

Abstract
Alpha/beta interferons (IFN-α/β) are key mediators of the innate immune response against viral infection. The ability of viruses to circumvent IFN-α/β responses plays a crucial role in determining the outcome of infection. In a previous study using subgenomic replicons of the Kunjin subtype of West Nile virus (WNVKUN), we demonstrated that the nonstructural protein NS2A is a major inhibitor of IFN-β promoter-driven transcription and that a single amino acid substitution in NS2A (Ala30 to Pro [A30P]) dramatically reduced its inhibitory effect (W. J. Liu, H. B. Chen, X. J. Wang, H. Huang, and A. A. Khromykh, J. Virol. 78:12225-12235). Here we show that incorporation of the A30P mutation into the WNVKUN genome results in a mutant virus which elicits more rapid induction and higher levels of synthesis of IFN-α/β in infected human A549 cells than that detected following wild-type WNVKUN infection. Consequently, replication of the WNVKUNNS2A/A30P mutant virus in these cells known to be high producers of IFN-α/β was abortive. In contrast, both the mutant and the wild-type WNVKUN produced similar-size plaques and replicated with similar efficiency in BHK cells which are known to be deficient in IFN-α/β production. The mutant virus was highly attenuated in neuroinvasiveness and also attenuated in neurovirulence in 3-week-old mice. Surprisingly, the mutant virus was also partially attenuated in IFN-α/βγ receptor knockout mice, suggesting that the A30P mutation may also play a role in more efficient activation of other antiviral pathways in addition to the IFN response. Immunization of wild-type mice with the mutant virus resulted in induction of an antibody response of similar magnitude to that observed in mice immunized with wild-type WNVKUN and gave complete protection against challenge with a lethal dose of the highly virulent New York 99 strain of WNV. The results confirm and extend our previous original findings on the role of the flavivirus NS2A protein in inhibition of a host antiviral response and demonstrate that the targeted disabling of a viral mechanism for evading the IFN response can be applied to the development of live attenuated flavivirus vaccine candidates.
Since its introduction into New York in 1999, West Nile virus (WNVNY99) has spread rapidly into all states of the United States. From 2002 to 2004, a total of 16,557 human cases of WNV disease have been reported in the United States, with 648 deaths (http://www.cdc.gov/ncidod/dvbid/westnile/qa/cases.htm). The virus has also been introduced into regions of Canada and Mexico, and evidence of bird infection has been detected in the Dominican Republic (20). In addition to human infections, the virus has caused significant morbidity and mortality in horses and birds, with more than 20,000 equine cases and hundreds of thousands of avian deaths. Relative to the WNVNY99 strain, an Australian subtype of WNV, Kunjin virus (WNVKUN), exhibits much reduced pathogenesis in humans, animals, and birds (3, 13). Only a small number of mostly mild human cases due to WNVKUN infection have been documented since it was first discovered in Australia more than 40 years ago, with no reported fatalities (13). Similarly, equine disease associated with WNVKUN infection is rare, and there is no evidence that WNVKUN causes significant disease in birds (3). At the genetic level, WNVKUN is more than 98% homologous in amino acid sequence to WNVNY99 (23, 36). The attenuated virulence phenotype of WNVKUN and its very close homology to the WNVNY99 strain makes WNVKUN an attractive candidate for development of a WNV vaccine. In a previous study, we showed that immunization with WNVKUN or with infectious WNVKUN cDNA results in complete protection of mice from lethal challenge with WNVNY99 (15). Despite the apparent low virulence in humans and domestic animals of naturally circulating WNVKUN, further virulence attenuation of this strain of WNV is desirable for its application as a safe live WNV vaccine candidate.
The interferon (IFN) response is one of the first lines of defense of the host against viral infections, and many viruses have developed strategies to inhibit the IFN response. Accordingly, the targeted disabling of a virus' ability to inhibit IFN responses is emerging as a promising approach for the development of attenuated viral vaccines (2, 5, 31, 39). We recently showed for the first time for a flavivirus that the nonstructural protein 2A (NS2A) is the major suppressor of beta interferon (IFN-β) transcription, and we have identified a single amino acid mutation in NS2A (alanine at position 30 to proline [A30P]) that abrogated this inhibition (24). The A30P mutation was originally identified as the mutation in WNVKUN replicon RNA, confirming its advantage in establishing persistent replication in BHK cells (24). Further studies showed that WNVKUN replicon RNA with the A30P mutation induced activation of IFN-β transcription ∼6- to 7-fold more efficiently than the wild-type (WT) WNVKUN replicon RNA (24). Similarly, individually expressed NS2A protein with the A30P substitution was shown to be ∼7- to 8-fold less efficient than the wt NS2A protein in suppression of IFN-β transcription in response to infection with Semliki Forest virus (SFV) (24). In this report, we extend our previous studies of the effect of the A30P substitution in WNVKUN replicon RNA on the efficiency of induction of the IFN response by incorporating the mutation into the WNVKUN full-length genome and comparing replicative and IFN-inducing properties of the mutant virus with those of the WT virus in vitro and in vivo.
MATERIALS AND METHODS
Cells and viruses.
BHK21, A549, L929, and HEp-2 cells were grown in Dulbecco's modification of minimal essential medium (DMEM) (Invitrogen, Carlsbad, Calif.) supplemented with 10% fetal bovine serum (FBS) at 37°C in a CO2 incubator. The establishment of HEp-2 cells stably expressing WNVKUN replicon RNAs encoding either the WT or NS2A/A30P NS2A protein was described previously (24), and the cells were maintained in the same medium supplemented with 5 μg of puromycin (Sigma Aldrich, St. Louis, Mo.) per ml. BHK21 cells (a hamster kidney cell line) are recognized to have a defect in interferon production (30), while A549 cells (a human lung epithelium cell line) are known to be potent IFN-α/β producers (40). SFV was kindly provided by Nigel McMillan (Center for Immunology and Cancer Research, University of Queensland) and was propagated and titrated in Vero cells.
Plasmid construction.
The construction of plasmid FLSDX (pro)_HDVr containing the WT WNVKUN full-length cDNA clone, followed by the hepatitis delta virus ribozyme, was reported previously (23). Plasmid FLSDX/NS2A/A30P (NS2A/A30P) containing the WNVKUN full-length cDNA clone with the A30P substitution in the NS2A gene was obtained by replacing the SphI-SphI fragments in FLSDX(pro)_HDVr with the SphI-SphI fragment derived from the intermediate plasmid repPAC-β-gal/NS2A/A30P (23); the presence of the mutation in the resulting construct was confirmed by sequencing.
Generation of virus stocks from full-length RNAs.
All RNA transcripts were prepared with SP6 RNA polymerase from XhoI-linearized plasmid DNAs and electroporated into BHK21 cells as described previously (19). Briefly, ∼10 μg of in vitro-transcribed RNA was electroporated into 2 × 106 BHK21 cells suspended in 400 μl of phosphate-buffered saline (PBS) using a 0.2-cm electrode gap cuvette (Bio-Rad) and a Bio-Rad Gene Pulser II apparatus. The electroporated cells were seeded into 100-mm dishes and incubated for 48 to 96 h until cytopathic effect (CPE) developed. Recovered viruses were amplified on Vero cells to generate virus stocks. The virus titers were determined by standard plaque assay on BHK21 cells. The virus stocks were stored in 0.5-ml aliquots at −80°C.
