

Abstract
The process by which transmissible spongiform encephalopathy agents, or prions, infect cells is unknown. We employed a new differentiable cell line (SN56) susceptible to infection with three mouse-adapted scrapie strains to gain insight into the cellular infection process. The effect of disease-associated PrP (PrP-res) association with microsomal membranes on infection efficiency was examined by comparing sustained PrP-res production in cells treated with either scrapie brain microsomes or purified, detergent-extracted PrP-res. When normalized for quantity of input PrP-res, scrapie brain microsomes induced dramatically enhanced persistent PrP-res formation compared to purified PrP-res. Infected SN56 cells released low levels of PrP-res into the culture supernatant, which also efficiently initiated infection in recipient cells. Interestingly, microsomes labeled with a fluorescent marker were internalized by SN56 cells in small vesicles, which were subsequently found in neuritic processes. When bound to culture wells to reduce internalization during the infection process, scrapie microsomes induced less long-term PrP-res production than suspended microsomes. Long-term differentiation of infected SN56 cells was accompanied by a decrease in PrP-res formation. Our observations provide evidence that infection of cells is aided by the association of PrP-res with membranes and/or other microsomal constituents.
The transmissible spongiform encephalopathies (TSEs), or prion diseases, are a group of neurodegenerative diseases affecting a wide variety of mammals from sheep and goats (scrapie) to humans (Creutzfeldt-Jakob disease). Some naturally occurring TSEs have been adapted to infect rodents as small-animal models, such as mouse-adapted scrapie (hereafter referred to as scrapie). TSE diseases are usually associated with the accumulation of a misfolded form of a glycosylphosphatidyl inositol (GPI)-anchored protein termed the prion protein (PrPC or PrP-sen) primarily in nervous and lymphoid tissues (for a review, see reference 1). The nature of the TSE agent has long been a subject of debate, but it has been proposed to be comprised of an abnormal, disease-associated conformation of PrP designated PrP-res or PrPSc. In spite of this debate, in the vast majority of systems PrP-res is a reliable marker for TSE infectivity (31).
Little is known about the mechanism by which TSE agents infect and are transmitted between cells, a process essential for trafficking of TSE infectivity from the periphery to the central nervous system. The recent development of a rapidly growing list of cell lines susceptible to TSE infection has opened new opportunities to investigate this subject (3, 8, 11, 20, 43, 52-54). Studies thus far have shown that cell-cell contact between infected and uninfected cells can be an efficient method of initiating infection (30). Similarly, others have found that TSE infectivity can be bound to steel wires and plastic cell culture plates and that these surfaces can also initiate infection in cells and animals with high efficiency (19, 57, 58). However, it is unclear in either of these systems whether infection is initiated without transfer of PrP-res molecules. Finally, both infected neuronal (this study) and nonneuronal (18) cells have been formally shown to release PrP-res into the culture supernatant which, when added to recipient cells, can initiate infection (18, 48; this study).
Previous studies from our laboratory using a cell-free conversion system that modeled the early interactions that occur during TSE infection of cells by using membrane-bound forms of PrPC and PrP-res showed that insertion of PrP-res into a membrane contiguous with PrPC was required for new PrP-res formation (5, 6). These observations led us to propose two scenarios for initiation and propagation of PrP-res synthesis (6). One mechanism involved the intercellular transfer of PrP-res via membrane microparticles (e.g., exosomes) released from infected cells which insert into the membranes of recipient cells, or perhaps exchange of membrane components between closely apposed cells, processes shown to mediate the intercellular transfer of other proteins (7, 36), including PrPC (35). A second possibility involved the release of PrP-res aggregates free of membranes with subsequent insertion into host cell membranes, a process called “GPI painting” (26, 41). Both brain membrane fractions containing membrane-associated forms of PrP-res (46) and purified PrP-res preparations that lack membranes (10) contain high titers of TSE infectivity. However, it is unclear which of these forms of PrP-res might be most important for initiating PrP-res and TSE agent formation in uninfected cells.
In this study, we introduce a new, highly susceptible neuronal cell model of mouse-adapted TSE infection and compare the relative infectivities of microsome-associated PrP-res and purified PrP-res. Our data show that microsome-associated PrP-res induces persistent PrP-res formation far more efficiently than membrane-free PrP-res, suggesting that membrane-associated forms of PrP-res may be the most efficient means of intercellular transfer of PrP-res and TSE infectivity.
MATERIALS AND METHODS
Cell culture.
SN56 cells are a mouse cholinergic septal neuronal cell line (9, 23) previously shown to express PrPC (34) that was generously provided by Bruce Wainer (Department of Pathology, Emory University School of Medicine, Atlanta, GA). The generation of pentosan polysulfate-cured, scrapie-susceptible N2a mouse neuroblastoma cells has been described elsewhere (32). The cells were maintained at 37°C in a humidified atmosphere of 5% CO2 in OptiMEM (Invitrogen) supplemented with 10% fetal bovine serum and penicillin-streptomycin. The SN56 cells were passed at 1:10 every 4 days. N2a cells were passed at 1:10 every 3 days. To induce differentiation, SN56 cells were cultured in serum-free OptiMEM supplemented with 1 mM dibutyryl cyclic AMP, and the medium was changed every day. These culture conditions induced differentiation-associated changes in essentially all the cells, including decreased cell division, extensive neurite outgrowth, and altered (neuron-like) cell body morphology, all consistent with previous studies of SN56 cells (4, 9).
PrP-res preparations used for infections.
Crude brain microsome fractions were prepared from brains of either normal or terminally ill mice infected with the Chandler (RML), ME7, or 87V strain of scrapie agent, as described previously (6), in sterile phosphate-buffered saline (PBS) (pH 7.4). Microsome fractions were also prepared from normal or 263K-infected hamsters. PrP-res was purified from terminally ill Chandler- and 22L-infected mice as described by Raymond and Chabry (45) without proteinase K (PK) digestion. Detailed methods for complete characterization of PrP-res preparations have been described elsewhere (45). Detergent-free PrP-res was prepared by diluting purified PrP-res 10-fold in sterile PBS, followed by centrifugation for 20 min at 21,000 × g. The pellet was resuspended in sterile PBS and pelleted again. The final pellet was resuspended in sterile PBS and sonicated briefly in a cuphorn sonicator. Cleared culture supernatants were prepared from cells grown from ∼30 to 40% to 100% confluence over a 2-day period. The conditioned medium was removed from the culture flasks and centrifuged at 1,800 × g for 10 min, and 80% of the volume of supernatant was carefully removed to avoid disturbing any pellet in the tubes. These cleared supernatants were then stored at 4°C until they were used. PK-digested PrP-res standards were used to quantify PrP-res levels in the various preparations by immunoblotting.
