

Abstract
The physiological properties of a hyd mutant of Desulfovibrio vulgaris Hildenborough, lacking periplasmic Fe-only hydrogenase, have been compared with those of the wild-type strain. Fe-only hydrogenase is the main hydrogenase of D. vulgaris Hildenborough, which also has periplasmic NiFe- and NiFeSe-hydrogenases. The hyd mutant grew less well than the wild-type strain in media with sulfate as the electron acceptor and H2 as the sole electron donor, especially at a high sulfate concentration. Although the hyd mutation had little effect on growth with lactate as the electron donor for sulfate reduction when H2 was also present, growth in lactate- and sulfate-containing media lacking H2 was less efficient. The hyd mutant produced, transiently, significant amounts of H2 under these conditions, which were eventually all used for sulfate reduction. The results do not confirm the essential role proposed elsewhere for Fe-only hydrogenase as a hydrogen-producing enzyme in lactate metabolism (W. A. M. van den Berg, W. M. A. M. van Dongen, and C. Veeger, J. Bacteriol. 173:3688–3694, 1991). This role is more likely played by a membrane-bound, cytoplasmic Ech-hydrogenase homolog, which is indicated by the D. vulgaris genome sequence. The physiological role of periplasmic Fe-only hydrogenase is hydrogen uptake, both when hydrogen is and when lactate is the electron donor for sulfate reduction.
Sulfate-reducing bacteria of the genus Desulfovibrio contain the genes for several hydrogenases, including the hynBA and hysBA genes for the NiFe- and NiFeSe-hydrogenases, the hydAB genes for Fe-only hydrogenase, and the hndABCD genes for NADP-reducing hydrogenase. The first three enzymes are translocated by the twin-arginine translocation (tat) system and are thus either periplasmic or membrane bound with the active site facing the periplasm. Only the NADP-reducing hydrogenase has been shown elsewhere to be cytoplasmic (11). Desulfovibrio vulgaris strain Hildenborough has the Fe-only hydrogenase as well as the NiFe- and NiFeSe-hydrogenases. The former is a soluble periplasmic enzyme, whereas the latter two are membrane bound. Searching of the database for the D. vulgaris genome at http://www.tigr.org indicated that D. vulgaris has an HndD homolog but lacks the hndA, hndB, and hndC genes. D. vulgaris is thus unlikely to have a cytoplasmic, NADP+-reducing hydrogenase.
The hydAB genes encode the 46-kDa α and the 10-kDa β subunit of Fe-only hydrogenase from D. vulgaris (25). The structures of Fe-only hydrogenase have been determined recently both for CpI, the cytoplasmic enzyme from Clostridium pasteurianum (16), and for the periplasmic enzyme from Desulfovibrio desulfuricans (14). The sequences of the α and β subunits of the periplasmic enzyme in D. desulfuricans and D. vulgaris form a contiguous, single polypeptide of 60 kDa in CpI. The splitting of the sequence into two polypeptides in Desulfovibrio spp. is for export: the β subunit has a long twin-arginine-type signal sequence for this purpose (24). The function of CpI in the fermentative metabolism of C. pasteurianum is to reoxidize reduced ferredoxin, using protons as the electron acceptor to produce H2 (2). A similar function has been proposed elsewhere for Fe-only hydrogenase in lactate metabolism by D. vulgaris (23). Reduction of the Fe-only hydrogenase content by expression of hydAB antisense RNA reduced the growth rate and growth yield of D. vulgaris in lactate- and sulfate-containing medium. Observation of a reduced H2 burst in the initial stages of growth on this medium also pointed to a decreased H2 production activity.
Desulfovibrio spp. are the only microorganisms with a periplasmic Fe-only hydrogenase. If the physiological function of this enzyme is indeed in H2 production, then the question arises why it is not simply cytoplasmic as in Clostridium spp. A cytoplasmic, H2-producing hydrogenase contributes to the transmembrane pH gradient, whereas a periplasmic, H2-producing hydrogenase consumes this gradient. Because Fe-only hydrogenase is the most abundant periplasmic hydrogenase in D. vulgaris (10), one expects it to also catalyze hydrogen uptake at least under certain metabolic conditions. We have now constructed a hydAB mutant with a modification of the sacB replacement mutagenesis method first used to mutate the dcrA gene of D. vulgaris (7). The physiological properties of the resulting hyd mutant are compared with those of the wild-type strain.
MATERIALS AND METHODS
Growth of bacteria.