Viral infection and growth kinetics analysis.
BHK and A549 cells in six-well plates were infected with the WT or mutant virus at a multiplicity of infection (MOI) of 1 for 2 h at 37°C, washed three times with PBS, and overlaid with 2 ml of DMEM containing 2% FBS. At the indicated times postinfection, cell culture fluids were harvested to determine virus titers by standard plaque assay on BHK-21 cells, and total RNA was isolated from infected cells by Trizol reagent (Invitrogen) for detection of WNVKUN viral RNA and IFN-β mRNA.
Northern blot and RT-PCR analysis.
Northern blot analysis of WNVKUN RNA and IFN-β mRNA in replicon-expressing HEp2 cells and in virus-infected A549 cells was performed as described previously (23, 24). Briefly, ∼10 μg of total cellular RNA was separated by 1% formaldehyde agarose gel electrophoresis, and the RNA was transferred to a nitrocellulose membrane. The nitrocellulose membrane was cut at 3 cm above the position of the bromophenol blue dye band into two parts. The upper part of the membrane was probed with a 32P-labeled WNVKUN RNA-specific probe. The lower part of the membrane was probed with a 32P-labeled IFN-β mRNA-specific probe. The lower part of the membrane was subsequently stripped and reprobed with a 32P-labeled β-actin mRNA-specific probe. The membranes were exposed to X-ray film for 16 to 72 h. The reverse transcription (RT)-PCR amplification of ∼1 μg of total cell RNA with primers specific for IFN-β or β-actin mRNA was performed using the One-Step Superscript RT-PCR kit (Invitrogen) essentially as described by the manufacturer. The cycling conditions were as follows: reverse transcription at 50°C for 30 min, followed by 20 cycles of PCR amplification: 94°C for 20 s, 52°C for 20 s, and 72°C for 40 s.
Detection of viral foci by immunofluorescence and by plaque assay.
For immunofluorescence, A549 and BHK21 cells grown on coverslips placed in 24-well plates were infected with either WT or NS2A/A30P mutated WNVKUN at an MOI of ∼1 for 2 h at 37°C, washed three times with PBS, and overlaid with 0.75% low-melting-temperature (LMT) agarose in DMEM containing 2% FBS. At 48 h postinfection, the cells were treated with 10% formadehyde-PBS for 20 min, the agarose was removed, and the cells were fixed with ice-cold acetone-methanol (1:1) for 20 min and stained with a WNV envelope (E) protein-specific monoclonal antibody. For plaque assay, BHK21 cells and A549 cells in six-well plates were infected by either WT or NS2A-mutated WNVKUN as described above, overlaid with 0.75% LMT agarose in DMEM containing 2% FCS, and incubated at 37°C for 5 days to allow plaque development. The cells were then fixed with 10% formaldehyde and stained with 0.2% crystal violet.
Detection of IFN-α/β in the culture fluid of infected A549 cells by bioassay.
The concentrations of IFN-α/β in the cell culture fluid were measured by a CPE inhibition bioassay using SFV infection of A549 cells. Briefly, A549 cells were seeded in 100 μl of complete medium at a concentration of 5 × 104/well in 96-well plates and incubated at 37°C for 6 to 8 h. Samples of cell culture fluid harvested from WT or mutant virus-infected A549 cells were exposed to UV light in a GS GENE LINKERCHAMBER (Bio-Rad) six times for 90 s each time to inactivate live virus. The UV-inactivated samples were then serially diluted (twofold dilutions) and added to A549 cells. After incubation for 16 h at 37°C, the cells were infected at an MOI of 0.5 with SFV, and the incubation continued for another 48 h. The cells were then fixed with 4% formaldehyde-PBS and stained with 0.2% crystal violet. Uninfected cells and cells infected with SFV only were used as blank and negative controls, respectively. The crystal violet was released by adding 100 μl of 100% methanol, and the absorbance was measured at a wavelength of 620 nm. The IFN-α/β concentrations in international units per ml were calculated according to the protection against SFV infection provided by the treatment with known concentrations of human IFN-α2A (Sigma). The sensitivity of the assays (50% protection against SFV infection at an MOI of 0.5) was 10 IU per ml.
Detection of IFN-α/β in the sera of infected mice by bioassay with L929 cells.
The IFN-α/β in the sera of mice infected with the WT and NS2A/A30P mutated viruses (pooled from five mice in each group) was detected by bioassay of L929 cells. The IFN-α/β levels were expressed as percentages of L929 cells protected against challenge with SFV at an MOI of 0.5 after pretreatment with 10 μl of UV-inactivated mouse sera. For the IFN-α/β neutralizing assay, 10 μl of UV-inactivated mouse sera was mixed with 400 units of each rabbit antibody against mouse IFN-α and IFN-β (PBL Biomedical Laboratories, Piscataway, NJ) for 30 minutes at 37°C prior to performing the bioassay. The percentage of protected L929 cells (the protection rate) was calculated according to the following formula: (optical density at 620 nm [OD620] of mouse serum-treated SFV-infected cells/OD620 of non-SFV-infected cells − OD620 of SFV-infected cells/OD620 of non-SFV-infected cells) × 100%.
Mouse neuroinvasiveness and neurovirulence assays.
Three-week-old Swiss outbred mice were used for virulence assays as described previously (14). Groups of 5 to 10 mice were inoculated intracerebrally (i.c.) with 10 μl or intraperitoneally (i.p.) with 100 μl of serial 10-fold dilutions of the WT or NS2A mutated WNVKUN and closely monitored for signs of paralysis or encephalitis. The mice were euthanized when signs of encephalitis were evident. The 50% lethal dose (LD50) for each virus was calculated using the method of Reed and Muench (32).
Measurement of antibody responses.
At 21 days postinoculation of 5-week-old Swiss mice with 103 infectious units of the WT or NS2A/A30P mutated WNVKUN, mouse sera were collected and assayed for virus-specific antibodies by enzyme-linked immunosorbent assay (ELISA) using antigens derived from lysates of Spodoptera frugiperda cells infected with a recombinant baculovirus expressing WNVKUN prM-E proteins (D. C. Clark and R. A. Hall, unpublished results). All sera were tested in duplicate.
Mouse protection studies.
Groups of 5-week-old BALB/c mice (n = 10) were immunized by i.p. injection of 104 infectious units per mouse of the WNVKUNNS2A/A30P mutant. Twenty control mice were left uninfected. Three weeks after immunization, the mice were challenged with WNVNY99 by the i.p. (100 infectious units per mouse) or i.c. (20 infectious units per mouse) route. The mice were monitored for 2 weeks for signs of encephalitis and euthanized when symptoms of encephalitis were evident.
RESULTS
The NS2A/A30P mutation enhances IFN-β mRNA transcription in response to infection with SFV in HEp2 cells stably expressing WNVKUN replicon RNA.
