

Abstract
FtsQ, a 276-amino-acid, bitopic membrane protein, is one of the nine proteins known to be essential for cell division in gram-negative bacterium Escherichia coli. To define residues in FtsQ critical for function, we performed random mutagenesis on the ftsQ gene and identified four alleles (ftsQ2, ftsQ6, ftsQ15, and ftsQ65) that fail to complement the ftsQ1(Ts) mutation at the restrictive temperature. Two of the mutant proteins, FtsQ6 and FtsQ15, are functional at lower temperatures but are unable to localize to the division site unless wild-type FtsQ is depleted, suggesting that they compete poorly with the wild-type protein for septal targeting. The other two mutants, FtsQ2 and FtsQ65, are nonfunctional at all temperatures tested and have dominant-negative effects when expressed in an ftsQ1(Ts) strain at the permissive temperature. FtsQ2 and FtsQ65 localize to the division site in the presence or absence of endogenous FtsQ, but they cannot recruit downstream cell division proteins, such as FtsL, to the septum. These results suggest that FtsQ2 and FtsQ65 compete efficiently for septal targeting but fail to promote the further assembly of the cell division machinery. Thus, we have separated the localization ability of FtsQ from its other functions, including recruitment of downstream cell division proteins, and are beginning to define regions of the protein responsible for these distinct capabilities.
While many fundamental cellular processes in bacteria, such as respiration, transcription, and translation, have clearly defined players and game plans, the team of molecular factors that carries out cytokinesis remains poorly characterized. In gram-negative bacterium Escherichia coli, at least nine genes (ftsA, ftsI, ftsK, ftsL, ftsN, ftsQ, ftsW, ftsZ, and zipA) are involved in constricting all three layers of the cell envelope at the septum and separating the two daughter cells (reviewed in references 26 and 33). Strains containing mutations in any one of these genes fail to divide but continue to elongate, forming filamentous cells. In wild-type cells, all nine cell division proteins appear to be recruited in hierarchical order into a septal ring structure at the midcell region (26, 33). Presumably, the septal ring is a multimeric protein complex that provides constrictive forces and synthesizes cell envelope components. Although the biochemical activities of certain cell division proteins are known, the detailed mechanisms by which these factors cooperatively promote septation are unclear.
FtsQ is an intermediate recruit to the septal ring with unknown function (10). It is a bitopic membrane protein with a short (24-amino-acid) cytoplasmic amino terminus, a single (25-amino-acid) transmembrane segment, and a relatively large (227-amino-acid) periplasmic domain (7, 20). This topology is shared by several other cell division proteins, such as DivIB, the FtsQ homolog in Bacillus subtilis (21, 23), and FtsN, FtsL, and FtsI in E. coli (3, 13, 18, 20). Since FtsI, also known as PBP3, is a transpeptidase specifically required for peptidoglycan synthesis at the septum (1, 22; reviewed in reference 30), FtsQ may play a role in cell wall synthesis as well. First, homologs of FtsQ are not found in bacterial species without a cell wall (31, 39). Second, FtsQ has low sequence identity (17%) to Mpl, an enzyme involved in the recycling of peptidoglycan for cell wall synthesis (5, 27). Finally, Katis and Wake (23) suggested that the greater abundance of DivIB in B. subtilis (5,000 molecules per cell [34]) than of FtsQ in E. coli (less than 50 molecules per cell [7]) might reflect the need of the bacterium to construct a thicker cell wall. However, there is no empirical evidence to implicate FtsQ in cell wall synthesis.
Genetic and cytological studies indicate that FtsQ functionally interacts with other cell division proteins. Overproduction of FtsQ causes filamentation of ftsA, ftsI, or ftsZ thermosensitive (Ts) mutants at the permissive temperature (11), and overexpression of ftsN partially suppresses the ftsQ1(Ts) mutation (12). Septal localization of FtsQ depends on the presence of FtsZ, FtsA, and FtsK but not FtsL, FtsW, FtsI, and FtsN (9, 10). Rather, FtsL, FtsW, FtsI, and FtsN depend on FtsQ for their recruitment into the septal ring (2, 17, 40; K. Mercer and D. Weiss, personal communication). Nevertheless, no evidence for direct physical interaction of FtsQ with other cell division proteins exists, despite various attempts to detect such an interaction.
To investigate the role of FtsQ in cell division and its interaction with other proteins, we analyzed mutant alleles of ftsQ. In a genetic screen for loss-of-function mutations in ftsQ, we identified four alleles that failed to complement the ftsQ1(Ts) mutation. We determined whether these mutant alleles complement a null mutation in ftsQ and whether the proteins that they encode localize to the division site. Such analyses suggest that mutant FtsQ proteins compete with wild-type FtsQ for sequestration to a limited number of target sites in the septal ring. In addition, our results show that, while the presence of residues 50 to 247 of FtsQ is sufficient for this sequestration process, the last 29 amino acids of the protein are necessary for function. One activity of FtsQ that requires this carboxy-terminal region is the recruitment of downstream cell division proteins, such as FtsL.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
Bacterial strains and plasmids used in this study are listed in Table 1. Rich media used were NZY liquid broth and agar, which differ from Luria broth (28) in that tryptone is replaced by NZ amine A (Quest International) and in that the salt concentration is 8 g of NaCl per liter. Concentrations of antibiotics used in rich media were as follows: ampicillin, 200 (for bla on plasmid) or 25 (for bla on chromosome) μg/ml; chloramphenicol, 10 μg/ml; kanamycin, 40 μg/ml; spectinomycin, 50 (in liquid) or 100 (in plate) μg/ml; tetracycline, 15 μg/ml. d-Glucose or l-arabinose was added at a final concentration of 0.2% to repress or induce, respectively, the expression of genes under the control of the PBAD promoter (19).
TABLE 1.
