O contains a hygromycin gene expressed from a pgk promotor. Therefore, hygromycin resistance is exhibited regardless of the site of integration
Next, Moynahan and colleagues tested the ability of Brca1-deficient cells to repair chromosomal double-strand breaks. They made use of a rare-cutting endonuclease, I-SceI, which has a unique 18-base-pair nonpalindromic recognition sequence that is not found in the mammalian genome [26]. A direct repeat recombination substrate containing this sequence was integrated at the pim1 and Rb loci. Introduction of the enzyme I-SceI would, therefore, create a double-strand break (DSB) in the direct repeat that consisted of two differentially mutated neomycin phosphotransferase (neo) genes separated by a thymidine kinase (tk) gene (Fig.2).It has already been demonstrated that the presence of a single DSB was a potent stimulus for homologous recombination [25]. By selecting for the loss of the tk gene, it would be possible to estimate the frequency of deletional homologous recombination (Fig. 2). In agreement with the previous assay, the Brca1-/-cell line exhibited a fivefold lower frequency of spontaneous and DSB-induced deletion compared with the heterozygous control. This was further supported by detailed examination of the substrates by polymerase chain reaction and Southern blotting.
Figure 2.
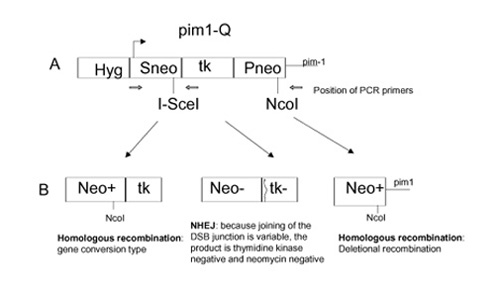
Outcomes of the repair of a double-strand break (DSB) introduced by I-SceI. (A) The integrated pim1-Q vector. Open arrows indicate the positions of PCR primers that are used to distinguish between the various repair products. (B) Possible repair products. NHEJ, nonhomologous end-joining.
Using primers flanking the DSB site and the NcoI site in the substrate (Fig. 2), Moynahan et al [12] were able to distinguish end-products from both homologous recombination and NHEJ. They concluded that Brca1-/-cells had a consistently fivefold to sixfold lower level of homologous recombination by both noncrossover gene conversion and deletion. On the contrary, NHEJ was intact and its frequency was even increased by 1.5-fold to 1.6-fold in Brca1-/-cells.
Conclusion
The work of Moynahan et al [12] is significant, because it is the first direct demonstration of the role of Brca1 in controlling homologous recombination. Cells deficient in Brca1 were found to have decreased gene targeting as well as impaired homologous recombination of an experimentally introduced DSB. Interestingly, the rate of homologous recombination in Brca1-/-embryonic stem cells was reduced by around fivefold, but was not absent. The authors proposed that this was due to the particular Brca1-/-cell line chosen. The mutation in the Brca1 gene apparently does not produce a complete null allele, because an in-frame fusion product is formed from expression of an exon 10 12 splice variant [12].In this Brca1-/-cell line, Rad51 focus formation following DNA damage is partly, but not totally defective. This may explain the relative, but not complete loss of homologous recombination in these cells. Alternatively, the partial Brca1 product expressed might not have any role in homologous recombination, but other as yet unidentified Brca1-independent pathways that govern homologous recombination could be responsible for the residual recombination observed.
This work also raises other interesting questions. For example, an increase in random integration of transfected DNA was observed. As Moynahan et al [12] suggested, this could be a direct result of increased NHEJ, or simply due to the elevated level of spontaneous DSBs in the genome into which transfected DNA could insert. Alternatively, other undefined mutagenic mechanisms may be at work, because evidence is now gathering that multiple DNA repair pathways can potentially compete for the same repair substrate, in this case the DSB [27,28].
It has been suggested that Brca1 plays a crucial role in maintaining genetic stability. Like Brca2-deficient cells [13], Brca1-deficient murine [11] and human [29] cells have been reported to exhibit spontaneous chromosomal aberrations. This is akin to the gross chromosomal aberrations that are typical of chromosome instability disorders such as Bloom syndrome, Werner syndrome, Nijmegen breakage syndrome and ataxia-telangiectasia [30]. The mechanism for the maintenance of genetic stability is not known, but it has been suggested that genes that are mutated in the chromosome instability syndromes function as 'caretakers' [31] to prevent the occurrence of gross chromosomal rearrangements. Most of these genes participate in some aspect of DNA damage recognition and/or repair, and Brca1 has been shown to interact with at least two of them. For example, Brca1 associates with the hRad50-hMre11-p95/nibrin complex (nibrin is the gene mutated in Nigmegen breakage syndrome) in response to irradiation [32]. These nuclear foci are distinct from the Rad51 foci described earlier, suggesting Brca1 may participate in more than one damage response pathway. Recently, Cortez et al [33] provided evidence that the DNA damage sensor ATM (the gene mutated in ataxia-telengiectasia) was required for the phosphorylation of Brca1 in response to DSBs. A model is now emerging in which phosphorylation of Brca1 by ATM recruits or regulates multiple repair pathways that may involve the hRad50-hMre11-p95/nibrin complex, transcription coupled repair, or Rad51-mediated homologous recombination [34].