We previously showed ∼7-fold more efficient activation of transcription from the IFN-β promoter in HEp2 cells stably expressing WNVKUN replicon RNA with a NS2A/A30P mutation than in cells stably expressing WT WNVKUN replicon RNA (24). The direct effect of the mutation in NS2A on the loss of suppression of IFN-β mRNA transcription mediated by the WNVKUN replicon RNA was determined by Northern blot and RT-PCR analyses. Both analyses showed higher levels of transcribed and accumulated IFN-β mRNA in cells stably expressing NS2A/A30P-mutated replicon RNA than in cells expressing WT replicon RNA (Fig. 1A and B). This difference was even more pronounced when IFN-β mRNA transcription was further induced by infection of the replicon-expressing cells with SFV (Fig. 1A and B). IFN-β mRNA transcription in cells expressing NS2A/A30P-mutated replicon RNA was induced in response to SFV infection to a level similar to that observed in SFV-infected control HEp2 cells, while the induction of IFN-β mRNA transcription in response to SFV infection was severely inhibited in the WT replicon-expressing cells (Fig. 1A and B). Note that the replication efficiencies of both WNVKUN replicon RNAs, WT and NS2A mutated, were similar (Fig. 1A). Analysis of SFV genomic RNA by Northern blotting showed no difference in the levels of accumulated SFV RNA between normal cells and WNVKUN replicon-expressing cells at 12 h postinfection, indicating that the initial infection with SFV was not affected by the replicating WNVKUN replicon RNAs.
FIG. 1.
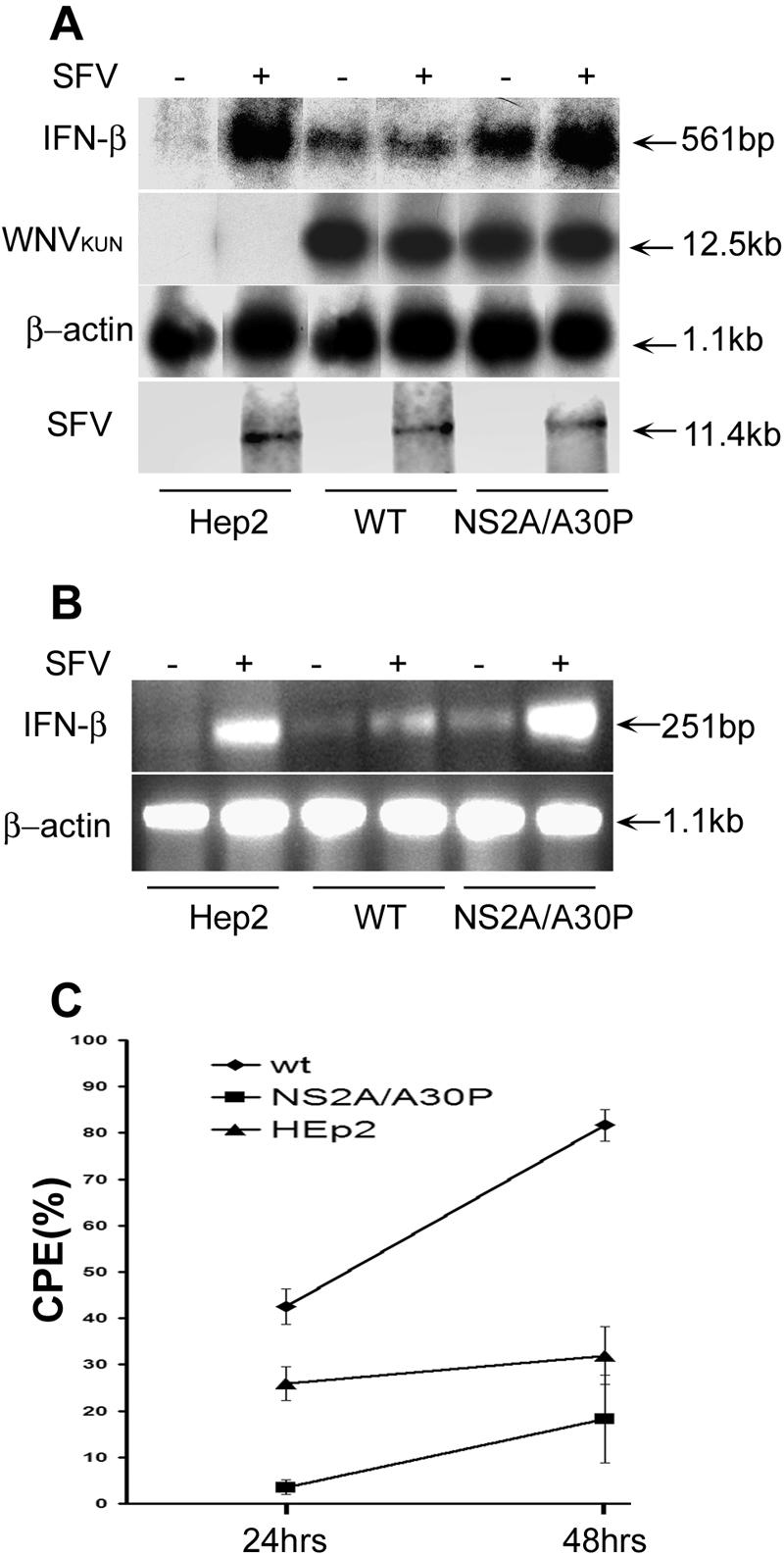
WNVKUN replicon RNA with an Ala30-to-Pro mutation in NS2A (NS2A/A30P) induced more IFN-β-specific mRNA in HEp2 cells and rendered them more resistant to SFV infection than WT replicon RNA. (A) Northern blot analysis. Normal HEp2 cells and HEp2 cells stably transfected with WNVKUN replicon RNAs encoding either the WT or Ala30-to-Pro-mutated (NS2A/A30P) NS2A gene were infected with SFV at an MOI of 1 or left uninfected. Twelve hours post-SFV infection, total cellular RNA was isolated, and ∼10 μg of total RNA was used for Northern blot hybridization with 32P-labeled probes specific for IFN-β mRNA, WNVKUN replicon RNA, β-actin mRNA, and SFV viral RNA. The membrane hybridized with the IFN-β mRNA-specific probe was exposed to X-ray film for 72 h, membranes hybridized with WNVKUN- or β-actin-specific probes were exposed to X-ray film overnight, and the membrane hybridized with SFV NSP4-specific probe was exposed to a phosphorimager screen and scanned by typhoon scanner (Amersham Biosciences, NSW, Australia). (B) RT-PCR analysis of total cellular RNA from the samples in panel A to detect IFN-β and control β-actin mRNAs was performed as described in Materials and Methods. (C) Cytopathogenicity of SFV in WNVKUN replicon-expressing HEp2 cells. Control HEp2 cells or HEp2 cells stably expressing WNVKUN replicon RNAs encoding either the WT or Ala30-to-Pro-mutated (NS2A/A30P) NS2A gene were infected with SFV at an MOI of 0.01. At 24 and 48 h after infection, the cells were fixed and stained with crystal violet. After the staining, the crystal violet was released by methanol treatment and measured at OD620. The percentage of CPE was calculated according to the following formula: 100% − (OD620 of infected sample/OD620 of uninfected sample) × 100%. The error bars indicate standard deviations.