Strains and plasmids
| Strain or plasmid | Relevant genetic marker(s) or features | Construction, source, or referencea |
|---|---|---|
| Strains | ||
| KS272 | F− ΔlacX74 galE galK thi rpsL ΔphoA(PvuII) | 37 |
| KS474 | KS272 degP41 (ΔPstI-Kanr) | 38 |
| MC4100 | F−araD139 ΔlacU169 relA1 rpsL150 thi mot flb5301 deoC7 ptsF25 rbsR | Laboratory collection |
| DH5αZ1 | DH5α attλ::(addA lacIqtetR) | 25 |
| DHB6521 | SM551 λInCh1 (Kanr) | 4 |
| JCB495 | MC1000 recD | 32 |
| JOE309 | MC4100 araD+ | 8 |
| JOE310 | MC4100 ΔaraBADAH33 | 8 |
| JOE326 | MC4100 ΔaraBADAH33attλ::(aadA lacIqtetR) | P1 (DH5αZ1) × JOE310, select Spcr |
| ftsQ1(Ts) strains | ||
| EC433 | MG1655 leu::Tn10 ftsQ1(Ts) | 10 |
| MJC129 | KS272 ftsQ1(Ts) recA::cat | 20 |
| JOE32 | MJC129/pLMG161 | Transform MJC129 with pLMG161 |
| JOE242 | MJC129/pMM2 | Transform MJC129 with pMM2 |
| JOE243 | MJC129/pMM6 | Transform MJC129 with pMM6 |
| JOE245 | MJC129/pMM15 | Transform MJC129 with pMM15 |
| JOE249 | MJC129/pMM65 | Transform MJC129 with pMM65 |
| JOE693 | MJC129/pBAD18 | Transform MJC129 with pBAD18 |
| JOE333 | JOE326 leu::Tn10 ftsQ1(Ts) | P1 (EC433) × JOE326, select Tetr |
| JOE714 | JOE333/pBAD18 | Transform JOE333 with pBAD18 |
| JOE715 | JOE333/pLMG161 | Transform JOE333 with pLMG161 |
| JOE716 | JOE333/pMM2 | Transform JOE333 with pMM2 |
| JOE719 | JOE333/pLD104 | Transform JOE333 with pLD104 |
| JOE720 | JOE333/pLD92 | Transform JOE333 with pLD92 |
| gfp fusion strainsb | ||
| EC439 | MC4100 Δ(λattL-lom)::bla lacIqP209-gfp-ftsL | Integrate pDSW237 via λInCh1 |
| EC442 | MC4100 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ | 10 |
| EC447 | MC4100 Δ(λattL-lom)::bla lacIqP210-ftsA-gfp | 40 |
| EC452 | MC4100 Δ(λatt-lom)::bla lacIqP207-gfp | 40 |
| JOE196 | MC4100 Δ(λattL-lom)::bla lacIqP207-gfp-FFQ | 10 |
| JOE257 | MC4100 Δ(λattL-lom)::bla lacIqP207-gfp-QQL | 10 |
| JOE299 | MC4100 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ2 | Integrate pJC19 via λInCh1 |
| JOE301 | MC4100 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ6 | Integrate pJC20 via λInCh1 |
| JOE303 | MC4100 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ15 | Integrate pJC21 via λInCh1 |
| JOE305 | MC4100 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ65 | Integrate pJC22 via λInCh1 |
| FtsQ depletion strains | ||
| JOE260 | JCB495 ftsQE14::kan/pLMG161 | See Materials and Methods |
| JOE417 | MC4100 araD+ftsQE14::kan/pJC10 | P1 (JOE424) × JOE309/pJC10, select Kanr |
| JOE424 | KS272 ftsQE14::kan/pJC10 | P1 (JOE260) × KS272/pJC10, select Kanr, screen Ara+ |
| JOE428 | JOE424 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ | P1 (EC442) × JOE424, select low Ampr |
| JOE429 | JOE424 Δ(λattL-lom)::bla lacIqP207-gfp | P1 (EC452) × JOE424, select low Ampr |
| JOE430 | JOE424 Δ(λattL-lom)::bla lacIqP207-gfp-FFQ | P1 (JOE196) × JOE424, select low Ampr |
| JOE431 | JOE424 Δ(λattL-lom)::bla lacIqP207-gfp-QQL | P1 (JOE257) × JOE424, select low Ampr |
| JOE432 | JOE424 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ2 | P1 (JOE299) × JOE424, select low Ampr |
| JOE433 | JOE424 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ6 | P1 (JOE301) × JOE424, select low Ampr |
| JOE434 | JOE424 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ15 | P1 (JOE303) × JOE424, select low Ampr |
| JOE435 | JOE424 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ65 | P1 (JOE305) × JOE424, select low Ampr |
| JOE436 | JOE417 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ | P1 (EC442) × JOE417, select low Ampr |
| JOE437 | JOE417 Δ(λattL-lom)::bla lacIqP207-gfp | P1 (EC452) × JOE417, select low Ampr |
| JOE438 | JOE417 Δ(λattL-lom)::bla lacIqP207-gfp-FFQ | P1 (JOE196) × JOE417, select low Ampr |
| JOE439 | JOE417 Δ(λattL-lom)::bla lacIqP207-gfp-QQL | P1 (JOE257) × JOE417, select low Ampr |
| JOE440 | JOE417 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ2 | P1 (JOE299) × JOE417, select low Ampr |
| JOE441 | JOE417 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ6 | P1 (JOE301) × JOE417, select low Ampr |
| JOE442 | JOE417 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ15 | P1 (JOE303) × JOE417, select low Ampr |
| JOE443 | JOE417 Δ(λattL-lom)::bla lacIqP207-gfp-ftsQ65 | P1 (JOE305) × JOE417, select low Ampr |
| JOE496 | JOE417 Δ(λattL-lom)::bla lacIqP210-ftsA-gfp | P1 (EC447) × JOE417, select low Ampr |
| JOE500 | JOE417 Δ(λattL-lom)::bla lacIqP209-gfp-ftsL | P1 (EC439) × JOE417, select low Ampr |
| JOE502 | JOE496/pJC53 | Transform JOE496 with pJC53 |
| JOE503 | JOE496/pJC54 | Transform JOE496 with pJC54 |
| JOE504 | JOE496/pJC57 | Transform JOE496 with pJC57 |
| JOE505 | JOE496/pAM238 | Transform JOE496 with pAM238 |
| JOE510 | JOE500/pJC53 | Transform JOE500 with pJC53 |
| JOE511 | JOE500/pJC54 | Transform JOE500 with pJC54 |
| JOE512 | JOE500/pJC57 | Transform JOE500 with pJC57 |
| JOE513 | JOE500/pAM238 | Transform JOE500 with pAM238 |
| Plasmids | ||
| pAM238 | Plac regulation, pSC101 origin, Spcr | 16 |
| pBAD18 | Arabinose regulation, pBR origin, Ampr | 19 |
| pBAD33 | Arabinose regulation, pACYC origin, Cmr | 19 |
| pTrc99a | Ptrc expression vector, pBR origin, Ampr | Pharmacia; GenBank accession no. U13872 |
| pDSW207 | pDSW204-gfp-MCS (fusion vector) | 40 |
| pDSW209 | pDSW206-gfp-MCS (fusion vector) | 40 |
| pDSW237 | pDSW209-ftsL | Weiss and Beckwith, unpublished |
| pDSW240 | pDSW207-ftsQ | 10 |
| pLD92 | pBAD18-QQL | 20 |
| pLD104 | pBAD18-FFQ | 20 |
| pLMG161 | pBAD18-ftsQ | 20 |
| pMM2 | pBAD18-ftsQ2 | See Materials and Methods |
| pMM6 | pBAD18-ftsQ6 | See Materials and Methods |
| pMM15 | pBAD18-ftsQ15 | See Materials and Methods |
| pMM65 | pBAD18-ftsQ65 | See Materials and Methods |
| pMM80 | pBAD18-ftsQ80 | EcoRI-XmaI fragment containing ftsQ15 from pMM15 replaced same fragment in pLMG161 |
| pMM81 | pBAD18-ftsQ81 | 3,883-bp BglI fragment from pLMG161 ligated with 1,614-bp BglI fragment from pMM6 |
| pMM82 | pBAD18-ftsQ82 | 3,883-bp BglI fragment from pMM6 ligated with 1,614-bp BglI fragment from pLMG161 |
| pMM83 | pBAD18-ftsQ83 | 3,883-bp BglI fragment from pLMG161 ligated with 1,614-bp BglI fragment from pMM80 |
| pMM84 | pBAD18-ftsQ84 | 3,883-bp BglI fragment from pMM80 ligated with 1,614-bp BglI fragment from pLMG161 |
| pJC10 | pBAD33-ftsQ | 10 |
| pJC17 | pZQ_E14 | See Materials and Methods |
| pJC19 | pDSW207-ftsQ2 | AgeI-HindIII fragment containing ftsQ2 from pMM2 replaced same segment in pDSW240 |
| pJC20 | pDSW207-ftsQ6 | AgeI-HindIII fragment containing ftsQ6 from pMM6 replaced same segment in pDSW240 |
| pJC21 | pDSW207-ftsQ15 | AgeI-HindIII fragment containing ftsQ15 from pMM15 replaced same segment in pDSW240 |
| pJC22 | pDSW207-ftsQ65 | AgeI-HindIII fragment containing ftsQ65 from pMM65 from pMM65 replaced same segment in pDSW240 |
| pJC53 | pAM238-ftsQ | EcoRI-HindIII fragment containing ftsQ from pLMG161 inserted into same sites of pAM238 |
| pJC54 | pAM238-ftsQ2 | EcoRI-HindIII fragment containing ftsQ2 from pMM2 inserted into same sites of pAM238 |
| pJC57 | pAM238-ftsQ65 | EcoRI-HindIII fragment containing ftsQ65 from pMM65 inserted into same sites of pAM238 |
| pJC91 | pTrc99a-ftsQ | EcoRI-HindIII fragment containing ftsQ from pLMG161 inserted into same sites of pTrc99a |
| pJC92 | pTrc99a-ftsQ2 | EcoRI-HindIII fragment containing ftsQ2 from pMM2 inserted into same sites of pTrc99a |
| pJC93 | pTrc99a-ftsQ6 | EcoRI-HindIII fragment containing ftsQ6 from pMM6 inserted into same sites of pTrc99a |
| pJC94 | pTrc99a-ftsQ15 | EcoRI-HindIII fragment containing ftsQ15 from pMM15 inserted into same sites of pTrc99a |
| pJC95 | pTrc99a-ftsQ65 | EcoRI-HindIII fragment containing ftsQ65 from pMM65 inserted into same sites of pTrc99a |
P1, P1 transduction. For example, JOE326 was constructed by infecting JOE310 with P1 lysate made from DH5αZ1.