All bacterial strains used are listed in Table 1. Tryptone-yeast extract medium was used for growth of Escherichia coli strains at 37°C (27). D. vulgaris strains were stored in Postgate’s medium B and grown in medium C and on solid medium E (17) at 32°C in a mixed-gas atmosphere (5% [vol/vol] H2, 10% CO2, and 85% N2) as described previously (7, 8). Antibiotics kanamycin (50 μg/ml) and chloramphenicol (CHL) (10 μg/ml) were added, when appropriate. Media B, C, and E contain 1 g of yeast extract/liter. D. vulgaris strains were also grown on defined BT media without yeast extract (4–6). Growth in these media was under conditions of exchange with the mixed-gas atmosphere of the anaerobic hood. BT media were made by combining, per 100 ml, 90 ml of minimal salt solution, 1.25 ml of trace element solution, 1 ml of CaCl2 solution, 1 ml of sodium lactate solution (for BT lactate-sulfate medium) or 1 ml of sodium acetate solution (for BT H2-sulfate medium; acetate and CO2 serve as the carbon source, and H2 serves as sole electron donor in this medium), 0.5 to 5 ml of 1 M Na2SO4, and 3 to 6 ml of water. All these were formulated as described before (6), except for the minimal salt solution. Because sulfate was a variable in the growth studies, it was removed from the minimal salt solution, which contained, per liter, 1.65 g of NH4Cl, 0.9 g of KH2PO4, and 0.36 g of MgCl2 · 6H2O at pH 7. Cultures of 100 ml were grown in 300-ml nephelometer (Erlenmeyer) flasks (Bellco Glass Inc., Vineland, N.J.) equipped with a 1.2- by 13-cm sidearm and closed with a sponge cap allowing gas exchange. Cell densities were measured with a Klett-Summerson colorimeter installed in the anaerobic chamber (Forma Scientific, Marietta, Ohio) or with a Shimadzu spectrophotometer at 600 nm. A cell density of 147 Klett units corresponded to an optical density at 600 nm (OD600) of 1, which in turn corresponded to a cell dry weight of 0.162 g/liter (6). BT medium plates were prepared by adding 15 g of agar per liter of medium.
TABLE 1.
Bacterial strains, primers, vectors and plasmids used
| Strain, primer, vector, or plasmid | Genotype, comment(s), and/or reference |
|---|---|
| Desulfovibrio vulgaris subsp. vulgaris Hildenborough | NCIMB 8303; isolated from clay soil near Hildenborough, United Kingdom (17) |
| D. vulgaris Hyd 3-2 | Plasmid pΔHydAB-CTB integrated into the chromosome through the downstream homologous regions; Sucs Cmr (this study) |
| D. vulgaris Hyd 3-7 | Plasmid pΔHydAB-CTB integrated into the chromosome through the upstream homologous regions; Sucs Cmr (this study) |
| D. vulgaris Hyd100 | ΔhydAB Sucr Cmr (this study) |
| E. coli TG2 | supE hsdΔ5 thi Δ(lac-proAB) Δ(srl-recA)306::Tn10(Tetr) F′ [traD36 proAB+lacIqlacZΔM15] (18) |
| E. coli S17-1 | thi pro hsdR hsdM+recA RP4-2 (Tc::Mu Km::Tn7) (20) |
| pUC19Cm | pUC19 containing the cat gene (7) |
| pNOT19 | Cloning vector pUC19; NdeI site replaced by a NotI site (19) |
| pMOB2 | Contains oriT of plasmid RP4 and Bacillus subtilis sacBR genes on a 4.5-kb NotI fragment; Kmr Cmr (19) |
| pHV15 | Contains the hydAB genes on a 4.7-kb SalI-EcoRI insert in pUC9 (26) |
| pHV15Not, pΔHydAB, pΔHydABcat, pΔHydAB-CTB | This study; see text |
| P173-r | 5′-ATGCATCTGCGTTGCCGGAG; 3827–3808a |
| P174-f | 5′-ATCACACGCGGGCACATGCT; 1751–1770a |
Position of primer in 4,678-bp HindIII-EcoRI insert of pHV15; the hydAB operon sequence (accession no. X02416 [25]) is located from nt 1904 to 3970 in this sequence. The deleted region extends from nt 1789 to 3796.
Growth in 500-ml closed serum bottles, containing 250 ml of headspace consisting of 90% (vol/vol) N2 and 10% (vol/vol) CO2, was in defined Widdel-Pfennig (WP) medium. Use of BT medium in this closed culture system led to formation of significant amounts of precipitated sulfides due to buildup of H2S, which interfered with the OD600 determination. WP media were formulated according to the work of Widdel and Bak (28) using 38 mM lactate and 28 mM sulfate.
Plasmid construction and replacement mutagenesis.
All plasmids and vectors used are listed in Table 1. The 4,678-bp HindIII-EcoRI insert from plasmid pHV15 was ligated to pNOT19, digested with HindIII and EcoRI, to give plasmid pHV15Not. pHV15Not was digested with SacII (nucleotide [nt] 1789) and MstII (nt 3796) to delete a 2-kb fragment containing the hydAB genes. The digested plasmid was end repaired with Klenow polymerase and deoxynucleoside triphosphates and ligated in the presence of BamHI linkers (5′-CCGGATCCGG) to give plasmid pΔHydAB. A 1.4-kb BamHI fragment from plasmid pUC19Cm, containing the cat gene, was ligated into the BamHI site of plasmid pΔHydAB to give plasmid pΔHydABcat. Plasmid pΔHydABcat was next digested with restriction endonuclease NotI and ligated with the 4.5-kb NotI fragment from pMOB2, containing the oriT locus and the sacBR genes, to give pΔHydAB-CTB. A map of this 11.2-kb plasmid is shown in Fig. 1.
FIG. 1.

Maps of the integration plasmid pΔHydAB-CTB and the DNA region containing the wild-type and mutated hydAB locus (WT and Hyd100, respectively). Positions of restriction sites for BamHI (B), EcoRI (E), HindIII (H), KpnI (K), MstII (M), NotI (N), PstI (P), SalI (S), and SacII (Sc) are shown. The 2-kb SacII-MstII fragment, containing the hydAB genes, in the wild-type strain was replaced by a 1.4-kb BamHI fragment containing the cat gene in D. vulgaris Hyd100.