Further incubation of cells after SFV infection showed earlier and substantially stronger CPE in the WT WNVKUN replicon-expressing cells than in cells expressing NS2A/A30P mutated replicon RNA or in control cells (Fig. 1C). The enhanced SFV infection in cells stably expressing WT WNVKUN replicon RNA detected by CPE assay (Fig. 1C) correlates well with the ability of WT WNVKUN replicon RNA to inhibit induction of IFN-β transcription in response to SFV infection detected by Northern blot and RT-PCR analyses (Fig. 1A and B). In contrast, the inability of NS2A/A30P mutated WNVKUN replicon RNA to inhibit induction of IFN-β transcription in response to SFV infection results in significant delay in virus spread and, as a consequence, a substantial decrease in the virus-induced CPE. Thus, we concluded from these results that NS2A/A30P mutation leads to a significant decrease in the ability of expressed WNVKUN replicon RNA to inhibit activation of IFN-β mRNA transcription in response to IFN-inducing viral stimuli.
The NS2A/A30P mutation results in abortive replication of WNVKUN in IFN-α/β-producing A549 cells but not in IFN-α/β-deficient BHK cells.
Toward development of an attenuated WNV vaccine defective in escape from the IFN-α/β response, the NS2A/A30P mutation was transferred into the WT full-length WNVKUN cDNA clone, FLSDX(pro)_HDVr (23), and the NS2A/A30P mutant virus was recovered from BHK cells electroporated with in vitro-transcribed RNA. Infection of BHK cells with the mutant or WT virus showed no significant difference between the two viruses in plaque morphology and in secreted virus titers (Fig. 2A, B, and D). In contrast, spread of the NS2A/A30P mutant, but not of the WT virus, was severely inhibited in A549 cells, as judged by (i) much smaller foci of infection detected by immunofluorescence analysis with anti-E antibodies (Fig. 2A), (ii) absence of detectable plaques under agar (Fig. 2B), and (iii) decline in the titers of secreted virus particles from 12 h postinfection onward (Fig. 2C). Even at the peak of virus growth at 12 h postinfection, the NS2A/A30P virus reached a titer of only 104 PFU per ml, with the titers declining to ∼102 PFU per ml later in infection (Fig. 2C). In contrast, the WT virus titers reached ∼107 PFU per ml by 24 to 36 h postinfection, and the titers remained at ∼106 PFU per ml until the end of the experiment (60 h postinfection) (Fig. 2C). Clearly, replication and spread of the mutant, but not of the WT virus, were severely compromised in IFN-α/β-producing A549 cells, while the two viruses replicated with similar efficiencies in IFN-α/β-deficient BHK cells.
FIG. 2.

Plaque morphology and growth kinetics of WT and NS2A/A30P mutated WNVKUN in BHK-21 and A549 cells. (A) Immunofluorescence analysis of viral foci. BHK-21 and A549 cells on coverslips in 24-well plates were infected with the WT or NS2A/A30P mutated WNVKUN and overlaid with 0.75% LMT agarose in DMEM containing 2% FBS. At 48 h after infection, the cells were fixed and stained using a monoclonal antibody specific for the WNV E protein. (B) Plaque assay. BHK and A549 cells in six-well plates were infected with the WT or NS2A/A30P-mutated WNVKUN, overlaid with 0.75% LMT agarose in DMEM with 2% FBS, and stained with crystal violet at 5 days postinfection. (C and D) Virus growth kinetics. BHK-21 and A549 cells in six-well plates were infected with the WT or NS2A/A30P-mutated WNVKUN at an MOI of ∼1. At the indicated times, cell culture fluid was collected and virus titers in the cell culture fluid were determined by plaque assays on BHK-21 cells.
The NS2A/A30P mutation enhances induction of IFN-β mRNA transcription and expression in A549 cells infected with WNVKUN.
To analyze the abilities of the NS2A/A30P mutant and the WT viruses to induce IFN-β mRNA transcription, total RNA from A549 cells isolated at different times after infection with the same MOI (MOI = 1) of these viruses was subjected to Northern blot analysis with an IFN-β mRNA-specific probe. Despite the lower levels of the NS2A/A30P mutated WNVKUN RNA at 24 h postinfection than in the WT virus-infected cells, the mutant virus induced transcription and accumulation of larger amounts of IFN-β mRNA (Fig. 3A). Quantitative analysis of labeled IFN-β, WNVKUN, and β-actin RNA bands in 24-h samples, followed by the normalization of IFN-β mRNA levels with respect to the levels of WNVKUN and β-actin RNAs, showed ∼7.7-fold-higher levels of accumulated IFN-β mRNA than for the same amount of WNVKUN RNA in cells infected with the mutant virus (Fig. 3A). Further comparative analysis of secreted IFN-α/β in the culture media of infected cells using a bioassay (see Materials and Methods) showed earlier induction of expression and larger amounts of secreted IFN-α/β in cells infected with the mutant virus (Fig. 3B). It should be noted, however, that the assay does not exclude the possibility of the presence in the culture fluid of other antiviral factors in addition to IFN-α/β.
FIG. 3.

Induction of IFN-α/β by WT and NS2A/A30P mutated WNVKUN in A549 cells. (A and C) Northern blot analysis of IFN-β mRNA. A549 cells were infected with the WT or NS2A/A30P mutated WNVKUN at an MOI of ∼1 (A) or at MOIs of 1 and 3, respectively (C). Total cellular RNA was isolated at the indicated times (A) or at 24 h postinfection (C) and subjected to Northern blot analysis with radiolabeled probes specific for IFN-β mRNA, WNVKUN RNA, or β-actin mRNA as in Fig. 1. The exposed X-ray films were scanned, and RNA bands in 24-h sample lanes were quantified using Qantity One software (Bio-Rad, Australia). (B and D) Detection of secreted IFN-α/β by bioassay. Cell culture fluids collected from infected A549 cells in corresponding experiments (B corresponds to A, and D corresponds to C) were analyzed for the presence of IFN-α/β by bioassay on A549 cells as described in Materials and Methods. The error bars indicate standard deviations.
In order to confirm the observed differences in the induction of IFN-β transcription and expression between the WT and the mutant viruses, the experiment was repeated, but this time the MOI for each virus was adjusted to allow the two viruses to reach similar levels of viral-RNA accumulation by 24 h postinfection (Fig. 3C and D). The WT virus was used at an MOI of 1, while the mutant virus was used at an MOI of 3. Indeed, Northern blot analysis showed accumulation of similar levels of viral RNA for both viruses at 24 h postinfection (Fig. 3C). At the same time, infection with mutant virus resulted in detection of much higher levels of accumulated IFN-β mRNA than infection with the wild-type virus (Fig. 3C). Quantitative analysis of labeled RNA bands confirmed the previously observed ∼7.7-fold difference in the amounts of accumulated IFN-β mRNA for the same amount of viral RNA between the mutant and the WT virus infections (Fig. 3A and C). Bioassay of the culture fluids of infected cells failed to detect secreted IFN-α/β in WT virus infection (the sensitivity of the assay was 10 IU per ml), while ∼400 IU of secreted IFN-α/β was detected in the mutant virus infection (Fig. 3D). Thus, all the in vitro results presented above demonstrate that the NS2A/A30P mutation substantially (by ∼7- to 8-fold) decreases the ability of the WNVKUN virus/replicon to inhibit activation of IFN-β transcription, which leads to increased production of secreted IFN-α/β.