Homologous recombination may be an important mechanism for maintaining genetic stability. Evidence already exists that cells that harbour mutations in other genes in the Rad51-family, XRCC2 and XRCC3, are defective in homologous recombination [35,36] and exhibit spontaneous chromosome instability [37,38]. It is not yet known what pathways are responsible for generating the abnormal chromosomes in the absence of normal homologous recombination. Cells that are defective in NHEJ per se do not exhibit abnormal chromosomes [39]. In Brca1-deficient cells, Moynahan et al [12] believe that nonhomologous processes may promote aberrant types of repair. Interestingly, Brca2-deficient cells that exhibit gross chromosomal aberrations also have intact NHEJ [13]. Although this idea is very plausible, further studies are required to clarify this issue.
Finally, the correlation between chromosomal instability and cancer has long been known, but we are just beginning to unravel the mechanisms responsible for maintaining genetic stability. As the number of potential candidates for homologous recombination increases, the strategy devised by Moynahan et al [12] remains a useful screening tool. Although our understanding of homologous recombination in mammalian cells is still in its infancy, the enormous research interest in the field means that our understanding will move at a considerable speed. At this juncture, one can only say 'watch this space'-knowledge of the caretakers of the genome may one day change the manner in which we manage cancers.
References
- Rajan JV, Wang M, Marquis ST, Chodosh LA. Brca2 is coordinately regulated with Brca1 during proliferation and differentiation in mammary epithelial cells. Proc Natl Acad Sci USA. 1996;93:13078–13083. doi: 10.1073/pnas.93.23.13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MS, Verma IM. Transcriptional activation by BRCA1. Nature. 1996;382:678–679. doi: 10.1038/382678a0. [DOI] [PubMed] [Google Scholar]
- Anderson SF, Schlegel BP, Nakajima T, Wolpin ES, Parvin JD. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nature Genet. 1998;19:254–256. doi: 10.1038/930. [DOI] [PubMed] [Google Scholar]
- Milner J, Ponder B, Hughes-Davies L, Seltmann M, Kouzarides T. Transcriptional activation functions in BRCA2 [letter]. Nature. 1997;386:772–773. doi: 10.1038/386772a0. [DOI] [PubMed] [Google Scholar]
- Yarden RI, Brody LC. BRCA1 interacts with components of the histone deacetylase complex. Proc Natl Acad Sci USA. 1999;96:4983–4988. doi: 10.1073/pnas.96.9.4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Milner J, Kouzarides T. BRCA2 associates with acetyltransferase activity when bound to P/CAF. Oncogene. 1998;17:2531–2534. doi: 10.1038/sj/onc/1202475. [DOI] [PubMed] [Google Scholar]
- Xu XL, Weaver Z, Linke SP, et al. Centrosome amplication and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon11 isoform-deficient cells. Mol Cell. 1999;3:389–395. doi: 10.1016/s1097-2765(00)80466-9. [DOI] [PubMed] [Google Scholar]
- Tutt A, Gabriel A, Bertwistle D, et al. Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr Biol. 1999;9:1107–1110. doi: 10.1016/s0960-9822(99)80479-5. [DOI] [PubMed] [Google Scholar]
- Harkin DP, Bean JM, Miklos D, et al. Induction of GADD45 and JNK/SAPK-dependent apoptosis following inducible expression of BRCA1. Cell. 1999;97:575–586. doi: 10.1016/s0092-8674(00)80769-2. [DOI] [PubMed] [Google Scholar]
- Gowen LC, Avrutskaya AV, Latour AM, Koller BH, Leadon SA. BRCA1 required for transcription-coupled repair of oxidative DNA damage. Science. 1998;281:1009–1012. doi: 10.1126/science.281.5379.1009. [DOI] [PubMed] [Google Scholar]
- Shen SX, Weaver Z, Xu XL, et al. A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene. 1998;17:3115–3124. doi: 10.1038/sj/onc/1202243. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. . Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- Patel KJ, Yu VP, Lee H, et al. Involvement of Brca2 in DNA repair. . Mol Cell. 1998;1:347–357. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- Gowen LC, Johnson BL, Latour AM, Sulik KK, Koller BH. Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. . Nature Genet. 1996;12:191–194. doi: 10.1038/ng0296-191. [DOI] [PubMed] [Google Scholar]