Cell infections.
Cells were plated at approximately 10% confluence in either 96-well plates (SN56) or 24-well plates (N2a) 2 days prior to infection. On the day of infection, the cells were carefully washed once with serum-deficient OptiMEM prior to addition of a minimal volume of microsomes or purified PrP-res (with or without normal microsomes) diluted in serum-free OptiMEM. After incubation for 4 to 5 h, 3 to 4 volumes of OptiMEM with serum was added. For infections with culture supernatants, 100 μl of cleared culture supernatant was added in duplicate both to wells with (plated as described above) and without cells, the latter to verify that the supernatants had been cleared of live cells. For infections with dried microsomes, microsomes were diluted in sterile PBS and added to wells of a separate 96-well plate. The plate was dried in a biosafety cabinet with the assistance of very gentle heat from a hot plate for ∼2 h. This heat-assisted drying was used because it accelerated the drying process, and microsomes immobilized on plates with gentle heat have been shown to exhibit significantly greater PrPC-converting activity than those bound by passive adsorption (38). The wells were carefully washed twice with sterile PBS prior to plating SN56 cells at 10% confluence on top of the dried microsomes. For all methods of infection, cells were grown to confluence (usually 2 to 4 days) and subsequently carried in 24-well plates, passaging the cells at a 1:10 dilution.
Immunoblotting.
To analyze PrP isoforms in cleared culture supernatants, 1 ml out of 5 ml of supernatant, prepared as described above from a confluent T-25 flask of cells, was first adjusted to a final concentration of 50 mM phosphate buffer (pH 7.4)-0.5% (vol/vol) Triton X-100-0.5% (wt/vol) deoxycholate (lysis buffer). Samples were then digested with 20 μg/ml PK at 37°C for 45 min. PK digestion was terminated by the addition of 1 mM Pefabloc SC (Roche) and incubation on ice for 5 min. Samples were then subjected to phosphotungstic acid (PTA) precipitation essentially as described by Wadsworth et al. (55) with the exception that the Benzonase digestion step was omitted. The PK digestion step was omitted for PK− samples. Pellets were resuspended in 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample buffer (2× NuPAGE LDS sample buffer [Invitrogen] with 10% dodecyl sulfate [final concentration] and 0.1 M dithiothreitol).
To assay for cell-associated PrP-res, cells were washed twice with phosphate-buffered balanced saline and then lysed in lysis buffer. Nuclei and cell debris were removed by centrifugation at 2,700 × g for 5 min at 4°C. The postnuclear supernatant fractions were assayed for total protein using a bicinchoninic acid assay (Pierce). Samples normalized for total protein (usually 100 to 120 μg/digest) were digested with 20 μg/ml PK for 1 h. PK digestion was terminated by the addition of 2 mM Pefabloc SC and incubation on ice for 5 min. PrP-res was recovered by PTA precipitation as described above or by ultracentrifugation as previously described (14). The pellets were resuspended in 1× sample buffer.
Samples were separated on 10% Bis-Tris NuPAGE gels in MES (morpholineethanesulfonic acid) running buffer (Invitrogen). Immunoblot detection of PrP was performed as described elsewhere (25) using a mouse-human recombinant anti-PrP monoclonal antibody Fab (D13; InPro Biotechnology) at a 1:10,000 dilution, followed by an alkaline phosphatase-conjugated goat anti-human immunoglobulin G Fab secondary antibody (Sigma) at a 1:10,000 dilution.
Cell blot analysis.
Cell blot analysis was performed essentially as described elsewhere (11, 16) with the primary and secondary antibodies described above. SN56 cells can grow as clumps of cells when undifferentiated, but these clumps disperse when the cells are differentiated. To facilitate cell blot analysis by inducing dispersion of the cells, cultures were differentiated for 1 day prior to cell blotting.
Cell viability assay.
SN56 cells were treated with various PrP-res preparations or buffer controls exactly as described for cell infections. After 48 h of treatment, the cultures were assayed for cell viability using a commercial assay for ATP (Vialight, Cambrex) according to the manufacturer's instructions. ATP concentrations in the cultures were determined from an ATP standard curve.
Fluorescence microscopy.
Fluorophore-tagged microsomes were prepared by incubating microsomes (13 mg/ml total protein; 100 μl) with 1 mg/ml of a primary amine-reactive fluorescent dye (Alexa Fluor 568; succinimidyl ester) diluted from a 10-mg/ml stock in dimethyl sulfoxide. The labeling reaction was incubated at room temperature in the dark for 1 h with periodic mixing and then stored overnight at 4°C in the dark. The reaction mixtures were then centrifuged for 30 min at 21,000 × g. The pellet was resuspended in sterile PBS with 10 mM glycine and incubated at room temperature in the dark for 10 min to wash the microsomes and quench any unreacted labeling reagent. The microsomes were recovered by centrifugation as described above and washed again with sterile PBS-glycine solution. After a final centrifugation, the microsomes were resuspended in sterile PBS. For scrapie brain microsomes, the PrP-res content was quantified as described above. Fluorophore-tagged purified PrP-res was prepared as described for microsomes with the exception that the protein concentration in the labeling reaction was 0.5 mg/ml.
SN56 cells plated at 5% confluence and differentiated for 1 day were treated with fluorescent microsomes essentially as described above. The cultures were differentiated to induce dispersion of the cells and increase neurite formation to facilitate microscopic analysis. Cultures subjected to these treatments have been shown to be susceptible to infection (37). Fluorescent scrapie microsomes containing 16 ng of PrP-res or an equivalent amount (based on total protein) of fluorescent normal microsomes were added to the cells. After incubation for 1 h, live cells were analyzed by confocal microscopy. After 4 h, the medium was replaced with serum-containing OptiMEM and the cells were analyzed every day thereafter for as long as 14 days. Imaging was preformed using a Perkin-Elmer UltraView Spinning-Disk confocal system connected to a Nikon Eclipse TE2000-S microscope with an oil immersion objective (60×; 1.4 numerical aperture) or a Bio-Rad MRC 1024 laser scanning confocal system coupled to a Zeiss microscope with a water immersion objective (40×; 1.2 numerical aperture). Image processing and analysis were performed with Lasersharp (Bio-Rad), Confocal Assistant, Adobe Photoshop, and Image J software.