WNVKUNNS2A/A30P induces more IFN-α/β than the WT virus in mice.
To determine whether the mutant virus also induces more IFN-α/β in vivo, 1- and 2-day-postinfection sera from 5-week-old Swiss mice injected with the mutant or the WT virus were analyzed for the presence of IFN-α/β by a bioassay of mouse L929 cells. At day 1 postinfection, substantially more IFN-α/β, measured as the percentage of cells protected from SFV infection, was present in the sera of mice immunized with the mutant virus than in the sera of mice immunized with the WT virus (Fig. 4). A difference in the levels of antiviral activity in the sera of mice immunized with the mutant or the WT virus was also seen at day 2 postinfection, albeit to a lesser extent (Fig. 4). To confirm that the protection from SFV infection was indeed generated by the IFN-α/β produced in the sera of infected mice, the sera were preincubated with the polyclonal antibodies against mouse IFN-α and IFN-β prior to the bioassay. Depletion of IFN-α/β with the antibodies significantly decreased the protection rate for both WT and mutant virus-infected sera (Fig. 4), indicating that the antiviral activity in the mouse sera was mainly due to the IFN-α/β produced (Fig. 4). The results demonstrate that the mutant virus induces IFN-α/β synthesis in infected mice more efficiently than the WT virus.
FIG. 4.

Induction of IFN-α/β by the WT and NS2A/A30P mutated WNVKUN in mice. Groups of 5-week-old Swiss outbred mice (n = 3) were inoculated i.p. with 104 infectious units per mouse of the WT or NS2A/A30P-mutated WNVKUN. Sera were collected at day 1 and day 2 postinfection, pooled, UV inactivated to eliminate infectious virus, and analyzed for the presence of IFN-α/β by bioassay on L929 cells as described in Materials and Methods. IFN-α/β depletion was performed by preincubating sera with 400 units of each rabbit antibody against mouse IFN-α and IFN-β (IFN-α/β antibodies) prior to the bioassay. The results shown on the y axis represent percentages of L929 cells protected against challenge with SFV at an MOI of 0.5 by pretreatment with IFN-α/β-depleted (+) or with intact (−) sera from WNVKUN-infected mice. Each assay was performed in duplicate; the error bars represent standard deviations.
WNVKUNNS2A/A30P is highly attenuated in weanling mice.
Taking into account the in vitro and in vivo results for the mutant virus showing enhanced activation of IFN-α/β expression, it was reasonable to assume that the virus is attenuated in animals in which IFN-α/β is important in recovery from virus infection. The mutant virus was highly attenuated when injected into 3-week-old Swiss outbred mice, a standard animal model for flavivirus virulence (26). Intraperitoneal injection of up to 105 infectious units of the mutant virus did not induce any signs of disease in the overwhelming majority of animals, with the exception of one mouse in the highest-dose (105-PFU) group, which developed signs of encephalitis (i.p. LD50 > 105) (Table 1). In contrast, only 1 infectious unit of the WT virus was sufficient to cause encephalitis in 60% of the animals, while 100 and 1,000 infectious units resulted in 80% mortality (i.p. LD50 = 100.63) (Table 1). In a more stringent test for virulence attenuation, none of the mice injected i.c. with up to 10 infectious units of the mutant virus developed encephalitis (i.c. LD50 = 101.5), while only 1 infectious unit of WT virus was sufficient to kill 60% of the mice, and 10 infectious units resulted in 100% mortality (i.c. LD50 = 100.32) (Table 1).
TABLE 1.
Comparison of neuroinvasiveness and neuroviolence of Kunjin WT and NS2A/A30P mutated viruses in Swiss outbred mice
| Virus | I.p. injection
|
I.c. injection
|
||||
|---|---|---|---|---|---|---|
| Dose (IU) | Mortality (%) | AST (days)a | Dose (IU) | Mortality (%) | AST (days) | |
| WT | 100,000 | 10/10 (100) | 6.64 | 1,000 | 5/5 (100) | 15 |
| 10,000 | 5/5 (100) | 7.8 | 100 | 5/5 (100) | 5.4 | |
| 1,000 | 4/5 (80) | 6.75 | 10 | 5/5 (100) | 6.4 | |
| 100 | 4/5 (80) | 7.75 | 1 | 3/5 (60) | 8 | |
| 10 | 3/5 (60) | 9 | 0.1 | 1/6 (16.7) | 8 | |
| 1 | 3/5 (60) | 6.67 | ||||
| NS2A/A30P | 100,000 | 1/10 (10) | 9 | 1,000 | 4/4 (100) | 5 |
| 10,000 | 0/5 (0) | 100 | 5/5 (100) | 5.6 | ||
| 1,000 | 0/5 (0) | 10 | 0/5 (0) | |||
| 100 | 0/5 (0) | 1 | 0/5 (0) | |||
| 10 | 0/5 (0) | 0.1 | 0/5 (0) | |||
| 1 | 0/5 (0) | |||||
AST, average survival time.
WNVKUNNS2A/A30P is partially attenuated in IFN-α/β/γ receptor knockout mice.
We were next interested to determine to what extent the attenuation of the mutant virus relates to its demonstrated ability to induce more IFN-α/β. For this purpose, IFN-α/β/γ receptor knockout mice (AG129) (41) were used. These mice are unable to mount efficient IFN-dependent antiviral responses due to their inability to amplify IFN-α/β production via the secondary loop through IFN-α/β receptor signaling and to generate IFN-γ-dependent cellular antiviral immune responses (41). The first, intracrine, loop of IFN response, however, is not affected in these mice. Mice of two different ages, 6 weeks and 12 weeks old, were used to account for age-dependent differences in susceptibility to WNVKUN infection. The mutant virus proved to be partially attenuated in these mice, as judged by a slight delay in the average time to death (ATD) (1 day in 6-week-old mice and 0.5 days in 12-week-old mice) (Fig. 5A and B) and by the lower virus titers detected in mouse serum at 2 days postinfection (1,000-fold lower in 6-week-old mice and 100-fold lower in 12-week-old mice) (Fig. 5C). While the difference between the two viruses in ATD in AG129 mice was not significant, the differences in viremia were highly significant. The latter may have been due to the still-present intracrine IFN antiviral response in these mice, which is expected to be induced in greater magnitude in mutant than in WT virus-infected mice. Interestingly, the differences in ATD and day 2 viremia became less pronounced with increased age of the mice; this contrasts with the expected amplification in older AG129 mice of virulence differences between virus strains that do not involve the IFN response. Thus, these results show that the WNVKUN NS2A mutant, which is highly attenuated in wild-type mice, also displays marginally reduced virulence in IFN-α/β/γ receptor knockout mice relative to the WT virus. While the marginal reduction in virulence is consistent with the interpretation that the mutant virus lacks the ability to efficiently down-modulate IFN production, the detection of any reduction at all suggests that the A30P mutation may more efficiently activate (or less efficiently down-regulate) another antiviral pathway(s) in addition to the IFN response.
FIG. 5.