gfp fusion strains, strains wit gfp fusions in a wild-type background.
Genetic and molecular biology procedures.
Standard techniques were used for cloning and analysis of DNA, PCR, electroporation, transformation, and P1 transduction (29, 35). Stable introduction of genes into the E. coli chromosome at the lambda attachment site via λInCh was performed as described previously (4). Enzymes used to manipulate DNA were from New England BioLabs or GIBCO Life Technologies. Oligonucleotides and deoxynucleoside triphosphates were obtained from GIBCO Life Technologies. DNA sequencing was performed by the Micro Core Facility at the Department of Microbiology and Molecular Genetics, Harvard Medical School.
Construction of ftsQE14::kan.
The new ftsQ null allele was derived from ftsQ::TnphoA80 (7). Primers ddl291f (5′-CTACGCAGCAAACTTCTATG-3′) and ftsQ_E14r (5′-AGAGGTAACCTATTCTTCGCTGTTTCG-3′; BstEII site underlined) were used to amplify the 5′ end of ftsQ from pZQ::TnphoA80 (pZQ containing the transposon insertion) (7). Codon 14 of ftsQ was mutated into a stop codon in the resulting PCR product with the help of ftsQ_E14r. This PCR product was digested with NsiI and BstEII and inserted into pZQ::TnphoA80 to replace the NsiI-BstEII fragment containing TnphoA and codons 14 to 80 of ftsQ. The resulting plasmid (pZQ_E14) contained ddl, the first 13 codons of ftsQ, kan (the gene coding for aminoglycoside-3′-O-phosphotransferase), the 3′ end of ftsQ (after codon 80), and the 5′ end of ftsA. The insert derived from PCR was confirmed by DNA sequencing. A linear fragment containing the region described above was obtained by digesting pZQ_E14 with XmnI and used to transform JCB495 (recD)/pLMG161. Transformants were selected on rich media containing ampicillin, kanamycin, and arabinose. Candidates were then screened by dependence on arabinose for growth. Allele replacement was confirmed by transduction with P1 and by PCR using primers Kan763f (5′-GCCTTCTATCGCCTTCTTGA-3′) and ftsA253r (5′-AGAAAGCGCCAGATATACCG-3′). JOE260 was one such isolate.
PCR mutagenesis and genetic screens.
The PCR mutagenesis protocol was modified from those previously described (15, 24). Primers used to amplify ftsQ from pLMG161 were ftsQ−31 (5′-GGCTAGCGAATTCTGGAACT-3′; EcoRI site underlined) and ftsQ3′+153 (5′-GACCGCTTCTGCGTTCTGAT-3′). A 100-μl reaction mixture included 10 ng of template DNA, 0.5 μM (each) primer, 5 U of Taq DNA polymerase, 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 1 mM (each) pyrimidine (dCTP and dTTP), 0.2 mM (each) purine (dATP and dGTP), and 1 to 5 mM MgCl2. The reactions were done with 25 amplification cycles (95°C for 1 min, 54°C for 1 min, and 72°C for 1 min) followed by a final extension at 72°C for 5 min. The resulting PCR products, 1,015 bp in length, were digested with EcoRI and HindIII and inserted into pLMG161 to replace the original ftsQ gene. Ligated plasmids were transformed into KS272 or MJC129. Transformants were selected on NZY plates containing ampicillin and glucose at 30°C and then replica plated onto NZY containing ampicillin plus glucose or arabinose. KS272 transformants were screened at 30 and 37°C, while MJC129 transformants were screened at 30 and 42°C. Candidates exhibiting mutant phenotypes were confirmed by restreaking. Plasmids of interest were purified and transformed into a fresh background to confirm their phenotypes. Plasmids derived from pLMG161 carrying mutant ftsQ were designated pMM. To detect expressed proteins, we grew candidate strains in rich media containing ampicillin and arabinose until early log phase and then harvested them for immunoblotting.
Growth conditions for complementation and expression analyses.
To determine complementation of the ftsQ1(Ts) mutation, we streaked cells carrying pBAD18-derived plasmids onto NZY plates containing ampicillin plus glucose or arabinose. Levels of growth were compared after overnight incubation at 30 or 42°C. Complementation of the null mutation by gfp fusion alleles was assessed as described previously (10).
We tested whether expression of the mutant FtsQ proteins caused a deleterious effect in ftsQ+ backgrounds by using procedures similar to those used for complementation analysis. Wild-type cells carrying pBAD18-derived plasmids were restreaked and incubated overnight at 37°C. No colony formation defect was observed. To increase the level of the mutant FtsQ protein, we introduced the plasmids into a degP ftsQ+ strain, which lacks a protease that degrades abnormal periplasmic proteins (37). Expression of the mutant alleles in that strain did not cause any effect either, but we did not check whether steady-state levels of mutant FtsQ in degP and degP+ strains were different. We then put the alleles into an expression vector (pTrc99a) under the control of an IPTG (isopropyl-β-d-thiogalactopyranoside)-regulated promoter. We streaked cells onto plates containing ampicillin and different concentrations of IPTG, ranging from 0 to 1 mM. Plates were incubated at 30, 37, or 42°C. At a high induction level (1 mM IPTG), expression of wild-type ftsQ from this plasmid was deleterious to cells; no colony formation was observed. Expression of mutant ftsQ at the same induction level led to smaller colonies but did not stop growth.
To examine the morphology of ftsQ1(Ts) cells expressing mutant ftsQ from pBAD18, we grew overnight cultures in rich media containing ampicillin and glucose. The cells were subcultured into NZY with ampicillin plus glucose or arabinose and grown until early log phase, after about 2 to 3 h, when they were harvested and chemically fixed for phase-contrast microscopy. Samples used to examine localization and expression levels of mutant green fluorescent protein (GFP)-FtsQ in wild-type cells (Fig. 2 and 3) were obtained with procedures similar to those described previously (10). All cultures were grown at 30°C.
FIG. 2.