E. coli S17-1(pΔHydAB-CTB) was conjugated with D. vulgaris, and single-crossover integrants were selected on medium E plates with CHL and kanamycin (7). Southern blot analysis was used to verify the integration of pΔHydAB-CTB into the D. vulgaris chromosome. A 2-kb upstream region probe (Fig. 1, up region) was prepared by digesting pHV15 with HindIII and SacII and agarose gel electrophoresis to isolate the 2-kb fragment and radioactive labeling with the random hexamer procedure using [α-32P]dCTP. The probe was hybridized with the blots under highly stringent conditions (18, 27).
Gene replacement was achieved by growing a mapped, single-crossover integrant, either D. vulgaris Hyd 3-2 or D. vulgaris Hyd 3-7, in 5 ml of medium C with CHL, followed by growth in 5 ml of BT H2-sulfate medium with CHL. Aliquots (50 to 100 μl) of the latter culture were then plated on BT H2-sulfate medium plates containing CHL and 5% (wt/vol) sucrose. Colonies, appearing after 3 to 4 weeks, were grown in 5 ml of medium C with CHL. DNA was isolated from these cultures and used to test their genotype by Southern blotting, yielding D. vulgaris Hyd100 for further study.
Analysis of the D. vulgaris Hyd100 phenotype.
For immunoblotting, the Hyd100 and wild-type strains were grown in 5 ml of medium C to stationary phase. Cells were pelleted and suspended in 250 μl of water. An equal volume of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) incubation buffer (8) was then added, and the samples were boiled immediately. Aliquots (30 μl; ca. 100 μg of protein) were loaded onto 12.5% (wt/vol) polyacrylamide gels together with molecular mass markers and a known amount of purified Fe-only hydrogenase. SDS-PAGE was performed according to the method of Laemmli (9). The separated proteins were electroblotted onto nitrocellulose (22). The blots were blocked with gelatin, incubated with a 2,000-fold dilution of anti-HydA antibodies for 4 h and then with a 2,000-fold dilution of the anti-rabbit, alkaline phosphatase-conjugated secondary antibody for 2 h, and then stained by incubation with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl-β-d-phosphopyranoside (BCIP).
Hydrogenase activity in the wild-type and Hyd100 strains was qualitatively evaluated by PAGE under nondenaturing conditions using the Phast System (Amersham Pharmacia Biotech). Following culturing of D. vulgaris Hyd100 and wild type in 100 ml of medium C to stationary phase, cells were pelleted and broken with a French press (590 MPa). After centrifugation for 20 min at 4,000 × g, a volume of supernatant containing 1 μg of total protein was loaded onto a PhastGel gradient gel of 8 to 25% polyacrylamide (pH 8.3). A 1-μl sample (2.5 μM) of pure Fe-only hydrogenase from D. vulgaris Hildenborough was also loaded. The gel was stained for hydrogenase activity according to the method of Ackrell et al. (1).
Measurement of hydrogenase activity by CV.
Enzyme activities were derived from enzyme-catalyzed electrochemical reactions (21) as measured by cyclic voltammetry (CV) An electrode modified with whole cells or cell fractions of D. vulgaris wild-type or Hyd100 strains produced a methyl viologen (MV)-mediated catalytic current (Ik) for evolution or consumption of H2. The catalytic scheme for H2 evolution can be summarized as follows:
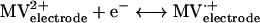 |
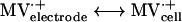 |
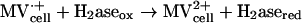 |
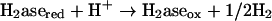 |
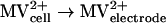 |
CV measurements were carried out using an EGG 263A potentiostat modulated by EGG PAR M270 software. Cyclic voltammograms were recorded at a sweep rate of 20 mV s−1 at room temperature. A three-electrode system consisting of a Metrohm Ag-AgCl-NaCl-saturated reference electrode, a platinum wire auxiliary electrode, and the glassy carbon membrane electrode (GCME) as the working electrode was used throughout. All potentials were relative to the Ag-AgCl-NaCl-saturated reference electrode. Potential versus the standard H2 electrode can be obtained by adding 0.210 V. For each H2 evolution measurement, the solution (100 mM acetate [pH 5.6], 0.1 mM MV2+) was flushed with high-purity nitrogen, whereas for each H2 uptake measurement the solution (100 mM Tris-HCl [pH 8.5], 0.1 mM MV2+) was flushed with H2 gas. Prior to use, the GCME (A = 0.07 cm2 from Tokay) was first polished with 0.05-μm alumina slurry. A 2-μl aliquot of either whole cells or cellular fraction suspensions of known protein concentration, prepared as described below, was then dropped onto the surface of the glassy carbon electrode. After the solvent had evaporated under N2 flow, the electrode was pressed against a square piece (1 by 1 cm) of dialysis membrane (Visking; molecular weight cutoff, 12,000 to 16,000; 30 μm thick). A rubber ring was fitted around the electrode body so that a uniform thin layer existed between the electrode surface and the membrane. The membrane electrode was then placed in the electrochemical cell, The dialysis membrane prevented loss of cells or protein molecules from the electrode surface, whereas small substrate and product molecules freely diffused through the membrane. The limiting current Ik was independent of the sweep rate, as expected for a catalytic process.