RESULTS
SN56 cells are susceptible to infection with three mouse scrapie strains.
SN56 cells are a mouse cholinergic septal neuronal cell line (9, 23). To evaluate SN56 cells as a new model for TSE infection, the cells were exposed to scrapie agent inocula in the form of crude brain microsome fractions or purified PrP-res prepared from mice infected with various strains of mouse-adapted scrapie agent. The microsome fraction contains membrane vesicles that may be derived from all types of cellular membranes, including plasma, synaptosomal, and endoplasmic reticulum membranes. For convenience, we refer to this fraction as “microsomal.” The cells were then passed at least five times, each at a 1:10 dilution, to allow dilution of the input PrP-res below the detection limit by immunoblotting and to verify measurement of persistent (i.e., maintained over multiple passes) as opposed to transient PrP-res formation. As shown in Fig. 1, persistent propagation of PrP-res was observed in cells treated with preparations containing the Chandler (Fig. 1A, lanes 1 to 4) and 22L (Fig. 1A, lanes 7 to 9) strains, indicating the cells supported scrapie agent replication. Levels of sustained PrP-res formation positively correlated with the quantity of PrP-res in the inocula (e.g., note the dose-dependent decrease in PrP-res signal for infections initiated with decreasing amounts of PrP-res in Fig. 1A, lanes 7 to 9), with the exception of cells treated with 100 ng or 20 ng of PrP-res, as contained in Chandler microsomes (Fig. 1A, lanes 1 and 2). Exposure to brain microsomes prepared from mice infected with the mouse-adapted strain 87V or hamsters infected with the 263K strain failed to induce persistent PrP-res formation (data not shown). PrP-res formation was stable in SN56 cells, as high PrP-res levels were detected by immunoblotting for Chandler-infected cells passaged at least 60 times (e.g., Fig. 1B, lane 10). Even for Chandler- and 22L-infected cultures initially producing low levels of PrP-res, we routinely observed an increase in PrP-res production with continued passage until the cultures reached an apparent plateau (∼2 to 4 ng PrP-res per 100 μg total lysate protein). A similar increase in the PrP-res level with passage was observed for ME7-treated cells (Fig. 1B, lane 2 versus lane 4), although the apparent plateau of PrP-res production occurred at a much lower level (78 pg PrP-res per 100 μg total protein at passage 17 [data not shown]; 50 pg PrP-res per 100 μg total protein at passage 27 [Fig. 1B, lane 7]), at least for our longest observation period thus far (27 passages). This shows that SN56 cells were also susceptible to ME7 infection, albeit at low efficiency of PrP-res formation. Semiquantitative cell blots (11, 16) of cells in cultures infected with 20 ng of PrP-res showed that the vast majority (>80%) of cells in Chandler- and 22L-infected cultures were producing PrP-res (Fig. 1C). Together, our data show SN56 cells are a new, highly susceptible model of TSE infection.
FIG. 1.

SN56 cells are susceptible to infection with three mouse-adapted scrapie strains. (A and B) SN56 cells were treated with various amounts of PrP-res as contained in crude brain microsome fractions (Chandler [A, lanes 1 to 4] or ME7 [B, lanes 1 to 4 and 7 and 8] or purified form (22L [A, lanes 7 to 9]) from mouse-adapted scrapie-infected animals. Mock-infected control cultures were treated with normal brain microsomes (A, lane 5) normalized for total protein versus a scrapie brain microsome sample (A, lane 2) or a buffer control (A, lane 6; B, lanes 5, 6, and 9). After the indicated number of passes, the cells were assayed for PrP-res by immunoblotting. Chandler-infected cells (from A, lane 2) were analyzed at passage 60 for comparison with ME7-infected cells in panel B (lane 10). Purified PrP-res standards (B, lanes 11 to 13) were used to provide semiquantitative estimates of PrP-res levels. The lanes in panel A represent cell equivalents from 1 well of a 24-well plate (∼150 to 160 μg total cell lysate protein). In panel B, the lanes represent 500 μg (lanes 1 to 6), 733 μg (lanes 7 to 9), and 100 μg (lane 10) of cell lysate protein/lane. Molecular mass markers are indicated in kDa. Brackets indicate PrP-res bands. (C) Cell blot analysis of infected cells. Cells infected with 20 ng of PrP-res from panel A (Chandler, passage 14; 22L, passage 11) were assayed for PrP-res (left). The membrane was then stained with ethidium bromide to visualize transfer of the cells (right).
Detection of PrP-res in the culture supernatant of infected SN56 cells.
Conditioned medium from other scrapie-infected neuroblastoma cell lines has been shown to initiate infection in uninfected cells (18, 48). To determine whether PrP-res was released from infected SN56 cells, we analyzed conditioned media from both Chandler-infected and uninfected cells with and without PK treatment, followed by concentration via PTA precipitation. The samples were also treated with detergent to solubilize any membranes that might be present. PrP was detected in the culture supernatants from both infected and uninfected cells prior to protease treatment, although additional lower-molecular-mass bands were present in the samples from infected cells (Fig. 2A, lanes 1 and 2). Although PTA was first reported to specifically precipitate PrP-res (47), we and others (55) have observed that PrPC can also be precipitated by PTA under certain conditions. However, protease-resistant PrP was only detected in culture media from infected cells (Fig. 2A, lanes 3 and 4). The PK-resistant PrP bands corresponded well in both apparent molecular mass and abundance to the lower-molecular-mass species present in the PK− sample (Fig. 2A, compare lanes 1 and 3), providing evidence that the PrP-res was secreted in an N-terminally truncated form. Quantitative analyses using purified PrP-res standards showed that the secreted PrP-res comprised approximately 2 to 4% of the cell-associated PrP-res (data not shown) and about 27% of the total culture supernatant PrP recoverable by PTA precipitation.
FIG. 2.

Culture supernatant from Chandler-infected cells contains PrP-res and infectivity. (A) Immunoblot detection of PrP-res in culture supernatants of infected cells. Culture supernatants of infected (Sc+, lanes 1 and 3) or uninfected (Sc−, lanes 2 and 4) cells (at passage 14) were assayed for total PrP (PK−, lanes 1 and 2) or PrP-res (PK+, lanes 3 and 4) using PTA precipitation. (B and C) Immunoblot analysis of cells infected by treatment with culture supernatants. Cells were infected in duplicate wells and passaged independently. After multiple passages, the cells were assayed for PrP-res. Lanes 1 and 2 (B), 1 to 4 (C), and 9 and 10 (C) represent cells treated with culture supernatants from Chandler-infected cells. Lanes 3 and 4 (B), 5 to 8 (C), and 11 and 12 (C) represent cells treated with culture supernatants from uninfected cells. Cells from 1 well of a 6-well plate were loaded per lane in panel B, while cells from 1 well of a 24-well plate were loaded per lane in panel C. Brackets indicate PrP-res bands. The results are representative of three independent experiments using three different culture supernatants, each performed in duplicate. The duplicate samples from one experiment are shown in panels B and C.