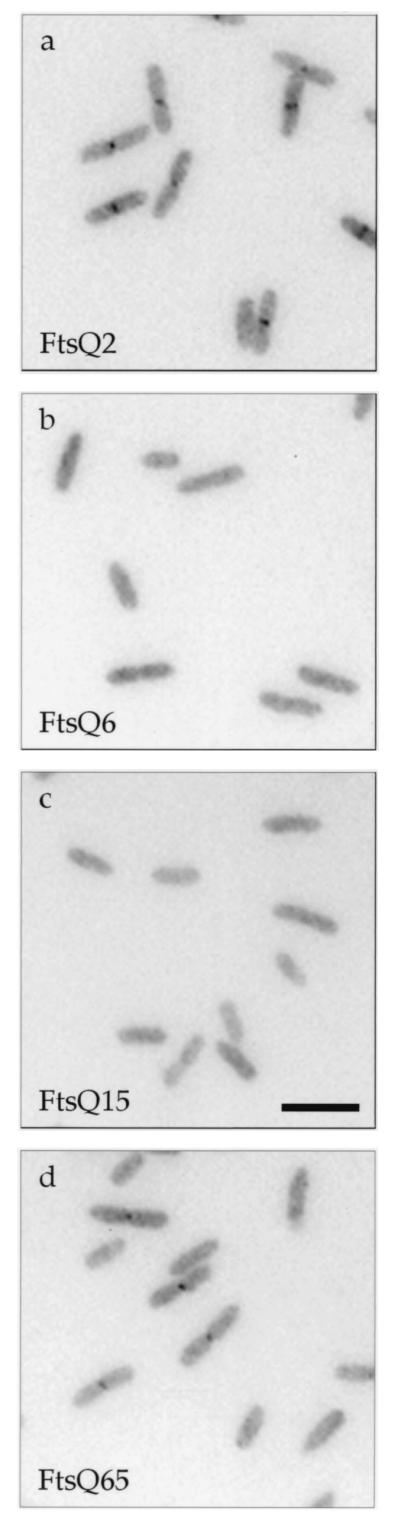
Localization of mutant GFP-FtsQ in ftsQ+ cells. Cells were grown to early log phase under induction at 30°C, chemically fixed, and prepared for microscopy as previously described (10). See Materials and Methods for details regarding fluorescence microscopy. Strains used (and their gfp fusion alleles): a, JOE299 (gfp-ftsQ2); b, JOE301 (gfp-ftsQ6); c, JOE303 (gfp-ftsQ15); d, JOE305 (gfp-ftsQ65). Bar, 5 μm.
FIG. 3.

Expression levels of GFP-FtsQ fusions in an ftsQ+ background. Preparation of cell samples and immunoblotting were performed essentially as described before (10). Equivalent amounts of samples were loaded except those for lanes 4 and 5; for these lanes the sample in lane 3 was diluted 1/10 and 1/20, respectively, with sodium dodecyl sulfate sample buffer. Expression of the gfp fusion allele was induced with 10 μM IPTG except in lane 7, for which induction was with 25 μM IPTG. The left blot was probed with anti-FtsQ antibodies, and the right blot was probed with anti-GFP antibodies. The positions of FtsQ and GFP-FtsQ and -FtsQ2 are indicated on the left, while numbers on the right indicate molecular mass standards in kilodaltons. Strains used: 1, JOE196; 2, EC452; 3 to 5, EC442; 6 and 7, JOE299; 8, JOE301; 9, JOE303; 10, JOE305.
Procedures to deplete cells of wild-type FtsQ were similar to or modified from those used previously (10). To localize mutant GFP-FtsQ in cells depleted of wild-type FtsQ, we grew overnight cultures with the appropriate antibiotics plus arabinose and used these to inoculate rich media containing chloramphenicol, arabinose, and 10 μM IPTG. The starter cultures were grown until the optical density at 600 nm (OD600) reached between 0.2 and 0.3; then cells were centrifuged, resuspended in rich media containing chloramphenicol, glucose, and 10 μM IPTG, and inoculated 1:80 to 1:100 into the same glucose media. Cultures were grown until early log phase, when the appropriate cells became filamentous, and then were harvested for microscopy or immunoblotting. Similar growth conditions were used to observe FtsA-GFP and GFP-FtsL localization when mutant ftsQ is expressed instead of wild-type ftsQ, except that spectinomycin was added to select for plasmid maintenance; in addition, IPTG was not used in the starter cultures but was added at 1 mM in the glucose media. All growth in liquid cultures was conducted at 30°C.
Phase-contrast and fluorescence microscopy.
Cells were harvested, chemically fixed, and prepared for microscopy as previously described (10). To examine cells expressing GFP, we used an Axioskop 2 microscope (Carl Zeiss) equipped with a 63× plan-Apochromat oil immersion objective (numerical aperture, 1.4) and a 100-W mercury lamp. The filter set for GFP fluorescence (HQ: fluorescein isothiocyanate/Bodipy/Fluo3) was from Chroma Technology Corp. Images were captured with an Orca, 12-bit, cooled charge-coupled device camera (C4742-95; Hamamatsu Photonics) and Carl Zeiss Axiovision software, with an exposure time of 1 min. Images were processed in Adobe Photoshop and CorelDRAW for presentation. Phase-contrast images for Fig. 1 were obtained using the same microscope, with a 40× CP-Achromat objective and the same image capture system.
FIG. 1.

Morphology of ftsQ1(Ts) cells expressing ftsQ or ftsQ2 at the permissive temperature. See Materials and Methods for growth conditions and microscopy techniques. Strain backgrounds are indicated on the top, while alleles being expressed from pBAD18 are on the left. Strains used: JOE32, -242, -693, and -714 to -716.
Immunoblotting.
Immunoblotting with anti-FtsQ or anti-GFP antibodies was done essentially as previously described (10).
RESULTS
Genetic screens for mutant ftsQ.
To facilitate our study of FtsQ and its function in cell division, we conducted a screen for trans-dominant-negative alleles of ftsQ. To perform the genetic screen, we first mutagenized ftsQ by PCR and inserted the resulting fragments into pBAD18 (19) such that ftsQ is under the control of the arabinose-dependent promoter. The pool of ligation products was transformed into wild-type cells under the repressing condition, with glucose in the media, and the transformants were replica plated onto rich media containing glucose or arabinose to repress or induce, respectively, expression from the PBAD promoter. Strains transformed with plasmids carrying trans-dominant-negative alleles of ftsQ should grow on glucose but not arabinose. The same plasmid carrying wild-type ftsQ did not have deleterious effects on normal cells under the inducing condition. We screened a total of 10,000 transformants but did not find any apparent trans-dominant mutants.
In a parallel screen, we used similarly mutagenized plasmids to look for missense mutations that failed to complement the ftsQ1(Ts) allele. We wanted to identify residues critical to FtsQ function through such mutations. Mutagenized plasmids were transformed into cells carrying the ftsQ1(Ts) allele at the permissive temperature, and the transformants were replica plated to compare levels of growth in three different situations: with glucose at 30°C (repressing, permissive), with arabinose at 30°C (inducing, permissive), and with arabinose at 42°C (inducing, restrictive). Wild-type ftsQ on the plasmid allowed growth of the temperature-sensitive strain in all three situations, while the alleles we were seeking should not support growth at the restrictive temperature. We replica plated onto rich media containing glucose at the permissive temperature in case a mutant allele is dominant negative, and, as discussed below, this precaution turned out to be warranted. Screening 1,200 transformants yielded 13 candidate plasmids, indicating that the PCR mutagenesis protocol worked and that the failure to identify a dominant-negative allele in the first screen was not a consequence of inefficient mutagenesis.