For sample preparation, all buffers were deaerated by flushing with argon. Cultures of D. vulgaris wild-type and Hyd100 strains, grown in 10 ml of medium C for 13 h, were centrifuged (5,700 × g, 15 min), and the cell pellets were resuspended in 1 ml of 0.1 M Tris-HCl-0.15 M NaCl (pH 7.6) and recentrifuged. The cell pellets were then resuspended in 1 ml of 0.01 M Tris-HCl, pH 7.6. For preparation of subcellular fractions, 50-ml cultures in medium C were centrifuged and the pellets were resuspended in 0.5 ml of 0.1 M Tris-HCl-0.1 M EDTA (pH 9.0) and incubated at 37°C for 30 min. After centrifugation (5,700 × g, 15 min, 4°C), periplasmic fractions were collected as the supernatants. These were concentrated and desalted by ultrafiltration on Centricon YM10 (Millipore) filters using 0.01 M Tris-HCl, pH 7.6. The pellets were resuspended in 4 ml of 0.01 M Tris-HCl, pH 7.6, and then passed twice through a French press unit at 590 MPa. After centrifugation (5,700 × g, 60 min, 4°C), the supernatant was centrifuged at 125,000 × g (12 h, 4°C). The cytoplasmic fractions were collected as the supernatants, and the membrane fractions were collected as the pellets from the latter centrifugation. The pellets were washed once by resuspension in 4 ml of 0.01 M Tris-HCl, pH 7.6, and centrifugation (125,000 × g, 12 h, 4°C). The membrane fractions were then resuspended in 500 μl of 0.01 M Tris-HCl buffer, pH 7.6. Purity of periplasmic and cytoplasmic fractions was verified by spectroscopic determination of the presence of cytochrome c3 and of desulfoviridin, respectively.
Analytical procedures.
H2 concentrations in the headspace of serum bottles containing cultures in WP media were analyzed by gas chromatography using a Shimadzu GC-8A gas chromatograph with a Molecular Sieve 5A column (80 to 100; 0.125 in. by 2 m; 32 ml of N2/min at 40°C) and an RGD2 reduction gas detector (Trace Analytical). Headspace samples of 1 ml were withdrawn and appropriately diluted in closed serum bottles filled with air. A 1-ml diluted sample was then withdrawn with a gastight syringe and injected into the gas chromatograph. H2 concentrations were calculated by comparing the peak area for the diluted sample with that of an H2 standard (20 ppm in helium).
Molecular biology reagents and (bio)chemicals.
DNA manipulation enzymes were obtained from Pharmacia or Roche Molecular Biochemicals. [α-32P]dCTP (3,000 Ci/mmol; 10 mCi/ml) was from ICN Biomedicals. Deoxyoligonucleotide primers P173-r and P174-f (Table 1) were obtained from University Core DNA Services of the University of Calgary. Rabbit antiserum generated against HydA purified from D. vulgaris was as described before (26). The chromogenic substrates nitroblue tetrazolium and BCIP, as well as anti-rabbit and anti-mouse immunoglobulin G-alkaline phosphatase conjugates, were from Promega. Nitrogen and mixed gas were from Praxair Canada. MV dichloride was purchased from Aldrich. All other chemicals were of reagent grade and used as received.
RESULTS
Construction of an hydAB deletion mutant.
Conjugation of E. coli S17-1(pΔHydAB-CTB) and D. vulgaris gave rise to single-crossover integrants in which the suicide plasmid recombined with the chromosome through either the upstream (Fig. 1, up region) or the downstream (Fig. 1, down region) homologous regions. Digestion of chromosomal DNA of these and of the wild type with EcoRI and hybridization of a Southern blot with the upstream region probe resulted in hybridizing fragments of 7.2 and/or 4.8 and/or 2.5 kb, as expected based on the maps shown in Fig. 1. It appeared impossible to achieve the second gene-replacing crossover by growing single-crossover integrants in medium C or medium E with CHL and sucrose. In view of this lack of success in rich media (both medium C and medium E contain 1 g of yeast extract per liter), we decided to attempt BT H2-sulfate medium. Growth in this medium is slow (μ = 0.050 h−1 [6]), and colonies emerged on solid BT H2-sulfate medium containing CHL and sucrose only after 3 to 4 weeks of incubation at 32°C. However, when these were subsequently cultured in liquid BT H2-sulfate medium with CHL or medium C with CHL a large fraction (50 to 100%) was found to have the required genotype both by Southern blot analysis and by PCR using primers P173-r and P174-f. One of these, D. vulgaris Hyd100, was selected for further study.
Immunoblotting and native PAGE.
Comparison of whole-cell extracts following SDS-PAGE and immunoblotting indicated the presence of the 46-kDa α subunit of Fe-only hydrogenase in D. vulgaris wild type (Fig. 2B, lane 2) and its absence in the Hyd100 strain (Fig. 2B, lane 3). Native gel electrophoresis also showed definitively that active Fe-only hydrogenase was present in the wild type but not in the Hyd100 strain. The membrane-bound NiFe- and NiFeSe-hydrogenases did not give an activity band under the conditions used. Some hydrogenase activity remained in the wells (Fig. 2C). In order to accurately evaluate the remaining hydrogenase activity in Hyd100 compared to that in wild-type cells, H2 uptake and evolution were quantitated in both whole cells and subcellular fractions using an electrochemistry approach.
FIG. 2.

Electrophoretic comparison of D. vulgaris wild-type and Hyd100 strains. (A) Gel stained with Coomassie blue following SDS-PAGE. Lane M, molecular mass markers; from top to bottom, 94, 67, 43, and 30 kDa; lane 1, 1 μg of purified Fe-only hydrogenase; lane 2, D. vulgaris wild type, 50 μg of protein; lane 3, D. vulgaris Hyd100, 50 μg of protein. (B) Immunoblot of gel as in panel A incubated with a polyclonal antiserum specific for the 46-kDa α subunit of Fe-only hydrogenase. (C) Native gel stained for hydrogenase activity. Lane 2, cell extract from D. vulgaris wild type, 4 μg of protein; lane 3, cell extract from D. vulgaris Hyd100, 4 μg of protein.