Culture supernatants from infected cells initiate TSE infection in recipient cells.
Having established that the culture supernatants of infected SN56 cells contain PrP-res, we next tested whether these supernatants could be used to initiate a new infection in uninfected SN56 cells. Recipient cells were incubated for 2 days with media previously conditioned for 2 days in cultures of either uninfected or Chandler-infected SN56 cells. The cells were passaged normally thereafter and periodically assayed for PrP-res by immunoblotting. In some experiments, weak PrP-res signals were detected at relatively early passes (<10), but it usually required assay of more cell equivalents (Fig. 2B, lanes 1 and 2). After several weeks of culturing (≥20 passes), high levels of PrP-res were detected only in recipient cells treated with culture supernatants from infected cells (Fig. 2C). Parallel wells without recipient cells that received only culture supernatants were consistently free of cells and debris, indicating that the supernatants were successfully cleared by our centrifugation treatment (data not shown). Based on the concentration of PrP-res in the culture media (data not shown), we estimate that these infections were initiated with extremely low levels of PrP-res (10 to 15 pg). As observed in other infection experiments described in this study, an increase in the PrP-res signal occurred with continued passage until a maximum level (usually 2 to 4 ng of PrP-res/∼100 μg of cell lysate protein for Chandler-infected cells) was achieved. These data demonstrate that infected SN56 cells release PrP-res that can be used to infect recipient cells and raise the possibility that released PrP-res may mediate the intercellular spread of TSE infection in SN56 cultures.
Enhanced induction of persistent PrP-res formation in SN56 cells by scrapie brain microsomes versus purified PrP-res.
To assess the effects of different inoculum preparations on TSE infection, we compared the efficiency of induction of persistent PrP-res formation by scrapie brain microsomes (as a source of membrane-associated PrP-res) with that of PrP-res purified from infected animals by multiple detergent extractions (as a source of membrane-free PrP-res). PrP-res concentrations in the samples were determined by immunoblot analysis using a previously characterized PrP-res preparation to create a standard curve for comparison (Fig. 3A). SN56 cells were treated with amounts of Chandler PrP-res equal to those contained in the two preparations and, after several passages, were monitored for PrP-res. Levels of PrP-res production were compared as an estimate of the infection level. Surprisingly, scrapie microsomes exhibited a substantially enhanced efficiency at initiating PrP-res formation per ng of PrP-res in the inoculum (Fig. 3B and C, compare lanes 1 to 4 with lanes 7 to 10). There were no detectable signals in cells infected with purified PrP-res at either passage 6 or 8, even when images were viewed with saturating intensity settings to detect any possible bands (data not shown). To obtain an estimate of the differential infection efficiencies of the two forms of PrP-res, we first compared the band intensities for equivalent amounts of input PrP-res at the earliest passage assayed that showed detectable amounts of PrP-res (Fig. 3D, compare lanes 1 and 7, 2 and 8, and 3 and 9). By this method of analysis, microsomes were 18- to 35-fold more efficient than purified PrP-res at inducing sustained new PrP-res formation (Fig. 3E, compare gray bars with open bars within each amount of input PrP-res). In addition, comparison on the basis of titration to determine the minimal amount of input PrP-res required to produce detectable PrP-res at an early pass (e.g., pass 6) suggested that microsomes are ≥20-fold more efficient (Fig. 3B, compare lane 3 with lane 7). Regardless of the method of analysis, our data show that scrapie microsomes have enhanced efficiency at initiating persistent PrP-res production compared with purified PrP-res.
FIG. 3.
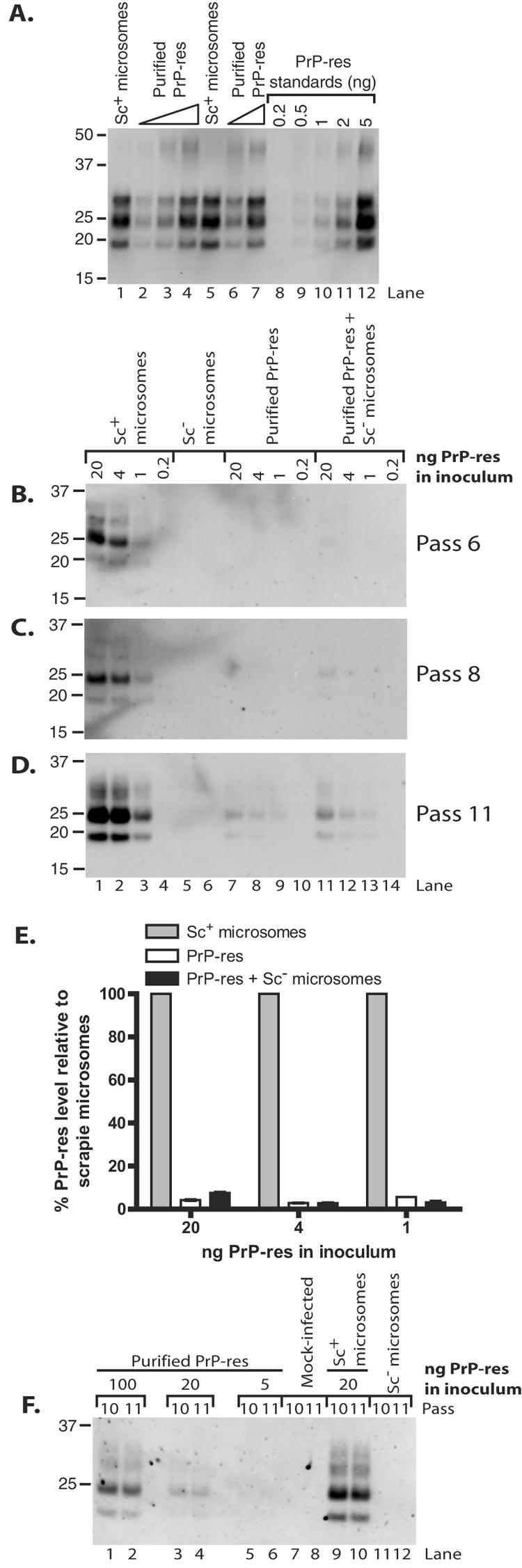
More efficient infection of SN56 and N2a cells by scrapie brain microsomes than by purified PrP-res. (A) Immunoblot quantitation of Chandler PrP-res in purified (lanes 2 to 4 and 6 and 7) and microsome (lanes 1 and 5) preparations. Twofold dilutions of purified PrP-res samples were loaded. Duplicate samples for each preparation are shown. A standard curve was plotted using PK-digested PrP-res standards that were characterized previously (lanes 8 to 12). (B to D) SN56 cells were treated with the indicated amounts of Chandler PrP-res as contained in scrapie (Sc+) microsomes (lanes 1 to 4) or purified from brains of infected animals (lanes 7 to 10). Some infections with purified PrP-res were supplemented with microsomes from normal (Sc−) animals (lanes 11 to 14) in quantities normalized for total protein to the corresponding scrapie microsome samples. The cells were treated with normal microsomes normalized for protein content to 20 ng (lane 5) or 4 ng (lane 6) of scrapie microsomes as a control. At the indicated passages (B to D), cells were assayed (100 μg protein/lane) by immunoblotting for PrP-res. Lane numbers in panel D apply to panels B and C also. (E) Quantitation of PrP-res in panel D. The PrP-res level for each scrapie microsome infection was set at 100%. The results are expressed as percent PrP-res signal for each purified PrP-res sample (lanes 7 to 9 and 11 to 13) relative to an infection initiated with the same amount of PrP-res as contained in scrapie brain microsomes. The error bars indicate the range (n = 2). (F) N2a cells were treated with the indicated amounts of Chandler PrP-res as contained in scrapie microsomes (lanes 9 and 10) or purified from brains of infected animals (lanes 1 to 6). Mock-infected control cells were treated with a buffer control (lanes 7 and 8) or normal microsomes (lanes 11 and 12). At the indicated passages, the cells were assayed by immunoblotting for PrP-res. Loading of comparable amounts of protein was verified by comparing total PrP levels in aliquots of the cell lysates not treated with PK (data not shown). The lanes correspond to protein in 99/100 cell equivalents from one well of a six-well plate.