To screen out alleles that did not generate protein products, including ones with nonsense mutations near the beginning of the gene, we performed immunoblot analysis on cells harboring the candidate plasmids grown under inducing conditions (data not shown). Polyclonal antibodies used in the analysis were made against the periplasmic domain of FtsQ (10). Four of the 13 candidates expressed protein products detectable above the endogenous level of FtsQ: ftsQ2, ftsQ6, ftsQ15, and ftsQ65. One of the four alleles (ftsQ2) produced a truncated protein. Expression levels from all four alleles on these plasmids were reduced compared to that of wild-type ftsQ on the same plasmid, giving expression levels similar to that of endogenous ftsQ on the chromosome (data not shown). The reduced steady-state levels of the mutant proteins suggest that they may be less stable than the wild-type protein.
DNA sequencing of the four independent mutant ftsQ alleles revealed that each allele contained two to four point mutations, all in the region corresponding to the periplasmic domain of FtsQ (Table 2). The mutations consisted of both transitions and transversions. The ftsQ2 allele contained two mutations: a nonsense mutation at codon 248, resulting in the production of a truncated FtsQ missing its last 29 amino acids, and a missense mutation at codon 249, which did not affect the protein product as it followed the nonsense codon. We separated the double and triple mutations in ftsQ6 and ftsQ15, respectively, generating four new alleles: ftsQ81 (L181R), ftsQ82 (F145L), ftsQ83 (Y227D), and ftsQ84 (Q108L, V111G). Each of these alleles was able to complement the ftsQ1(Ts) mutation at the restrictive temperature. Thus, at least for ftsQ6 and ftsQ15, the component mutations were required to be present together in the same gene to yield the nonfunctional phenotype.
TABLE 2.
Sequences of ftsQ mutations
| Allele | Amino acid change | Base change (s) | Type of mutationa |
|---|---|---|---|
| ftsQ1(Ts)b | Glu125 → Lys | GAA → AAA | S |
| ftsQ2 | Tyr248 → ochre | TAT → TAA | V |
| Asp249 → Gly | GAC → GGC | S | |
| ftsQ6 | Phe145 → Leu | TTC → TTA | V |
| Leu181 → Arg | CTG → CGG | V | |
| ftsQ15 | Gln108 → Leu | CAG → CTG | V |
| Val111 → Gly | GTC → GGC | V | |
| Tyr227 → Asp | TAT → GAT | V | |
| ftsQ65 | Ile207 → Phe | ATT → TTT | V |
| Gln232 → Arg | CAG → CGG | S | |
| Val244 → Asp | GTT → GAT | V | |
| Leu259 → Ser | TTG → TCG | S |
S, transition; V, transversion.
DNA sequencing confirmed the mutation in ftsQ1(Ts), as reported by Storts and Markovitz (36).
Characterization of mutant ftsQ in ftsQ1(Ts) and ftsQ+ backgrounds.
During the course of the genetic screen, we noticed that ftsQ2 behaved as a dominant-negative mutant in the ftsQ1(Ts) background (Table 3). The diploid strain formed tiny colonies when expression of the FtsQ2 protein was induced at the permissive temperature. The small colony size was reflected in the lower growth rate in liquid culture at 30°C (data not shown). The ftsQ65 allele also exhibited a dominant-negative effect in the ftsQ1(Ts) background, albeit weaker than that of ftsQ2. The two other alleles, ftsQ6 and ftsQ15, showed no trans-dominant effects.
TABLE 3.
Phenotypes of cells expressing different ftsQ alleles and localization frequencies of mutant GFP-FtsQ
| Allele | Description of gene product | Phenotype in Ts backgrounda at:
|
Complementation in null backgroundb at:
|
% Localization frequency (n)c | |||
|---|---|---|---|---|---|---|---|
| 30°C | 42°C | 30°C | 37°C | 42°C | |||
| Vector | None | ++ | − | − | − | − | 0 (200) |
| ftsQ | Wild type | ++ | ++ | ++ | ++ | ++ | 72 (2,668) |
| ftsQ2 | Y248*, D249G | −/+ | − | − | − | − | 30 (3,114), 37 (1,977) |
| ftsQ6 | F145L, L181R | ++ | − | ++ | + | − | <1 (1,608) |
| ftsQ15 | Q108L, V111G, Y227D | ++ | − | + | − | − | <1 (1,768) |
| ftsQ65 | I207F, Q232R, V244D, L259S | + | − | − | − | − | 12 (3,377) |
| FFQ | Membrane anchor from MalF, periplasmic domain of FtsQ | ++ | ++ | ++ | + | +/− | 35 (599) |
| QQL | Membrane anchor of FtsQ, periplasmic domain of FtsL | ++ | − | − | − | − | 0 (150) |
Growth of ftsQ1(Ts) cells expressing mutant ftsQ was assessed with strains MJC129 or JOE333 as described in Materials and Methods. Plasmids used were pBAD18, pLMG161, pMM2, pMM6, pMM15, pMM65, pLD104, and pLD92. Scoring ranged from good growth (++) to no growth (−).
Complementation of ftsQE14::kan null allele by mutant gfp-ftsQ was determined as described previously (10). Strains used were derived from KS272 (JOE428 to -435) or MC4100 (JOE436 to -443). MC4100-derived strains tended to grow better than their KS272 counterparts under the same conditions. Complementation ranged from good (++) to none (−).
Septal localization was scored in wild-type background as described previously (10); n, number of cells scored. All localization frequencies were measured when cells were grown with 10 μM IPTG, except for the second, higher percentage of ftsQ2, which was measured with 25 μM IPTG induction. Frequencies listed for vector, FFQ, and QQL were obtained previously with a different microscope (10). Strains used were EC452, EC442, JOE299, JOE301, JOE303, JOE305, JOE196, and JOE257.
We examined ftsQ1(Ts) cells expressing ftsQ2 to determine whether the dominant-negative effect manifested itself in a cell division defect. While strains carrying the ftsQ1(Ts) mutation already exhibited some degree of filamentation at the permissive temperature, expression of ftsQ2 exacerbated the filamentation phenotype (Fig. 1). Thus, FtsQ2 appears to inhibit division in ftsQ1(Ts) cells. However, expression of mutant alleles ftsQ2 and ftsQ65 from pBAD18 did not cause any observable dominant effect in ftsQ+ cells. We tested expression from a different plasmid and in different genetic backgrounds (see Materials and Methods) but did not find a significant effect.
Next, we used fusions to GFP to determine whether the ftsQ mutations were affecting the abilities of the mutant proteins to localize to the division site. Constructs expressing GFP fused to the amino termini of the mutant FtsQ proteins were placed under the control of an IPTG-regulated promoter and integrated into the chromosome in single copy (Table 1 and Materials and Methods). In merodiploid cells expressing both wild-type FtsQ and either GFP-FtsQ2 or -FtsQ65, fluorescent dots or bands were observed in the midcell region, indicating septal localization of both mutant proteins (Fig. 2a and d). In contrast, mutant proteins encoded by the ftsQ6 and ftsQ15 alleles did not localize to the division site (Fig. 2b and c). Immunoblotting indicated that the mutant GFP-FtsQ fusions were expressed at similar levels (Fig. 3, lanes 6 and 8 to 10). These results suggest that the noncomplementing phenotype of ftsQ6 and ftsQ15 might be caused by the failure of the proteins to localize to the division site.