H2 uptake and production activity by bioelectroanalysis.
CV curves for measurement of H2 production and consumption activities are shown in Fig. 3A and B. Figure 3C indicates a reversible electrochemical wave with a midpoint potential of −630 mV, representing the MV2+-MV·+ electrochemical couple. This signal turned into an S-shaped pH-dependent wave when using the D. vulgaris cell-modified GCME (Fig. 3A and B). The value of the limiting current Ik is directly proportional to the enzyme activity in the linear part of the curve of Ik versus protein concentration. This is illustrated in Fig. 3D for whole cells of the D. vulgaris wild-type and Hyd100 strains. It appeared that Ik first linearly increased with protein concentration and then leveled off as protein concentration was increased further. Estimates of the specific hydrogenase activity (relative units) were obtained by dividing the limiting current Ik by the current obtained at the GCME with MV2+ alone and by the protein concentration. The results are summarized in Table 2, which shows that the specific hydrogenase activity of the Hyd100 strain is about 50% less than that of the wild-type strain in both H2 uptake and H2 evolution reactions. A large difference was observed in the periplasmic fraction, which is in agreement with the deletion of the gene encoding periplasmic Fe-only hydrogenase in the Hyd100 strain. On the other hand, no significant changes were observed in either the cytoplasmic or the membrane fractions (Table 2).
FIG. 3.

Determination of hydrogenase activities with CV. (A to C) Cyclic voltammograms obtained with a GCME. (A) D. vulgaris cell-modified GCME (1 μg of protein) in N2-saturated 0.1 M Na-acetate buffer, pH 5.6, with 100 μM MV2+; (B) D. vulgaris cell-modified GCME (1 μg of protein in H2-saturated 0.1 M Tris-HCl buffer [pH 8.5] with 100 μM MV2+); (C) bare GCME under same conditions as in panel A. (D) Limiting catalytic current Ik plotted against protein concentration (milligrams per milliliter) for experiments with cell-modified GCME under the same conditions as for panel A. Data are for whole cells of D. vulgaris wild-type (closed circles) and Hyd100 (open circles) strains.
TABLE 2.
Specific hydrogenase activities derived from CV experiments on whole cells or subcellular fractions prepared from D. vulgaris wild-type and Hyd100 strains
| Cell or fraction | Hydrogenase activity (relative units)/protein concn (mg/mL) for strain
|
|||
|---|---|---|---|---|
| Uptake
|
Production
|
|||
| Wild type | Hyd100 | Wild type | Hyd100 | |
| Whole cells | 47 | 24 | 33 | 16 |
| Periplasm | 36 | NDa | 13 | 3 |
| Membrane | 1 | 0.5 | 8 | 11 |
| Cytoplasm | ND | ND | 6 | 6 |
ND, not detected.
Comparison of growth yields of the wild-type and Hyd100 strains in BT media.
Growth of D. vulgaris Hyd100 colonies on solid BT H2-sulfate medium already indicated that Fe-only hydrogenase is not critically important for H2 uptake. Colonies of the wild-type and Hyd100 strains also grew similarly on medium E plates, in which lactate serves as the electron donor for sulfate reduction. The Hyd100 strain is thus capable of using both lactate and H2 as the electron donor for sulfate reduction.
Growth rates and yields were compared quantitatively in BT media in which either H2 or lactate and H2 served as the electron donor for sulfate reduction. H2 was in principle never limiting, because growth was in an atmosphere containing 5% (vol/vol) H2 under conditions of gas exchange. These culture conditions allow a specific growth rate (μ) of 0.050 h−1 and a molar growth yield, Ysulfate, of 5.1 g of cells mol−1 with H2 (6), similar to values observed by Badziong and Thauer (3) for the same medium. Growth studies were carried out at different sulfate concentrations (5 to 50 mM). Growth rates of D. vulgaris wild-type and Hyd100 strains were similar. However, differences in final cell densities were found (Fig. 4). Final cell densities were constant (Fig. 4A) or tended to display a maximum (Fig. 4B). The average of four determined values at or near the maximum (Fig. 4B; 70 to 100 h) was taken as the final cell density in these cultures. Comparison of final cell densities as a function of sulfate concentration indicated that the hyd mutant grew less well than did the wild-type strain when only H2 was available, especially at a high sulfate concentration (Fig. 5A). Statistical analysis of the data indicated that the ratio (R) of final cell densities of the wild-type and Hyd100 strains was R = (1.10 ± 0.16) for sulfate concentrations from 5 to 20 mM and R = (1.30 ± 0.15) for sulfate concentrations from 25 to 50 mM. The limiting slope at low sulfate concentration was the same for both strains and indicated a Ysulfate of 4.4 g of cells mol−1 for H2 as electron donor, in reasonable agreement with previously reported values. With lactate (38 mM) and hydrogen as the electron donor, no differences in final cell density between the two strains were found at sulfate concentrations of 0 to 15 mM [R = (0.98 ± 0.10)]. The final cell density increased linearly with sulfate concentration in this range (Fig. 5B). Above 15 mM sulfate, this increase ceased abruptly, and final cell densities decreased with increasing sulfate concentration. Because the transition point is close to 19 mM sulfate, where the culture goes from sulfate to lactate limitation, one can conclude that lactate serves as the main electron donor in the cultures with 5 to 15 mM sulfate. The data in this range indicate a Ysulfate of 6.2 g of cells mol−1 for lactate as the electron donor. Above 19 mM sulfate, the wild-type strain grew to a higher final cell density than did the Hyd100 strain [Fig. 5B, R = (1.13 ± 0.08)]. The reasons why final cell densities decrease in cultures with increasing sulfate concentration in the range 20 to 50 mM are not clear.