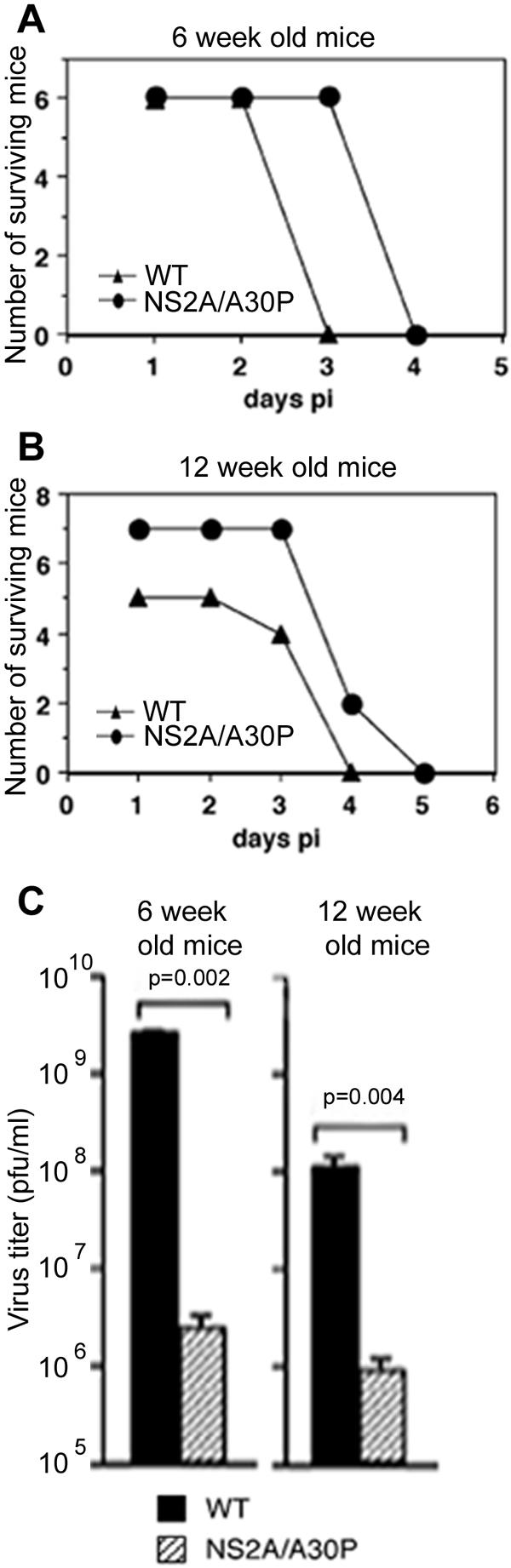
Partial attenuation of WNVKUNNS2A/A30P in IFN-α/β/γ receptor knockout mice. (A and B) Six- or 12-week-old AG129 mice were inoculated intravenously with 1,000 PFU of WNVKUN or WNVKUNNS2A/A30P, and morbidity and mortality were recorded daily. (C) Sera were collected on day 2 postinfection, and viremia titers were determined by plaque assay on Vero cells. Differences were analyzed for significance using the Mann-Whitney test. The error bars represent the standard errors of the mean.
WNVKUNNS2A/A30P induces antibody response in mice and protects them from lethal challenge with WNVNY99.
Intraperitoneal immunization of 5-week-old Swiss outbred mice with 103 infectious units of the WT virus or the mutant WNVKUN resulted in induction of WNV-specific antibody responses of a magnitude similar to that detected by ELISA using recombinant WNVKUN prM-E antigens. The ELISA titers were 1:320 in all five animals immunized with WT virus and in three out of five animals immunized with the mutant virus. The ELISA titers in the remaining two mice immunized with the mutant virus were only twofold lower.
We then examined the protective efficacy of WNVKUNNS2A/A30P immunization against challenge with highly virulent WNVNY99 in 5-week-old BALB/c mice. Immunization with 104 infectious units of the mutant virus resulted in 100% protection from i.p. challenge with 100 infectious units and 44% protection against i.c. challenge with 20 infectious units of WNVNY99 (Fig. 6). The same doses of WNVNY99 produced 60% mortality after i.p. challenge and 90% mortality after i.c. challenge in control unimmunized mice (Fig. 6).
FIG. 6.

Immunization with WNVKUNNS2A/A30P protects BALB/c mice against lethal challenge with WNVNY99. Groups of 5-week-old BALB/c mice (n = 10) were immunized i.p. with 104 infectious units of WNVKUNNS2A/A30P or left unimmunized. Three weeks after immunization, the mice were challenged with WNVNY99 by the i.p. route (100 infectious units per mouse) (A) or by the i.c. route (20 infectious units per mouse) (B). The mice were monitored for 2 weeks for signs of encephalitis.
DISCUSSION
In this study, we have extended our previous observations on the unique role of the flavivirus NS2A protein in inhibition of IFN-β induction by demonstrating the effect of an Ala30-to-Pro substitution in NS2A on attenuation of virus replication in IFN-producing A549 cells. Further analysis of IFN-β mRNA transcription in infected A549 cells and of secreted IFN-α/β in the cell culture fluid showed at least ∼6- to 7-fold more efficient activation of IFN-α/β induction by the WNVKUNNS2A/A30P mutant than by the WT virus. Importantly, despite the poor growth of the mutant virus in A549 cells, the level of IFN-α/β detectable in the cell culture supernatant exceeded that secreted from WT virus-infected cells even late in infection. Given the limited spread of the mutant virus in the IFN-α/β-producing cell line as a result of a deficiency in suppressing IFN induction, it is likely that the prolonged IFN-α/β production by mutant virus-infected A549 cells is maintained by IFN-α/β receptor-mediated signaling in uninfected cells. Thus, it appears that a low level of mutant virus replication is sufficient for the continuing production and release of IFN-α/β due to its paracrine effect on uninfected cells. In contrast, early inhibition of IFN-β induction by the WT virus allows it to spread efficiently. As the number of WT virus-infected cells increases, a corresponding increase in IFN-α/β production is detectable late in infection, most likely due to its amplification via IFN-α/β receptor signaling in the remaining noninfected cells. We and others have demonstrated that IFN-α/β signaling is severely inhibited in cells in which replication of flavivirus RNA is established (12, 17, 22, 25, 29), thus making infected cells an unlikely source of efficient IFN-α/β production via autocrine signaling.
The delay in activation of expression of antiviral genes during WNV infection was recently shown to be caused by a delay in activation of the transcription factor, IFN regulatory factor 3 (IRF-3) (6). However, IRF-3 is only one of many transcription factors involved in activation of IFN-β transcription by viral infections (10, 18, 35), and a number of viral proteins from other viruses have been reported to block IRF-3 and/or other transcription factors (reviewed in references 10, 18, and 35; for more recent reviews, see references 1, 4, 16, and 42). Given the dominant effect of the NS2A protein on suppression of IFN-α/β transcription shown here and in our previous study (24), it is reasonable to speculate that NS2A may inhibit the function of one or more of these transcription factors. Studies of the identification of the transcription factor(s) involved in IFN-β activation whose function is affected by NS2A protein are in progress.