Although GFP-FtsQ2 and -FtsQ65 were recruited to the division site, the relative intensities of their localization signals were reduced compared to that of GFP-FtsQ; fluorescent dots or bands in the cell center were less frequently observed (Table 3). This may be explained by the fact that the mutant GFP fusions were expressed at levels lower than that of endogenous FtsQ (Fig. 3). Higher induction slightly increased the localization frequency of GFP-FtsQ2 (Table 3) but not those of the other three mutant fusions (data not shown). Immunoblotting revealed that while more full-length fusion proteins were produced at higher induction (25 μM IPTG and above), more breakdown products were also generated (Fig. 3, lane 7, and data not shown). Since an increase in breakdown products could obscure the localization signal by elevating the background fluorescence, higher induction did not necessarily improve detection of septal recruitment.
Nevertheless, septal localization of FtsQ2 and FtsQ65 was detected, indicating that the proteins were defective in an aspect of their function other than localization. Notably, the ability to localize appeared to correlate with the severity of the dominant-negative phenotype: FtsQ2 was more deleterious than FtsQ65 when expressed in ftsQ1(Ts) cells, and it localized to the division site better. One explanation for these results is that the FtsQ2 and FtsQ65 proteins interacted well with other cell division components, sequestering them away from FtsQ1 at the septum. Replacing FtsQ1 with these mutant FtsQ proteins, which were defective in other functions, resulted in an inhibition of cell division.
Characterization of mutant ftsQ in strains with ftsQ null allele.
We have previously studied the complementation properties of various derivatives of FtsQ using a “null” allele that expressed a fusion of alkaline phosphatase to FtsQ (10). While the product encoded by the gene fusion was missing essentially the entire periplasmic domain of FtsQ, it still contained the cytoplasmic and transmembrane domains of the protein. We were concerned that complementation studies in such a background might be complicated by the continued expression of these two domains. Therefore, we constructed an ftsQ null allele (ftsQE14::kan) on the chromosome; in this allele codons 14 through 80 are replaced by a stop codon and the kanamycin resistance gene from TnphoA (see Materials and Methods). With this new, more complete null allele, we confirmed previous results (10) showing that the specific sequences of the cytoplasmic and transmembrane domains of FtsQ were not required for FtsQ function: when these sequences were replaced by comparable domains from an unrelated protein (maltose transport protein MalF), the “swap” protein (FFQ) was still active in promoting cell division (Table 3). Furthermore, the swap protein localized to the septum in cells depleted of wild-type FtsQ (Fig. 4a). On the other hand, a swap protein in which the periplasmic domain of FtsQ was replaced by the periplasmic domain of FtsL (QQL) was nonfunctional and failed to localize to the division site (Table 3 and Fig. 4b).
FIG. 4.

Localization of GFP fusions in cells depleted of wild-type FtsQ. Cells were depleted of FtsQ at 30°C, chemically fixed, and prepared for fluorescence microscopy as described in Materials and Methods. Strains used (and their gfp fusion alleles): a, JOE438 (gfp-FFQ); b, JOE439 (gfp-QQL); c, JOE440 (gfp-ftsQ2); d, JOE441 (gfp-ftsQ6); e, JOE442 (gfp-ftsQ15); f, JOE443 (gfp-ftsQ65). Bar, 10 μm.
Fusions of gfp to each of the mutant ftsQ alleles were used to test for complementation of the ftsQE14::kan null mutation to assess the functionality of the mutant GFP-FtsQ proteins at temperatures other than 42°C (Table 3). Surprisingly, ftsQ6 and ftsQ15 complemented the null allele at 30°C and ftsQ6 was even slightly active at 37°C; they did not complement at 42°C (Table 3). Considering that GFP-FtsQ6 and -FtsQ15 did not localize to the division site at 30°C in the presence of wild-type FtsQ, we wondered how they were able to complement the null mutation at the same temperature. So we examined localization of the GFP fusions in cells depleted of wild-type FtsQ at 30°C. GFP-FtsQ6 and -FtsQ15 now localized to the division site, and the cells exhibited fairly normal morphology (Fig. 4d and e). Immunoblotting confirmed that wild-type FtsQ was depleted in these cells and that the levels of the GFP fusions before and after depletion were comparable (data not shown). The ability of FtsQ6 and FtsQ15 to localize in FtsQ-depleted cells but not in wild-type cells suggests that the mutant proteins competed less well against wild-type FtsQ for septal recruitment. However, they were able to localize to the division site and function there once the competition disappeared. FtsQ-depleted cells expressing gfp-ftsQ15 appeared to be longer, possibly due to reduced activity of the mutant protein.
On the other hand, ftsQ2 and ftsQ65 failed to complement the null mutation at all temperatures tested (Table 3). GFP-FtsQ2 and -FtsQ65 were able to localize in the wild-type background, and they continued to localize to potential division sites in filamentous cells depleted of FtsQ (Fig. 4c and f). These results suggest that localization of the two mutant proteins in merodiploid cells was not due to interaction with and septal recruitment by wild-type FtsQ. The results also indicate that the last 29 amino acids of FtsQ, truncated in FtsQ2, are not involved in localization but are required for function. Depletion of wild-type FtsQ in these cells was confirmed by immunoblotting as well, and the levels of the GFP fusions before and after depletion were comparable (data not shown).
Since FtsQ2 and FtsQ65 localized to potential division sites without wild-type FtsQ and since their dominant-negative effects in ftsQ1(Ts) cells hinted at interactions with other division machinery components, we asked whether they are able to recruit downstream cell division proteins. Specifically, we investigated whether FtsQ2 or FtsQ65 can recruit FtsL, which depends on FtsQ for septal localization, to the division site. First, we constructed pAM238-derived plasmids with ftsQ(pJC53), ftsQ2(pJC54), or ftsQ65(pJC57) under the control of the lac promoter (Table 1). We tested these low-copy-number plasmids by introducing them into an FtsQ depletion strain carrying lacIq. In the absence of arabinose, the strain containing pJC53 required at least 100 μM IPTG for colony formation, while the other strains did not grow under the same conditions (data not shown).
We then transformed these plasmids into FtsQ depletion strains carrying ftsA-gfp or gfp-ftsL; expression of both fusions was regulated by a weak IPTG-dependent promoter (P206) (40). These strains were grown without arabinose to deplete FtsQ but with IPTG to induce expression of the gfp fusion from the chromosome and wild-type or mutant ftsQ from pAM238-derived plasmids. Cells expressing ftsQ were normal in morphology, while those expressing ftsQ2 or ftsQ65 became filamentous (Fig. 5). These cells were chemically fixed and examined by fluorescence microscopy to determine the localization patterns of FtsA-GFP and GFP-FtsL. Both fusion proteins localized to the septa of normally dividing cells expressing FtsQ (Fig. 5a and b). Since it does not depend on the presence of FtsQ for septal recruitment, GFP-FtsA localized to potential division sites in filaments expressing FtsQ2 or FtsQ65 (Fig. 5c and e). On the other hand, GFP-FtsL did not localize in the mutant filaments (Fig. 5d and f). Immunoblot analysis indicated that mutant FtsQ proteins in these strains were produced at levels similar to or higher than those of the corresponding GFP fusions in wild-type cells (Fig. 6; GFP-FtsQ6 in lane 9 served as a standard for comparing expression from pAM238 versus expression from P207 on the chromosome). Hence, the lack of GFP-FtsL localization was not caused by insufficient expression of mutant FtsQ. These results indicated that FtsQ2 and FtsQ65 could not recruit cell division proteins that normally depended on FtsQ for localization.
FIG. 5.