FIG. 4.

Growth of D. vulgaris wild-type (□) and Hyd100 (◊) strains in BT medium. (A) H2 as sole electron donor for sulfate reduction; 40 mM sulfate. (B) Lactate (38 mM) and H2 as electron donor for sulfate reduction; 30 mM sulfate.
FIG. 5.

Final cell density (OD600) for cultures of D. vulgaris wild-type (□) and Hyd100 (◊) strains in BT medium as a function of the sulfate concentration. All data points are for single cultures, except where the vertical bars indicate the spread of data obtained for duplicate cultures. Cultures were with H2 only (A) or with lactate (38 mM) and H2 as the electron donor for sulfate reduction (B). Lactate becomes limiting at sulfate concentrations above 19 mM (↓). Final cell densities for wild type were (13 ± 8)% larger than for Hyd100 cells at 25 to 50 mM sulfate. The downward-sloping lines drawn are best fits to the data in this range of sulfate concentrations and reflect this difference in cell density.
Comparison of growth and hydrogen production in a closed culture system.
Cultures of the wild-type and Hyd100 strains in 500-ml serum bottles containing 250 ml of WP medium (38 mM lactate and 28 mM sulfate) and 250 ml of a 90% (vol/vol) N2 and 10% (vol/vol) CO2 headspace were compared. The Hyd100 strain grew to a significantly lower cell density than did the wild type (Fig. 6A) while transiently producing significantly larger amounts of hydrogen (Fig. 6B). The levels of hydrogen indicated in Fig. 6B were reproducible in two other experiments (data not shown). The fact that the absence of Fe-only hydrogenase leads to increased headspace hydrogen concentrations points to a role of this enzyme in hydrogen consumption, not hydrogen production during lactate metabolism.
FIG. 6.

Growth of wild-type and hyd mutant strains in WP medium containing lactate (38 mM), sulfate (28 mM), and a headspace of 10% (vol/vol) CO2 and 90% N2. Plotted as a function of time (hours) are cell density (OD600) (A) and H2 concentration in the headspace (parts per million) (B).
DISCUSSION
The genes for Fe-only hydrogenase were the first to be cloned and sequenced for D. vulgaris (25, 26). However, the isolation of a deletion mutant proved to be problematic. van den Berg et al. (23) were unsuccessful in their attempts to construct a mutant and resorted to antisense RNA expression technology to reduce hydAB gene expression. Following transfer of pWB1151, a broad-host-range plasmid that constitutively expresses hydAB antisense mRNA, a two- to threefold-reduced content of Fe-only hydrogenase of D. vulgaris was found. This decreased both growth rates and growth yields when lactate (70 mM) was the electron donor for sulfate (53 mM) reduction. The strain with reduced Fe-only hydrogenase content showed a reduced H2 concentration in the headspace compared to that for the wild type. This H2 was transient and was eventually all used for sulfate reduction. The effects of reducing Fe-only hydrogenase content on growth with H2 as electron donor were not reported by these authors.
We have reduced expression of Fe-only hydrogenase to zero (Fig. 2), but we cannot confirm the phenotype suggested by van den Berg et al. (23). With lactate as the electron donor and H2 in the headspace, there was no difference in growth yield between the Hyd100 and wild-type strains when sulfate was limiting (Fig. 5B; 5 to 20 mM). Because lactate (38 mM) was in excess (two lactate molecules reduce one sulfate molecule), it served as the main electron donor for sulfate reduction under these conditions. At sulfate concentrations in excess of available lactate, the wild-type strain reached a higher final cell density than did the Hyd100 strain (Fig. 5B, 20 to 50 mM), possibly indicating more efficient H2 metabolism. Indeed, when H2 was the sole electron donor for sulfate reduction, larger differences in final cell density were seen, especially at high sulfate concentrations (Fig. 5A). When strains were cultured on lactate- and sulfate-containing media, in the absence of H2 (Fig. 6) the hyd mutant was found to generate more, not less, H2 than that generated by the wild-type strain. Therefore, we cannot support the conclusion (23) that Fe-only hydrogenase has an important role in H2 production from lactate when sulfate is in excess. Instead, the data in Fig. 5 and 6 suggest a role of Fe-only hydrogenase in H2 uptake under these conditions. It should be pointed out that the culture conditions are not completely comparable, as van den Berg et al. (23) used rich medium, containing 1 g of yeast extract per liter, whereas we used defined medium in which lactate was the only organic molecule present. The strains used by van den Berg et al. and in this study are identical.
Reduced efficiency of growth on H2 has also been found by Malki et al. (12), who described the effects of single and double mutations in hydrogenase genes of Desulfovibrio fructosovorans. This organism has hyn, hyd, and hnd genes, encoding a periplasmic NiFe-, a periplasmic Fe-only, and a cytoplasmic NADP+-reducing hydrogenase, respectively. The latter is absent from D. vulgaris. Strains with deletions in either the hnd or the hyn gene, as well as a strain with deletions in both the hyn and hnd genes, were still able to grow on H2 as the sole electron donor for sulfate reduction, which was credited to the presence of the remaining Fe-only hydrogenase. The double mutant also had reduced growth efficiency when lactate or pyruvate was used as the electron donor for sulfate reduction.