- Liu CY, Flesken-Nikitin A, Li S, Zeng Y, Lee WH. Inactivation of the mouse Brca1 gene leads to failure in the morphogenesis of the egg cylinder in early postimplantation development. . Genes Dev. 1996;10:1835–1843. doi: 10.1101/gad.10.14.1835. [DOI] [PubMed] [Google Scholar]
- Hakem R, de la Pompa JL, Sirard C, et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. . Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- Sharan SK, Morimatsu M, Albrecht U, et al. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. . Nature. 1997;386:804–810. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A. Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. . Genes Dev. 1997;11:1226–1241. doi: 10.1101/gad.11.10.1226. [DOI] [PubMed] [Google Scholar]
- Wong AKC, Pero R, Ormonde PA, Tavtigian SV, Bartel PL. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. . J Biol Chem. 1997;272:31941–31944. doi: 10.1074/jbc.272.51.31941. [DOI] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. . Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Silver DP, Walpita D, et al. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. . Mol Cell. 1998;2:317–328. doi: 10.1016/s1097-2765(00)80276-2. [DOI] [PubMed] [Google Scholar]
- Scully R, Chen JJ, Ochs RL, et al. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. . Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- Liang F, Han M, Romanienko PJ, Jasin M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. . Proc Natl Acad Sci USA. 1998;95:5172–5177. doi: 10.1073/pnas.95.9.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchlow SE, Jackson SP. DNA end-joining: from yeast to man. . Trends Biochem Sci. 1998;23:394–398. doi: 10.1016/s0968-0004(98)01284-5. [DOI] [PubMed] [Google Scholar]
- Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. . Trends Genet. 1996;12:224–228. doi: 10.1016/0168-9525(96)10019-6. [DOI] [PubMed] [Google Scholar]
- Colleaux L, d'Auriol L, Betermier M, et al. Universal code equivalent of a yeast mitochondrial intron reading frame is expressed into E. coli as a specific double strand endonuclease. . Cell. 1986;44:521–533. doi: 10.1016/0092-8674(86)90262-x. [DOI] [PubMed] [Google Scholar]
- Chen C, Umezu K, Kolodner RD. Chromosomal rearrangements occur in S-cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. . Mol Cell. 1998;2:9–22. doi: 10.1016/s1097-2765(00)80109-4. [DOI] [PubMed] [Google Scholar]
- Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. . Nature Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- Tomlinson G, Chen TT, Stastny VA, et al. Characterization of a breast cancer cell line derived from a germ-line BRCA1 mutation carrier. . Cancer Res. 1998;58:3237–3242. [PubMed] [Google Scholar]
- Meyn MS. Chromosome instability syndromes: lessons for carcinogenesis. . Curr Top Microbiol Immunol. 1997;221:71–148. doi: 10.1007/978-3-642-60505-5_6. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers [news]. . Nature. 1997;386:761–763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- Zhong Q, Chen CF, Li S, et al. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. . Science. 1999;285:747–750. doi: 10.1126/science.285.5428.747. [DOI] [PubMed] [Google Scholar]
- Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM dependent phosphorylation of Brca1 in the DNA damage response to double-strand breaks. . Science. 1999;286:1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- Venkitaraman AR. Breast cancer genes and DNA repair. . Science. 1999;286:1100–1102. doi: 10.1126/science.286.5442.1100. [DOI] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. . Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RD, Jasin M. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. . Nature. 1999;401:397–399. doi: 10.1038/43935. [DOI] [PubMed] [Google Scholar]
- Jones NJ, Cox R, Thacker J. Isolation and cross-sensitivity of X-ray sensitive mutants of V79-4 hamster cells. . Mutat Res. 1987;183:279–286. doi: 10.1016/0167-8817(87)90011-3. [DOI] [PubMed] [Google Scholar]
- Liu N, Lamerdin JE, Tebbs RS, et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. . Mol Cell. 1998;1:783–793. doi: 10.1016/s1097-2765(00)80078-7. [DOI] [PubMed] [Google Scholar]
- Kemp LM, Jeggo PA. Radiation-induced chromosome damage in X-ray-sensitive mutants (xrs) of the Chinese hamster ovary cell line. . Mutat Res. 1986;166:255–263. doi: 10.1016/0167-8817(86)90025-8. [DOI] [PubMed] [Google Scholar]