Localization of FtsA-GFP and GFP-FtsL in cells expressing different ftsQ alleles. These cells carry the ftsQE14::kan null mutation on the chromosome, wild-type ftsQ under the control of an arabinose-dependent promoter on a plasmid, and mutant or wild-type ftsQ under the control of the lac promoter on another plasmid. Growth in the absence of arabinose depleted these cells of wild-type FtsQ, but addition of IPTG induced expression from the lac promoter and allowed substitution with wild-type or mutant FtsQ. See text for further details. The GFP fusion being examined is indicated above, while the FtsQ protein being produced is indicated to the left. Strains used: a, JOE502; b, JOE510; c, JOE503; d, JOE511; e, JOE504; f, JOE512. Bar, 10 μm.
FIG. 6.

Expression levels of ftsQ, ftsQ2, and ftsQ65 from pAM238 in ftsQE14::kan null cells. Cells were grown as for localization in Fig. 5 and as described in Materials and Methods. Immunoblotting was done with anti-FtsQ antibodies as described previously (10). Lysates from cells carrying ftsA-gfp (JOE502 to -505) were loaded into lanes 1 through 4, while those from cells carrying gfp-ftsL (JOE510 to -513) were loaded into lanes 5 through 8. Lane 9 is similar to lane 8 in Fig. 3, containing lysate from JOE301, which is an ftsQ+ strain carrying gfp-ftsQ6 at the lambda att site. Positions of FtsQ, FtsQ2, and GFP-FtsQ6 are indicated on the left, while molecular mass standards in kilodaltons are on the right.
DISCUSSION
In this paper we report the isolation of ftsQ mutants that illuminate aspects of the protein’s activity, including possible interactions with other substrates at the division site. First, there appears to be a limited amount of the recruiting factor that attracts FtsQ to the division site; variant forms of FtsQ compete for this target when expressed in the same cell. Evidence from complementation and localization studies indicates that FtsQ, FtsQ2, and FtsQ65 have higher affinities for the recruiting factor than FtsQ1, FtsQ6, and FtsQ15. This hierarchy of affinities explains why FtsQ2 and FtsQ65 can localize to the division site in the presence of endogenous FtsQ, whereas FtsQ6 and FtsQ15 cannot, even when expressed at comparable or higher levels.
In addition, one possible explanation for the trans-dominant-negative effect of ftsQ2 in the ftsQ1(Ts) background is that FtsQ2 may compete with FtsQ1 at the septal sites. Site occupation by this inactive protein results in a lack of productive interactions and inhibits cell division in the ftsQ1(Ts) background. FtsQ2 does not exert its dominant-negative effect in the ftsQ+ background because wild-type FtsQ competes better for the septal sites, or at least sufficiently for cytokinesis to take place. Consistent with the idea that only a limited number of proteins can be incorporated into the septal ring, overproduction of GFP-FtsQ led to diffuse fluorescence throughout the cell membrane (4). The nature of the septal recruiter for FtsQ remains to be determined, but sequestration must take place in the periplasm, where the essential domain of FtsQ is located. A possible candidate is another cell division protein such as FtsK, which contains a periplasmic region (14) and which is required for FtsQ localization (9).
Another aspect of FtsQ activity revealed by the mutants is that the endogenous protein level is above the minimal requirement for effective cell division. In the absence of wild-type FtsQ, GFP-FtsQ6 and -FtsQ15 could localize to the division site and function reasonably well in cytokinesis at low temperature. Immunoblotting indicated that they were expressed at levels below that of endogenous FtsQ (Fig. 3 and data not shown). Thus, those lower levels were sufficient for growth under the conditions tested. At elevated temperatures, when ftsQ6 and ftsQ15 failed to complement mutations in ftsQ efficiently, the number of protein molecules required might be higher. Alternatively, FtsQ6 and FtsQ15 might be less stable at higher temperatures, or they might not localize to the division site or interact as well with other components of the cell division machinery.
In this study we also constructed a new ftsQ null allele for complementation analysis and confirmed that FtsQ could still function with its cytoplasmic and transmembrane domains replaced. Previous studies had suggested that the periplasmic region of FtsQ is the only essential part of the protein (6, 10, 13), but the leakiness of the ftsQ mutations used to assess complementation was a concern. The present analysis dispels that concern. There have been reports of similar findings regarding domain requirements for two other cell division proteins with similar topology: FtsN (13) and DivIB, the FtsQ homolog of B. subtilis (23).
Analysis of the mutant alleles identified some of the structural determinants of FtsQ activity as well. We deduced that the septal targeting signal resides within amino acids 50 to 247. The first 49 and the last 29 residues are not involved because both FFQ and FtsQ2 can localize to the division site, independently of wild-type FtsQ. In addition, amino acids mutated in FtsQ6 and FtsQ15 appear to be important for septal localization, since these variant forms compete less well against FtsQ for the recruiting factor. All five mutations are located in the region containing the targeting signal (Table 2). Although the five residues may contribute to interaction with the recruiter, none of them appears to be critical on its own. The noncomplementing phenotype of ftsQ6 and ftsQ15 was lost once the individual point mutations were separated. We must also consider the possibility that these mutations simply disrupt the structural integrity of the protein. This change in structure might destabilize interactions of other residues with the recruiting factor. When the degree of disruption was lessened at lower temperature, the noncomplementing phenotype was also suppressed.
Finally, we have separated the structural determinants within FtsQ that affect its localization from those that affect its other activities. While the last 29 amino acids of FtsQ are not required for septal targeting, they are important for function. Regardless of the expression level, ftsQ2 could not complement Ts or null mutations in ftsQ. Similarly, one or a combination of the mutations in ftsQ65 appears to interfere with FtsQ function. Like the 29-amino-acid truncation, these mutations did not abolish FtsQ’s localization activity. One of the protein activities disrupted by the nonsense mutation in ftsQ2 and the missense mutations in ftsQ65 is septal recruitment of cell division proteins downstream. For instance, when cells produced FtsQ2 or FtsQ65 instead of FtsQ, FtsL failed to localize to the division site. How FtsQ mediates this recruitment activity remains to be determined. The mutant proteins may be defective because they simply lost the ability to interact physically with downstream proteins, or they may have lost a biochemical activity that is necessary for recruitment to occur.
Acknowledgments
We thank members of the Beckwith laboratory for assistance and encouragement.
J.C.C. was partially supported by a National Science Foundation predoctoral fellowship. M.M., a participant of the Summer Honors Undergraduate Research Program at Harvard Medical School, was supported by NSF Research Experience for Undergraduates grant DBI-9322334. J.B. was supported by an American Cancer Society Research Professorship. This work was funded by a grant from the National Institute of General Medical Sciences, GM38922.
REFERENCES
- 1.Adam, M., C. Fraipont, N. Rhazi, M. Nguyen-Disteche, B. Lakaye, J. M. Frere, B. Devreese, J. Van Beeumen, Y. van Heijenoort, J. van Heijenoort, and J. M. Ghuysen. 1997. The bimodular G57–V577 polypeptide chain of the class B penicillin-binding protein 3 of Escherichia coli catalyzes peptide bond formation from thiolesters and does not catalyze glycan chain polymerization from the lipid II intermediate. J. Bacteriol. 179:6005–6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Addinall, S. G., C. Cao, and J. Lutkenhaus. 1997. FtsN, a late recruit to the septum in Escherichia coli. Mol. Microbiol. 25:303–309. [DOI] [PubMed] [Google Scholar]
- 3.Bowler, L. D., and B. G. Spratt. 1989. Membrane topology of penicillin-binding protein 3 of Escherichia coli. Mol. Microbiol. 3:1277–1286. [DOI] [PubMed] [Google Scholar]
- 4.Boyd, D., D. S. Weiss, J. C. Chen, and J. Beckwith. 2000. Towards single-copy gene expression systems making gene cloning physiologically relevant: lambda InCh, a simple Escherichia coli plasmid-chromosome shuttle system. J. Bacteriol. 182:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buddelmeijer, N. 1997. Functional analysis of cell division protein FtsQ of Escherichia coli. Ph.D. thesis. University of Amsterdam, Amsterdam, The Netherlands.