The data in Fig. 6B indicate that hydrogen production and consumption are an important feature of the use of lactate as the electron donor for sulfate reduction. However, the hydrogen-producing hydrogenase is not the periplasmic Fe-only hydrogenase, as we have shown. A good candidate is a homolog of the Ech-hydrogenase from Methanosarcina barkeri, which appears to be present in the D. vulgaris genome. This enzyme consists of six subunits (EchA to EchF), which together form a membrane-bound hydrogenase of which the active site faces the cytoplasm. The enzyme from M. barkeri has been purified previously as a six-subunit complex following its solubilization from membranes (13). The function ascribed to Ech-hydrogenase, based on biochemical studies of the purified enzyme, is to reoxidize reduced ferredoxin (Fdred) formed during oxidative conversion of acetyl coenzyme A to methyl-tetrahydrosarcinapterin (CH3-H4SPT) and CO2. Ech-hydrogenase reoxidizes Fdred and uses the electrons together with cytoplasmic protons for hydrogen production. The produced hydrogen may then be captured by an externally located NiFe-hydrogenase of M. barkeri, with the electrons being used for cytoplasmic reduction reactions. This hydrogen cycling mechanism contributes to the proton gradient formed across the membrane by M. barkeri, when it metabolizes acetate to methane.
An H2 cycling mechanism was first proposed for Desulfovibrio spp. by Odom and Peck (15). However, a cytoplasmic hydrogenase has so far not been identified. The Ech-hydrogenase homolog of D. vulgaris is encoded by an operon with the same gene order as that in the ech operon from M. barkeri. All subunits have the corresponding sequences from M. barkeri as their closest homologs. Thus, D. vulgaris is likely to have a very similar cytoplasmic, energy-conserving Ech-hydrogenase. Fdred is formed during lactate metabolism by D. vulgaris in the oxidative conversion of pyruvate to acetyl coenzyme A and CO2, catalyzed by pyruvate-ferredoxin oxidoreductase. Fdred may be reoxidized by the Ech-hydrogenase homolog, with the H2 formed by this enzyme diffusing across the membrane to be captured by the periplasmic Fe-only and nickel-containing hydrogenases. The absence of the main periplasmic hydrogenase results in less efficient hydrogen capture and thus in higher hydrogen headspace concentrations. These are transient due to the presence of excess sulfate under the conditions of the experiment shown in Fig. 6.
Why is H2 production and subsequent capture by the Hyd100 mutant bioenergetically less efficient than the proposed H2 cycling catalyzed by the wild-type strain? One factor to consider is that thermodynamically the energy content of H2 per mole is proportional to the logarithm of its concentration. During lactate metabolism, a high H2 concentration may be maintained in the periplasm. Yet, little escapes due to the high periplasmic hydrogenase activity of wild-type D. vulgaris (Fig. 6B). In the case of the hyd mutant, H2 escaping into the headspace is considerably diluted. Although this headspace H2 is used for sulfate reduction, its availability at a reduced concentration leads to less energy conservation. This could be one of the factors contributing to the large difference in cell densities seen in Fig. 6A and could also explain why the presence of 5% (vol/vol) H2 in the headspace reduces this difference (Fig. 5B). Another factor that may contribute to the larger difference in final cell densities between the cultures in Fig. 6A than between those in Fig. 5B is the buildup of H2S, but this remains to be investigated.
Acknowledgments
This work was supported by a Research Grant from the Natural Science and Engineering Research Council of Canada to G. Voordouw, who also acknowledges the support of a fellowship from the Hanse Wissenschaftskolleg in Bremen, Germany, and of a visiting professorship of the CNRS in Marseille, France. A. Dolla was supported by a NATO postdoctoral fellowship during part of this work. A database of the D. vulgaris Hildenborough genome was searched at the website of The Institute for Genomic Research at http://www.tigr.org. Sequencing of this genome is financially supported by the U.S. Department of Energy.
We thank Marie-Claire Durand, Mireille Bruschi, Daniela Lange, Ramona Appel, Christina Probian, Ralf Rabus, and Fritz Widdel for discussions and technical assistance. We thank Reiner Hedderich for drawing our attention to the presence of Ech-hydrogenase in the D. vulgaris genome.