Interestingly, the Ala30-to-Pro mutation in NS2A that disables the virus' ability to inhibit activation of IFN-β transcription was identified by selection of replicon RNA molecules able to persistently replicate in BHK cells (24). In similar studies with Sindbis virus, a mutation in the nsP2 protein was identified by selection of persistently replicating replicon RNA in BHK cells. This mutation resulted in a dramatic increase in IFN-α/β production in NIH 3T3 cells when they were infected with the mutant Sindbis virus in comparison to WT Sindbis virus-infected cells. It also decreased virus-induced cytopathogenicity in various cell lines and resulted in virulence attenuation in suckling mice after intracranial injection (7-9). Accordingly, the phenotype of the Sindbis mutant resembles that of the WNVKUN NS2A mutant described here, suggesting that the mutations were selected (by establishment of efficient replicon persistence in BHK cells) for modulation of the same host pathway(s). It seems unlikely that the IFN pathway was targeted directly by the mutations, since BHK cells have a defect in IFN production (30) and the benefit of less efficient escape from the IFN response for replicon persistence in these cells is not obvious. Interestingly, microarray analysis of cells infected with the WT and the nsP2 mutated Sindbis viruses showed dramatic differences between the two viruses in the expression of virus-induced stress-dependent genes, with some of the genes induced by the mutant virus identified to be related to apoptosis (9, 11). The up-regulation of antiapoptotic pathways leading to cell survival would be obviously advantageous for establishment of replicon persistence. If this was the primary mechanism of adaptation of the Sindbis virus and WNVKUN replicons, the intriguing question of the downstream impact of the mutations on the IFN pathway remains. Lee et al. (21) have recently shown that Japanese encephalitis virus and dengue virus activate antiapoptotic PI3K-Akt signaling early in infection, and others have shown that hepatitis C virus replicon RNA activates the N-Ras-PI3K-Akt-mTOR pathway to promote cell survival and the establishment of persistent RNA replication (28). The antiapoptotic PI3K-Akt pathway may provide the link with the IFN response pathways, given that the former has recently been shown to be involved in TLR3-dependent activation of IRF-3 (33).
This speculation may be supported by the intriguing finding of this study that the WNVKUNNS2A/A30P mutant was partially attenuated in IFN-α/β/γ receptor knockout (AG129) mice. It was envisaged from the observed differences in activation of IFN response between the WT and mutant viruses that their pathogeneses in AG129 mice would be comparable. However, a significant difference in viremia (∼1,000-fold in 6-week-old mice and ∼100-fold in 12-week-old mice) was found at 2 days postinfection between the two viruses. The still-functioning intracrine antiviral activity of IFN-α/β in the AG129 mice may account for the less efficient growth of WNVKUNNS2A/A30P than of the WT virus in these mice. However, we cannot exclude the possibility that the differences in activation in vivo of other factors and pathways in virus-host interactions (e.g., the antiapoptotic pathway) exist between the WT and NS2A mutant viruses which become apparent in this exquisitely sensitive animal model of flavivirus pathogenesis. The high susceptibility of adult IFN-α/β receptor-deficient mice to extraneural infection with encephalitic flavivirus contrasts with the much greater resistance of WT mice (27, 37). Thus, despite the apparent ability of flaviviruses to suppress the induction of IFN-α/β, the IFN-α/β response clearly remains essential for recovery from encephalitic flavivirus infection.
As expected, mutant virus was highly attenuated in the 3-week-old wild-type mice. The difference in LD50 between the mutant and the WT viruses after extraneural infection was almost 100,000-fold, clearly demonstrating a high degree of attenuation, probably caused by the limited virus spread due to the induction of a robust IFN-α/β response. Although we have not tested virus titers in the sera of these mice, it is highly likely that the mutant virus titers would be much lower than the WT virus titers. Limited extraneural virus replication would probably preclude the virus from invading the central nervous system and causing encephalitis. Interestingly, the mutant virus was also attenuated when injected directly into the brain, although the difference in LD50 between the mutant and the WT viruses was less profound. Encephalitic viral infections have been shown to induce IFN response in the brain (34, 38); thus, the virulence attenuation of the WNVKUN mutant after intracranial injection is not surprising. Importantly, despite the high degree of attenuation of WNVKUNNS2A/A30P in wild-type mice, immunization with a single dose of 103 to 104 infectious units induced a strong humoral response that protected all animals against peripheral challenge with a lethal dose of highly virulent WNVNY99. Clearly, the mutation in NS2A did not markedly compromise the magnitude and protective value of the WNV-specific immune response elicited and, therefore, should find application in the development of safe and efficient live attenuated flavivirus vaccines.
Acknowledgments
We thank Megan Pavy for expert technical help.
The studies were supported by grants to A.A.K. from the National Health and Medical Research Council of Australia.
REFERENCES
- 1.Bonjardim, C. A. 2005. Interferons (IFNs) are key cytokines in both innate and adaptive antiviral immune responses—and viruses counteract IFN action. Microbes Infect. 7:569-578. [DOI] [PubMed] [Google Scholar]
- 2.Bossert, B., and K. K. Conzelmann. 2002. Respiratory syncytial virus (RSV) nonstructural (NS) proteins as host range determinants: a chimeric bovine RSV with NS genes from human RSV is attenuated in interferon-competent bovine cells. J. Virol. 76:4287-4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brault, A. C., S. A. Langevin, R. A. Bowen, N. A. Panella, B. J. Biggerstaff, B. R. Miller, and K. Nicholas. 2004. Differential virulence of West Nile strains for American crows. Emerg. Infect. Dis. 10:2161-2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conzelmann, K. K. 2005. Transcriptional activation of alpha/beta interferon genes: interference by nonsegmented negative-strand RNA viruses. J. Virol. 79:5241-5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donelan, N. R., C. F. Basler, and A. Garcia-Sastre. 2003. A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J. Virol. 77:13257-13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fredericksen, B. L., M. Smith, M. G. Katze, P. Y. Shi, and M. Gale, Jr. 2004. The host response to West Nile virus infection limits viral spread through the activation of the interferon regulatory factor 3 pathway. J. Virol. 78:7737-7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frolov, I., E. Agapov, T. A. Hoffman, Jr., B. M. Pragai, M. Lippa, S. Schlesinger, and C. M. Rice. 1999. Selection of RNA replicons capable of persistent noncytopathic replication in mammalian cells. J. Virol. 73:3854-3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frolov, I., and S. Schlesinger. 1994. Comparison of the effects of Sindbis virus and Sindbis virus replicons on host cell protein synthesis and cytopathogenicity in BHK cells. J. Virol. 68:1721-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frolova, E. I., R. Z. Fayzulin, S. H. Cook, D. E. Griffin, C. M. Rice, and I. Frolov. 2002. Roles of nonstructural protein nsP2 and alpha/beta interferons in determining the outcome of Sindbis virus infection. J. Virol. 76:11254-11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodbourn, S., L. Didcock, and R. E. Randall. 2000. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J. Gen. Virol. 81:2341-2364. [DOI] [PubMed] [Google Scholar]