- 6.Buddelmeijer, N., M. E. Aarsman, A. H. Kolk, M. Vicente, and N. Nanninga. 1998. Localization of cell division protein FtsQ by immunofluorescence microscopy in dividing and nondividing cells of Escherichia coli. J. Bacteriol. 180:6107–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carson, M. J., J. Barondess, and J. Beckwith. 1991. The FtsQ protein of Escherichia coli: membrane topology, abundance, and cell division phenotypes due to overproduction and insertion mutations. J. Bacteriol. 173:2187–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, J. C. 2001. The role of an essential membrane protein (FtsQ) in Escherichia coli cell division. Ph.D. thesis. Harvard University, Cambridge, Mass.
- 9.Chen, J. C., and J. Beckwith. 2001. FtsQ, FtsL and FtsI require FtsK, but not FtsN, for co-localization with FtsZ during Escherichia coli cell division. Mol. Microbiol. 42:395–413. [DOI] [PubMed] [Google Scholar]
- 10.Chen, J. C., D. S. Weiss, J. M. Ghigo, and J. Beckwith. 1999. Septal localization of FtsQ, an essential cell division protein in Escherichia coli. J. Bacteriol. 181:521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dai, K., and J. Lutkenhaus. 1992. The proper ratio of FtsZ to FtsA is required for cell division to occur in Escherichia coli. J. Bacteriol. 174:6145–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dai, K., Y. Xu, and J. Lutkenhaus. 1993. Cloning and characterization of ftsN, an essential cell division gene in Escherichia coli isolated as a multicopy suppressor of ftsA12(Ts). J. Bacteriol. 175:3790–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai, K., Y. Xu, and J. Lutkenhaus. 1996. Topological characterization of the essential Escherichia coli cell division protein FtsN. J. Bacteriol. 178:1328–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dorazi, R., and S. J. Dewar. 2000. Membrane topology of the N-terminus of the Escherichia coli FtsK division protein. FEBS Lett. 478:13–18. [DOI] [PubMed] [Google Scholar]
- 15.Fromant, M., S. Blanquet, and P. Plateau. 1995. Direct random mutagenesis of gene-sized DNA fragments using polymerase chain reaction. Anal. Biochem. 224:347–353. [DOI] [PubMed] [Google Scholar]
- 16.Ghigo, J. M., and J. Beckwith. 2000. Cell division in Escherichia coli: role of FtsL domains in septal localization, function, and oligomerization. J. Bacteriol. 182:116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghigo, J. M., D. S. Weiss, J. C. Chen, J. C. Yarrow, and J. Beckwith. 1999. Localization of FtsL to the Escherichia coli septal ring. Mol. Microbiol. 31:725–737. [DOI] [PubMed] [Google Scholar]
- 18.Guzman, L. M., J. J. Barondess, and J. Beckwith. 1992. FtsL, an essential cytoplasmic membrane protein involved in cell division in Escherichia coli. J. Bacteriol. 174:7716–7728. [PMC free article] [PubMed] [Google Scholar]
- 19.Guzman, L. M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guzman, L. M., D. S. Weiss, and J. Beckwith. 1997. Domain-swapping analysis of FtsI, FtsL, and FtsQ, bitopic membrane proteins essential for cell division in Escherichia coli. J. Bacteriol. 179:5094–5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harry, E. J., S. R. Partridge, A. S. Weiss, and R. G. Wake. 1994. Conservation of the 168 divIB gene in Bacillus subtilis W23 and B. licheniformis, and evidence for homology to ftsQ of Escherichia coli. Gene 147:85–89. [DOI] [PubMed] [Google Scholar]
- 22.Ishino, F., and M. Matsuhashi. 1981. Peptidoglycan synthetic enzyme activities of highly purified penicillin-binding protein 3 in Escherichia coli: a septum-forming reaction sequence. Biochem. Biophys. Res. Commun. 101:905–911. [DOI] [PubMed] [Google Scholar]
- 23.Katis, V. L., and R. G. Wake. 1999. Membrane-bound division proteins DivIB and DivIC of Bacillus subtilis function solely through their external domains in both vegetative and sporulation division. J. Bacteriol. 181:2710–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin-Goerke, J. L., D. J. Robbins, and J. D. Burczak. 1997. PCR-based random mutagenesis using manganese and reduced dNTP concentration. BioTechniques 23:409–412. [DOI] [PubMed] [Google Scholar]
- 25.Lutz, R., and H. Bujard. 1997. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 25:1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Margolin, W. 2000. Themes and variations in prokaryotic cell division. FEMS Microbiol. Rev. 24:531–548. [DOI] [PubMed] [Google Scholar]
- 27.Mengin-Lecreulx, D., J. van Heijenoort, and J. T. Park. 1996. Identification of the mpl gene encoding UDP-N-acetylmuramate: l-alanyl-γ-d-glutamyl-meso-diaminopimelate ligase in Escherichia coli and its role in recycling of cell wall peptidoglycan. J. Bacteriol. 178:5347–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 29.Miller, J. H. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 30.Nguyen-Disteche, M., C. Fraipont, N. Buddelmeijer, and N. Nanninga. 1998. The structure and function of Escherichia coli penicillin-binding protein 3. Cell. Mol. Life Sci. 54:309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nikolaichik, Y. A., and W. D. Donachie. 2000. Conservation of gene order amongst cell wall and cell division genes in Eubacteria, and ribosomal genes in Eubacteria and eukaryotic organelles. Genetica 108:1–7. [DOI] [PubMed] [Google Scholar]
- 32.Rietsch, A., D. Belin, N. Martin, and J. Beckwith. 1996. An in vivo pathway for disulfide bond isomerization in Escherichia coli. Proc. Natl. Acad. Sci. USA 93:13048–13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rothfield, L., S. Justice, and J. Garcia-Lara. 1999. Bacterial cell division. Annu. Rev. Genet. 33:423–448. [DOI] [PubMed] [Google Scholar]
- 34.Rowland, S. L., V. L. Katis, S. R. Partridge, and R. G. Wake. 1997. DivIB, FtsZ and cell division in Bacillus subtilis. Mol. Microbiol. 23:295–302. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 36.Storts, D. R., and A. Markovitz. 1991. A novel rho promoter::Tn10 mutation suppresses an ftsQ1(Ts) missense mutation in an essential Escherichia coli cell division gene by a mechanism not involving polarity suppression. J. Bacteriol. 173:655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strauch, K. L., and J. Beckwith. 1988. An Escherichia coli mutation preventing degradation of abnormal periplasmic proteins. Proc. Natl. Acad. Sci. USA 85:1576–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strauch, K. L., K. Johnson, and J. Beckwith. 1989. Characterization of degP, a gene required for proteolysis in the cell envelope and essential for growth of Escherichia coli at high temperature. J. Bacteriol. 171:2689–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang, X., and J. Lutkenhaus. 1996. Characterization of the ftsZ gene from Mycoplasma pulmonis, an organism lacking a cell wall. J. Bacteriol. 178:2314–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiss, D. S., J. C. Chen, J. M. Ghigo, D. Boyd, and J. Beckwith. 1999. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J. Bacteriol. 181:508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]