REFERENCES
- 1.Ackrell, B. A. C., R. N. Asato, and H. F. Mower. 1966. Multiple forms of bacterial hydrogenases. J. Bacteriol. 92:828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams, M. W. W., and E. I. Stiefel. 1998. Biological hydrogen production: not so elementary. Science 282:1842–1843. [DOI] [PubMed] [Google Scholar]
- 3.Badziong, W., and R. K. Thauer. 1978. Growth yields and growth rates of Desulfovibrio vulgaris (Marburg) growing on hydrogen plus sulfate and hydrogen plus thiosulfate as the sole energy sources. Arch. Microbiol. 117:209–214. [DOI] [PubMed] [Google Scholar]
- 4.Badziong, W., B. Ditter, and R. K. Thauer. 1979. Acetate and carbon dioxide assimilation by Desulfovibrio vulgaris (Marburg), growing on hydrogen and sulfate as sole energy source. Arch. Microbiol. 123:301–305. [Google Scholar]
- 5.Brandis, A., and R. K. Thauer. 1981. Growth of Desulfovibrio species on hydrogen and sulphate as sole energy source. J. Gen. Microbiol. 126:249–252. [Google Scholar]
- 6.Dolla, A., B. K. J. Pohorelic, J. K. Voordouw, and G. Voordouw. 2000. Deletion of the hmc-operon of Desulfovibrio vulgaris subsp. vulgaris Hildenborough hampers hydrogen metabolism and low-redox potential niche establishment. Arch. Microbiol. 174:143–151. [DOI] [PubMed] [Google Scholar]
- 7.Fu, R., and G. Voordouw. 1997. Targeted gene-replacement mutagenesis of dcrA encoding an oxygen sensor of the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Microbiology 143:1815–1826. [DOI] [PubMed] [Google Scholar]
- 8.Keon, R. G., R. Fu, and G. Voordouw. 1997. Deletion of two downstream genes alters expression of the hmc operon of Desulfovibrio vulgaris subsp. vulgaris Hildenborough. Arch. Microbiol. 167:376–383. [DOI] [PubMed] [Google Scholar]
- 9.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. [DOI] [PubMed] [Google Scholar]
- 10.Lissolo, T., E. S. Choi, J. LeGall, and H. D. Peck, Jr. 1986. The presence of multiple intrinsic nickel-containing hydrogenases in Desulfovibrio vulgaris Hildenborough. Biochem. Biophys. Res. Commun. 139:701–708. [DOI] [PubMed] [Google Scholar]
- 11.Malki, S., I. Saimmaime, G. De Luca, M. Rousset, Z. Dermoun, and J.-P. Belaich. 1995. Characterization of an operon encoding an NADP-reducing hydrogenase in Desulfovibrio fructosovorans. J. Bacteriol. 177:2628–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malki, S., G. De Luca, M. L. Fardeau, M. Rousset, J.-P. Belaich, and Z. Dermoun. 1997. Physiological characteristics and growth behavior of single and double hydrogenase mutants of Desulfovibrio fructosovorans. Arch. Microbiol. 167:38–45. [DOI] [PubMed] [Google Scholar]
- 13.Meuer, J., S. Bartoschek, J. Koch, A. Künkel, and R. Hedderich. 1999. Purification and catalytic properties of Ech hydrogenase from Methanosarcina barkeri. Eur. J. Biochem. 265:325–335. [DOI] [PubMed] [Google Scholar]
- 14.Nicolet, Y., C. Piras, P. Legrand, C. E. Hatchikian, and J. C. Fontecilla-Camps. 1999. Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center. Structure 7:13–23. [DOI] [PubMed] [Google Scholar]
- 15.Odom, J. M., and H. D. Peck, Jr. 1981. Hydrogen cycling as a general mechanism for energy coupling in the sulfate-reducing bacteria Desulfovibrio sp. FEMS Microbiol. Lett. 12:47–50. [Google Scholar]
- 16.Peters, J. W., W. N. Lanzilotta, B. J. Lemon, and L. C. Seefeldt. 1998. X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 Angstrom resolution. Science 282:1853–1858. [DOI] [PubMed] [Google Scholar]
- 17.Postgate, J. R. 1984. The sulphate-reducing bacteria, 2nd ed. Cambridge University Press, Cambridge, United Kingdom.
- 18.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 19.Schweizer, H. P. 1992. Allelic exchange in Pseudomonas aeruginosa using novel ColE1-type vectors and a family of cassettes containing a portable oriT and the counter-selectable Bacillus subtilis sacB marker. Mol. Microbiol. 6:1195–1204. [DOI] [PubMed] [Google Scholar]
- 20.Simon, R., U. Priefer, and A. Pühler. 1983. A broad-host-range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Bio/Technology 1:784–791. [Google Scholar]
- 21.Tatsumi, H., K. Takagi, M. Fujita, K. Kano, and T. Ikeda. 1999. Electrochemical study of reversible hydrogenase reaction of Desulfovibrio vulgaris cells with methyl viologen as an electron carrier. Anal. Chem. 71:1753–1759. [DOI] [PubMed] [Google Scholar]
- 22.Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van den Berg, W. A. M., W. M. A. M. van Dongen, and C. Veeger. 1991. Reduction of the amount of periplasmic hydrogenase in Desulfovibrio vulgaris (Hildenborough) with antisense RNA: direct evidence for an important role of this hydrogenase in lactate metabolism. J. Bacteriol. 173:3688–3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voordouw, G. 2000. A universal system for the transport of redox proteins: early roots and latest developments. Biophys. Chem. 86:131–140. [DOI] [PubMed] [Google Scholar]
- 25.Voordouw, G., and S. Brenner. 1985. Nucleotide sequence of the gene encoding the hydrogenase from Desulfovibrio vulgaris (Hildenborough). Eur. J. Biochem. 148:515–520. [DOI] [PubMed] [Google Scholar]
- 26.Voordouw, G., J. E. Walker, and S. Brenner. 1985. Cloning of the gene encoding the hydrogenase from Desulfovibrio vulgaris (Hildenborough) and determination of the NH2-terminal sequence. Eur. J. Biochem. 148:509–514. [DOI] [PubMed] [Google Scholar]
- 27.Voordouw, G., J. D. Strang, and F. R. Wilson. 1989. Organization of genes encoding [Fe] hydrogenase in Desulfovibrio vulgaris subsp. oxamicus Monticello. J. Bacteriol. 171:3881–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Widdel, F., and F. Bak. 1992. Gram-negative mesophilic sulfate-reducing bacteria, p.3352–3378. In A. Balows, H. G. Truper, M. Dworkin, W. Harder, and K. H. Schleifer (ed.), The prokaryotes, 2nd ed., vol. 4. Springer-Verlag, New York, N.Y.