Addition of normal microsomes to purified PrP-res does not alter levels of long-term PrP-res formation.
These results raised the question of whether the enhanced activity of scrapie microsomes was simply due to the presence of microsomes during infection. To address this question, we conducted infections in which purified PrP-res preparations were supplemented with brain microsomes prepared from normal mice in amounts normalized for total protein to those contained in the corresponding scrapie microsome samples. The samples were vortexed, but no treatment was intentionally applied to facilitate the incorporation of the PrP-res into the microsomal membranes. As seen in Fig. 3, addition of normal microsomes to infections with purified PrP-res samples did not dramatically alter the infection efficiency (Fig. 3B to D, compare lanes 7 to 10 with lanes 11 to 14). We did note that addition of normal microsomes to the infection with 20 ng of purified PrP-res gave a small increase in infection efficiency, allowing a weak but detectable PrP-res signal as early as passage 6 (Fig. 3B, lane 11). However, this may have been attributable to microsome-dependent alleviation of a low level of cytotoxicity induced by detergent (sulfobetaine) in the PrP-res storage buffer that was observed only in infections with 20 ng of purified PrP-res. Again, based on comparison of the band intensities for equivalent amounts of input PrP-res (Fig. 3D, compare lanes 1 and 11, 2 and 12, and 3 and 13), microsomes were ∼13- to 36-fold more efficient than purified PrP-res (Fig. 3E) and ≥20-fold more efficient when analyzed on the basis of titration (Fig. 3B). No obvious effect on cell viability was observed using ≤4 ng of PrP-res or in wells treated with microsomes alone (data not shown). Thus, these data provide evidence that it is primarily the context of PrP-res as associated with scrapie microsomes rather than the presence of microsomes themselves that enhances the ability of microsomal PrP-res to induce long-term PrP-res production.
Enhanced induction of persistent PrP-res formation in N2a cells by scrapie brain microsomes versus purified PrP-res.
To determine whether the elevated ability of microsomes to induce sustained PrP-res formation was applicable to other cells susceptible to TSEs, we conducted similar studies in a highly susceptible N2a cell line generated by pentosan polysulfate-mediated curing of a Chandler-infected clone that produces high levels of PrP-res (32). As observed in SN56 cells, infections initiated with scrapie microsomes were far more efficient at inducing PrP-res formation than those with purified PrP-res when normalized for input PrP-res (Fig. 3F, lanes 3 versus 9 and 4 versus 10). Addition of normal microsomes to infections with purified PrP-res also did not affect the levels of long-term PrP-res production induced in this cell model (data not shown). For 20 ng of PrP-res in the inoculum, microsomes were ∼16-fold more efficient as measured at passage 5 (data not shown), but this difference decreased somewhat with further passage (12-fold at passage 10 and 10-fold at passage 11), suggesting that a small relative increase in the PrP-res signal of the purified PrP-res-infected cells may have occurred. Also, as seen above, infection with increased amounts of purified PrP-res induced higher levels of persistent PrP-res formation (Fig. 3F, lanes 1 to 6) even in spite of some cytotoxicity occurring at the highest amount of PrP-res tested (data not shown). In any case, these studies show that the enhanced induction of persistent PrP-res formation by microsome-associated PrP-res is common to at least two cell culture models of TSE infection.
Removal of detergent from purified PrP-res does not improve infection efficiency.
The initial experiments described above were performed without removing the sulfobetaine from the purified PrP-res due to the potentially beneficial role of detergents in GPI painting (2, 41, 56). To address the potential inhibitory effects of this detergent, infection experiments were performed using purified PrP-res that had been prewashed with PBS to remove the detergent. Removal of the detergent did not increase the levels of sustained PrP-res formation induced by purified PrP-res in the presence or absence of normal microsomes and suggested at least a 20-fold difference compared to scrapie microsomes on the basis of titration (Fig. 4). Effective removal of the bulk of the detergent by the washing procedure was suggested by the absence of cytotoxicity in the PBS-washed PrP-res preparation (Fig. 5). Therefore, our data demonstrate that the low efficiency of purified PrP-res in initiating persistent PrP-res formation is not related to the presence of sulfobetaine but rather is a property of the purified PrP-res itself.
FIG. 4.