- 11.Gorchakov, R., E. Frolova, and I. Frolov. 2005. Inhibition of transcription and translation in Sindbis virus-infected cells. J. Virol. 79:9397-9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo, J. T., J. Hayashi, and C. Seeger. 2005. West Nile virus inhibits the signal transduction pathway of alpha interferon. J. Virol. 79:1343-1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall, R. A., A. K. Broom, D. W. Smith, and J. S. Mackenzie. 2002. The ecology and epidemiology of Kunjin virus. Curr. Top. Microbiol. Immunol. 267:253-269. [DOI] [PubMed] [Google Scholar]
- 14.Hall, R. A., A. A. Khromykh, J. M. Mackenzie, J. H. Scherret, T. I. Khromykh, and J. S. Mackenzie. 1999. Loss of dimerisation of the nonstructural protein NS1 of Kunjin virus delays viral replication and reduces virulence in mice, but still allows secretion of NS1. Virology 264:66-75. [DOI] [PubMed] [Google Scholar]
- 15.Hall, R. A., D. J. Nisbet, K. B. Pham, A. T. Pyke, G. A. Smith, and A. A. Khromykh. 2003. DNA vaccine coding for the full-length infectious Kunjin virus RNA protects mice against the New York strain of West Nile virus. Proc. Natl. Acad. Sci. USA 100:10460-10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hengel, H., U. H. Koszinowski, and K. K. Conzelmann. 2005. Viruses know it all: new insights into IFN networks. Trends Immunol. 26:396-401. [DOI] [PubMed] [Google Scholar]
- 17.Jones, M., A. Davidson, L. Hibbert, P. Gruenwald, J. Schlaak, S. Ball, G. R. Foster, and M. Jacobs. 2005. Dengue virus inhibits alpha interferon signaling by reducing STAT2 expression. J. Virol. 79:5414-5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katze, M. G., Y. He, and M. Gale, Jr. 2002. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2:675-687. [DOI] [PubMed] [Google Scholar]
- 19.Khromykh, A. A., and E. G. Westaway. 1997. Subgenomic replicons of the flavivirus Kunjin: construction and applications. J. Virol. 71:1497-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komar, O., M. B. Robbins, K. Klenk, B. J. Blitvich, N. L. Marlenee, K. L. Burkhalter, D. J. Gubler, G. Gonzalvez, C. J. Pena, A. T. Peterson, and N. Komar. 2003. West Nile virus transmission in resident birds, Dominican Republic. Emerg. Infect. Dis. 9:1299-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee, C. J., C. L. Liao, and Y. L. Lin. 2005. Flavivirus activates phosphatidylinositol 3-kinase signaling to block caspase-dependent apoptotic cell death at the early stage of virus infection. J. Virol. 79:8388-8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin, R. J., C. L. Liao, E. Lin, and Y. L. Lin. 2004. Blocking of the alpha interferon-induced Jak-Stat signaling pathway by Japanese encephalitis virus infection. J. Virol. 78:9285-9294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu, W. J., H. B. Chen, and A. A. Khromykh. 2003. Molecular and functional analyses of Kunjin virus infectious cDNA clones demonstrate the essential roles for NS2A in virus assembly and for a nonconservative residue in NS3 in RNA replication. J. Virol. 77:7804-7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu, W. J., H. B. Chen, X. J. Wang, H. Huang, and A. A. Khromykh. 2004. Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A in inhibition of beta interferon promoter-driven transcription. J. Virol. 78:12225-12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu, W. J., X. J. Wang, V. V. Mokhonov, P. Y. Shi, R. Randall, and A. A. Khromykh. 2005. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J. Virol. 79:1934-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lobigs, M., I. D. Marshall, R. C. Weir, and L. Dalgarno. 1988. Murray Valley encephalitis virus field strains from Australia and Papua New Guinea: studies on the sequence of the major envelope protein gene and virulence for mice. Virology 165:245-255. [DOI] [PubMed] [Google Scholar]
- 27.Lobigs, M., A. Mullbacher, Y. Wang, M. Pavy, and E. Lee. 2003. Role of type I and type II interferon responses in recovery from infection with an encephalitic flavivirus. J. Gen. Virol. 84:567-572. [DOI] [PubMed] [Google Scholar]
- 28.Mannova, P., and L. Beretta. 2005. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: control of cell survival and viral replication. J. Virol. 79:8742-8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munoz-Jordan, J. L., G. G. Sanchez-Burgos, M. Laurent-Rolle, and A. Garcia-Sastre. 2003. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 100:14333-14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otsuki, K., J. Maeda, H. Yamamoto, and M. Tsubokura. 1979. Studies on avian infectious bronchitis virus (IBV). III. Interferon induction by and sensitivity to interferon of IBV. Arch. Virol. 60:249-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinlivan, M., D. Zamarin, A. Garcia-Sastre, A. Cullinane, T. Chambers, and P. Palese. 2005. Attenuation of equine influenza viruses through truncations of the NS1 protein. J. Virol. 79:8431-8439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reed, L. J., and H. Muench. 1938. A simple method for estimating fifty percent end points. Am. J. Hyg. 27:493-497. [Google Scholar]
- 33.Sarkar, S. N., K. L. Peters, C. P. Elco, S. Sakamoto, S. Pal, and G. C. Sen. 2004. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat. Struct. Mol. Biol. 11:1060-1067. [DOI] [PubMed] [Google Scholar]
- 34.Schoneboom, B. A., J. S. Lee, and F. B. Grieder. 2000. Early expression of IFN-alpha/beta and iNOS in the brains of Venezuelan equine encephalitis virus-infected mice. J. Interferon Cytokine Res. 20:205-215. [DOI] [PubMed] [Google Scholar]
- 35.Sen, G. C. 2001. Viruses and interferons. Annu. Rev. Microbiol. 55:255-281. [DOI] [PubMed] [Google Scholar]
- 36.Shi, P. Y., M. Tilgner, M. K. Lo, K. A. Kent, and K. A. Bernard. 2002. Infectious cDNA clone of the epidemic West Nile virus from New York City. J. Virol. 76:5847-5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shresta, S., J. L. Kyle, H. M. Snider, M. Basavapatna, P. R. Beatty, and E. Harris. 2004. Interferon-dependent immunity is essential for resistance to primary dengue virus infection in mice, whereas T- and B-cell-dependent immunity are less critical. J. Virol. 78:2701-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silvia, O. J., L. Pantelic, J. S. Mackenzie, G. R. Shellam, J. Papadimitriou, and N. Urosevic. 2004. Virus spread, tissue inflammation and antiviral response in brains of flavivirus susceptible and resistant mice acutely infected with Murray Valley encephalitis virus. Arch. Virol. 149:447-464. [DOI] [PubMed] [Google Scholar]
- 39.Solorzano, A., R. J. Webby, K. M. Lager, B. H. Janke, A. Garcia-Sastre, and J. A. Richt. 2005. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J. Virol. 79:7535-7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanabe, M., M. Kurita-Taniguchi, K. Takeuchi, M. Takeda, M. Ayata, H. Ogura, M. Matsumoto, and T. Seya. 2003. Mechanism of up-regulation of human Toll-like receptor 3 secondary to infection of measles virus-attenuated strains. Biochem. Biophys. Res. Commun. 311:39-48. [DOI] [PubMed] [Google Scholar]
- 41.van den Broek, M. F., U. Muller, S. Huang, M. Aguet, and R. M. Zinkernagel. 1995. Antiviral defense in mice lacking both alpha/beta and gamma interferon receptors. J. Virol. 69:4792-4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weber, F., G. Kochs, and O. Haller. 2004. Inverse interference: how viruses fight the interferon system. Viral Immunol. 17:498-515. [DOI] [PubMed] [Google Scholar]