Removal of detergent from purified PrP-res does not improve infection efficiency. SN56 cell infections were conducted and analyzed as described in the legend to Fig. 3 with the exception that the purified PrP-res was prewashed with PBS to remove the detergent. SN56 cells were treated with the indicated amounts of Chandler PrP-res as contained in scrapie (Sc+) microsomes (lanes 1 to 4) or purified from brains of infected animals (lanes 7 to 10). Some infections with purified PrP-res were supplemented with microsomes from normal (Sc−) animals (lanes 11 to 14) in quantities normalized for total protein to the corresponding scrapie microsome samples. The cells were treated with normal microsomes normalized for protein content to 20 ng (lane 5) or 4 ng (lane 6) of scrapie microsomes as a control. At the indicated passages (A and B), the cells were assayed (100 μg protein/lane) by immunoblotting for PrP-res. Lane numbers in panel B apply to panel A also. (C) Quantitation of PrP-res in panel B. The results are expressed as percent PrP-res signal for each purified PrP-res sample (lanes 7 and 8 and 11 to 13) relative to an infection initiated with the same amount of PrP-res as contained in scrapie brain microsomes. The PrP-res level for each scrapie microsome infection was set at 100%. The error bars indicate the range (n = 2).
FIG. 5.

Scrapie microsomes and purified PrP-res are not cytotoxic to SN56 cells during acute infection. SN56 cells were treated with either 20 ng (open bars) or 4 ng (black bars) of PrP-res in the form of scrapie microsomes or purified PrP-res preparations as described for cell infections in the legends to Fig. 3 and 4. Purified PrP-res preparations without (PBS) and with (Det) detergent were tested. Mock treatments with either PBS or PBS with the detergent sulfobetaine (0.00055% [open bar] and 0.00011% [black bar]) were included as buffer controls. After 48 h, the cultures were assayed for ATP as a measure of cell viability. The values indicate the mean ± standard deviation of triplicate wells. The results are representative of three independent experiments, each performed in triplicate.
Purified PrP-res and scrapie microsomes do not affect SN56 cell viability during infection.
Although the mechanism of neurodegeneration in TSE diseases is not understood, there is some evidence that purified PrP-res may be directly cytotoxic to cells, albeit using higher concentrations of PrP-res than employed in the current study (24). To determine whether the different efficiency of induction of PrP-res formation by scrapie microsomes versus purified PrP-res was due to differential cytotoxicity, cell viability was assayed in cultures treated with the PrP-res preparations as described for the infection experiments above. The results were completely consistent with those based on visual inspection of the cultures, showing no evidence of a significant effect on cell viability for any treatment condition, with the exception of cells treated with 20 ng of purified PrP-res containing low levels of detergent (Fig. 5). However, a similar reduction in cell viability was observed for cells treated with an equal concentration of the detergent alone [Fig. 5, Mock (Det)], suggesting that the cytotoxic effect was directly mediated by the detergent. Thus, these observations show that the enhanced induction of sustained PrP-res formation by scrapie microsomes versus purified PrP-res could not be attributed to differences in cytotoxicity of the two preparations.
SN56 cells bind aggregates of purified PrP-res.
Since the purified PrP-res may be more highly aggregated than that associated with microsomes, we considered the possibility that the reduced induction of sustained PrP-res formation by purified PrP-res was attributable to inefficient binding/uptake by SN56 cells. To investigate this issue, we labeled purified PrP-res with a primary amine-reactive Alexa Fluor dye and used this labeled PrP-res to infect SN56 cells as described above. Analysis of the cultures after 2 days by fluorescence microscopy showed that ∼80% of the cells in the culture were positive for at least one fluorescent aggregate, but most cells contained several aggregates (Fig. 6A to C). Even at 4 days postinfection, the majority of the cells in the culture (∼70%) still contained at least one fluorescent aggregate (data not shown). After further passage of the cells, we verified that cultures treated with Alexa Fluor-labeled PrP-res were infected (data not shown). These data suggest that at least for infections with the greatest amounts of purified PrP-res we tested (20 ng), most of the cells were intimately exposed to the input PrP-res and thus had the potential to become infected directly.
FIG. 6.

SN56 cells bind aggregates of purified PrP-res. SN56 cells were treated with 20 ng of purified PrP-res either labeled with a primary amine-reactive dye (Alexa Fluor 568; succinimidyl ester) (A to C) or unlabeled (D and E) and examined by fluorescence microscopy after 2 days. Panels A and D are fluorescent images. Panels B and E are phase-contrast images. Panel C is a merged image of panels A and B.
Infection with immobilized microsomes induces sustained PrP-res formation less efficiently than microsomes in suspension.
Data from our previous cell-free conversion studies (38), in addition to infection studies (19, 57, 58), have shown that PrP-res/TSE infectivity immobilized onto solid surfaces with the assistance of drying can efficiently induce conversion of PrPC and initiate infections in cell culture and animals. We were interested in comparing the infection efficiencies of similarly immobilized forms of PrP-res with PrP-res added in suspension. For this purpose, various amounts of scrapie microsomes were dried in a 96-well cell culture plate over several hours. SN56 cells were then plated in these wells, cultured for 5 days, and then passaged every 4 days thereafter. Parallel infections were conducted using scrapie microsomes or purified PrP-res in suspension as described above. As shown in Fig. 7, suspended scrapie microsomes were more efficient than dried microsomes at initiating long-term PrP-res formation. This distinction was most evident at an early passage in the experiment, which suggested that microsomes in suspension were 6- to 15-fold more efficient based on semiquantitative estimates of the PrP-res bands (Fig. 7A, compare lanes 1 to 3 with lanes 8 to 12). The differences tended to decrease with extended passage of the infected cells, indicating that relative PrP-res production was increasing in the cells infected with dried microsomes, perhaps due to the spread of infection to uninfected cells (Fig. 7B to D). A similar effect was observed in cells infected with purified PrP-res (Fig. 7A to C, compare lanes 1 and 2 with lanes 5 and 6, and D). However, even after 17 passages, cells infected with 1 ng of dried microsome-associated PrP-res exhibited ∼26-fold lower levels of PrP-res versus the positive control microsomes in suspension (Fig. 7D). Nevertheless, the dried microsomes induced sustained PrP-res formation more efficiently than purified PrP-res for all quantities of PrP-res used in the inocula (Fig. 7A to C, compare lanes 5 to 7 with 8 to 10, and D). Altogether, of the three methods of infection tested, we found that treating cells with scrapie microsomes in suspension induces the most efficient PrP-res production, suggesting that immobilizing microsome-associated PrP-res to surfaces reduces infection efficiency in this model.
FIG. 7.

Infection with dried microsomes is less efficient than with microsomes in suspension. SN56 cells were plated onto various amounts of scrapie (Sc+) microsomes (lanes 8 to 12) or normal (Sc−) microsomes (lane 13) which had been immobilized by drying onto a tissue culture plate. The cells were passaged normally thereafter. Control infections with scrapie microsomes (lanes 1 to 3), normal microsomes (lane 4), or purified PrP-res (lanes 5 to 7) were conducted as described in the legend to Fig. 3. At the indicated passages (A to C), the cells were assayed (panel A, 93 μg protein/lane; panel B, 113 μg protein/lane; panel C, 120 μg protein/lane) by immunoblotting for PrP-res. The lane numbers in panel C also apply to panels A and B. (D) Semiquantitative estimation of PrP-res in panels B and C. The PrP-res level for each infection with scrapie microsomes in suspension was set at 100%. The results are expressed as percent PrP-res signal for each purified PrP-res sample (lanes 5 and 6) or dried microsome sample (lanes 8 to 10) versus an infection initiated with the same amount of PrP-res as contained in scrapie brain microsomes in suspension. The error bars indicate standard deviations (n = 3).
Efficient internalization and trafficking of fluorescent scrapie microsomes in SN56 cells.
Having observed the increased infection efficiency of scrapie microsomes, we became interested in the fate of the microsomes after addition to the cells. To monitor the uptake of microsomes, we labeled scrapie microsomes with a primary amine-reactive Alexa Fluor dye to covalently label microsomal molecules. Analysis of both normal and scrapie brain fluorescent microsomes by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by fluorescent imaging on a phosphorimaging system, verified covalent labeling of many microsomal proteins (data not shown). In labeled scrapie microsome samples enriched for PrP-res by PK digestion and PTA precipitation, weak bands of apparent molecular mass similar to that of PrP-res were detected, suggesting at least some of the PrP-res molecules were labeled, although they represented a small minority of the total labeled proteins in the sample (data not shown). We then added the labeled microsomes to SN56 cells and analyzed their subsequent distribution over several days by confocal microscopy. Within 1 h after addition to the cells, fluorescent microsome material associated with the surface of the cells in large highly fluorescent particles that likely corresponded to aggregates of microsomal vesicles (Fig. 8A). A small amount of fluorescent material was internalized in small vesicles (Fig. 8A), which were verified as internal by analysis of serial optical sections (data not shown). With further incubation for 1 to 3 days, the relative distribution and appearance of the fluorescent material changed. We observed a progressive increase in the amount of internalized fluorescence in small vesicles with a concomitant decrease in the proportion of large, cell surface microsomal aggregates, suggesting that the size of the microsomal aggregates had been reduced in some way prior to and/or during internalization (Fig. 8C and E). By 3 days, most of the fluorescent material was found internalized in vesicles, many of which were located in neuritic processes. An example of this pattern of distribution is shown in Fig. 8E for cells 4 days after treatment with microsomes. Similar results were obtained using Alexa Fluor-labeled normal microsomes, indicating that the distribution observed was not scrapie dependent (data not shown). Beyond 3 days of incubation, no further change in the distribution of fluorescent material was observed, although there was a slow decrease in the intensity of the fluorescence in cultures maintained longer than 7 days (data not shown). Thus, microsomes are efficiently internalized and processed by SN56 cells, a property that may contribute to the higher infection efficiency of microsome-associated PrP-res.
FIG. 8.

Efficient internalization and trafficking of fluorescent scrapie microsomes. Scrapie microsomes were labeled with Alexa Fluor 568 as in Fig. 6 and added to SN56 cells. The cells were imaged by laser scanning confocal microscopy. Representative images after 1 h (A and B), 1 day (C and D), and 4 days (E and F) are shown. Panels A, C, and E are fluorescent images. Panels B, D, and F are the corresponding differential interference contrast images. In panels A, C, and E, the large arrows indicate probable aggregates of microsomal material, while the small arrows indicate small internalized microsomal material. Some unbound microsomal material is also visible in panel A. Scale bar, 20 μm. The results shown are representative of cumulative data from three independent infection experiments with at least 49 individual cells examined on each day within the first 5 days of infection.
Decreased PrP-res formation in infected SN56 cells during differentiation.
Since we were interested in using differentiated SN56 cells for studies of PrP-res formation and trafficking, we tested the effects of differentiation on PrP-res and total PrP formation in infected and uninfected SN56 cells. Surprisingly, in three separate experiments (each assayed in triplicate with a standard deviation of <10%), PrP-res formation decreased by approximately 1.6- to 2.6-fold for Chandler-infected cells differentiated for 4 days compared to undifferentiated controls (Fig. 9, lanes 1 versus 2 and 3 versus 4). This effect was not due to a change in the level of PrPC expression, as infected and uninfected cultures produced similar amounts of total PrP irrespective of the differentiation state (Fig. 9, compare lanes 5 and 6 and lanes 7 and 8). Also, we saw no overt effect of differentiation on the viability of either scrapie-infected or uninfected cells (data not shown). Differentiation was observed to induce a slightly increased heterogeneity within the population of fully glycosylated PrP molecules (Fig. 9, lanes 5 to 8), suggesting there was a change in the glycosylation and/or trafficking of PrPC. Differentiation-associated changes to PrPC glycoform heterogeneity have been observed in another neuroblastoma cell line (42). These findings suggest that SN56 cells may be a useful model to investigate scrapie infection-associated alterations in neuronal cells at multiple stages of development.
FIG. 9.

Reduced PrP-res formation in differentiated, infected SN56 cells. Chandler-infected (lanes 1 to 6) or uninfected (lanes 7 and 8) SN56 cells were incubated in complete medium (−) or in serum-free medium with cyclic AMP (+) to induce differentiation. The cells were plated to achieve similar densities at the time of harvest (as verified by protein assays). After 4 days, the cells were assayed by immunoblotting for PrP-res (PK+) (lanes 1 to 4) and total PrP (PK−) (lanes 5 to 8). Lanes 1 to 4 contained 80 μg of protein/lane, and lanes 5 to 8 contained 10 μg of protein/lane. Brackets indicate PrP-res bands (left) or total PrP bands (right). The arrow indicates differentiation-associated increase in heterogeneity of the fully glycosylated PrP band. The results are representative of three independent cultures, each assayed in triplicate. Examples from two separate experiments are shown in lanes 1 to 4.
DISCUSSION
Our previous work provided very limited data indicating SN56 cells could be infected with the Chandler strain of the TSE agent (37). However, the present study constitutes the first detailed characterization of scrapie infection of the SN56 neuronal cell line. SN56 cells are a cholinergic mouse septal neuronal cell line (9, 23). The cells are well characterized and possess a number of neuronal features, including synaptic vesicle proteins (4), neuronal-type calcium channels (33), and production of vast neuritic networks (9), which are induced upon differentiation of the cells. In contrast to the commonly used N2a cell line (11), no subcloning was required to isolate susceptible cells. The discovery that SN56 cells are susceptible to scrapie infection allowed their use as a model system more closely related to neurons to examine the process of infection and intercellular spread of TSE agents. Although our data to this point do not allow us to exclude the possibility that the reduction of PrP-res signal on differentiation is due to specific death of PrP-res-producing cells (which would be highly interesting as a model of PrP-res-induced neurotoxicity), it seems difficult to believe, given the high percentage of infected cells in the cultures and the lack of a substantial and concomitant scrapie infection-specific loss of cells on differentiation. Our studies have also shown that microsome-associated PrP-res induces much greater levels of persistent PrP-res formation than purified PrP-res.
Our previous cell-free conversion experiments (5, 6) led us to consider that PrP-res aggregates or membrane microparticles (e.g., exosomes) released from infected cells might mediate transfer of TSE infectivity to neighboring uninfected cells. This also led us to consider models of TSE infection that involved the transfer of PrP-res in either membrane-associated forms or as free aggregates via a process described as “painting” (41). Consistent with this notion is the observation that culture supernatants from infected neuroblastoma cells can initiate infection in new cells, but these studies did not determine whether PrP-res was present in the culture supernatants (48). Our data here show that very small amounts of PrP-res released from infected SN56 cells (Fig. 2A) are sufficient to initiate sustained PrP-res formation in recipient cells (Fig. 2B), suggesting the secreted PrP-res may have a very high infectivity per amount of PrP-res.
This raised the question of the biophysical nature of the secreted PrP-res and whether it might be associated with released membrane particles. One recently described form of released membrane particle is called an exosome (for reviews, see references 15, 17, 50, and 51). Exosomes are small membrane vesicles released from cells by the fusion of multivesicular bodies (MVBs) with the plasma membrane. MVBs are late endosome-like compartments in which vesicles destined to become exosomes form by invagination of the MVB-limiting membrane. During the preparation of this paper, Fevrier et al. (18) reported that exosomes containing PrPC and PrP-res are released from epithelial and glial cell lines. Culture supernatants, as well as exosome preparations, from the culture media were infectious for cultured cells and animals (18). Culture supernatants/exosomes might then serve as a tractable source of material for the characterization of TSE agents in a naturally generated, biologically significant form without the use of detergents. It is possible that the PrP-res released from infected SN56 cells is also associated with exosomes. These findings are interesting given a proposed role for MVBs in PrP-res biosynthesis several years ago (39).
Why might microsome-associated PrP-res be more infectious than membrane-extracted PrP-res? One possible explanation is related to the different aggregation states of PrP-res in the two preparations. When associated with membranes, either naturally as produced in the brain (40) or after reconstitution of purified material into synthetic liposomes (21, 22), PrP-res molecules form diffuse aggregates that can be detected by immunoelectron microscopy (27-29). However, detergent extraction of these membranes in combination with limited proteolysis results in the formation of larger rod-like polymers of PrP-res (21, 22, 40). Although we did not use proteases in the purification of the PrP-res used in this study, electron micrographs of our preparations do show both fibrillar and amorphous aggregates (data not shown). In the context of nucleated polymerization models of PrP conversion (for a review, see reference 12), PrP-res preparations containing smaller aggregates would have a higher concentration of seeds per unit of PrP-res to initiate conversion than those comprised of larger aggregates, and thus, the former might be expected to have a higher specific infectivity. Biochemical and infectivity data supporting this proposition have recently been obtained (49). Also consistent with this notion is the observation that infectivity titers of purified PrP-res are 10- to 100-fold higher when the PrP-res is reconstituted into liposomes (21, 22). Unfortunately, we were unable to generate similarly reconstituted PrP-res using protein purified/enriched by a variety of methods for use in our studies. Nevertheless, our data now provide a biochemical explanation for the observations of Gabizon and coworkers.
Another possibility is that there is a more efficient binding and/or internalization of microsomes than with purified PrP-res. The microsome-associated PrP-res might associate with cells more efficiently via interactions of microsomal molecules with cell-surface ligands. Such a comparison would be difficult to perform in this system, though we did verify by using fluorophore-tagged PrP-res that SN56 cells were capable of binding significant quantities of purified PrP-res (Fig. 6A to C) and we have visualized the uptake and trafficking of fluorescent PrP-res coincident with infection (37). Likewise, SN56 cells also bound, internalized, and somehow processed microsomal material in small vesicles and redistributed it to neuritic processes. This process may result in the delivery of microsomes to a compartment in which PrP conversion occurs (13, 53).
Considering our models of the infection process, it is also possible that membrane-associated PrP-res is more efficiently inserted into host cell membranes, perhaps via membrane fusion, than is membrane-free PrP-res, which would be restricted to a “GPI-painting” mechanism that is known to be poorly efficient in vitro (41). This would position the PrP-res in the same membrane as the host cell membrane-associated PrPC, a prerequisite for efficient conversion of membrane-bound PrPC (5, 6). This does not exclude the possibility that infection can also occur as a result of intimate contact of recipient cells with infected cells or surfaces coated with TSE infectivity without transfer of PrP-res (30, 57). However, aggregation of PrPC-containing membranes with separate membranes containing PrP-res in the absence of membrane fusion did not allow efficient formation of new PrP-res (6). We also cannot rule out the possibility that some important component of the TSE agent is lost during purification of the PrP-res. In this event, the infection assay described here might help to identify non-PrP molecules that contribute to TSE infectivity.
Intrigued by the observation that brain homogenate-derived TSE infectivity bound to culture wells could efficiently initiate infection in N2a cells (57), we also compared the infection efficiency of scrapie microsomes bound to culture wells with those added in suspension. As shown in Fig. 7, scrapie microsomes in suspension were significantly more efficient at initiating sustained PrP-res propagation than the dried microsomes, again perhaps due to more efficient internalization or delivery to host cell membranes as outlined above. It should be noted that we did not verify the quantity of PrP-res that remained bound to the culture wells after the microsomes were dried and washed, and thus, it is possible that these cells were exposed to less PrP-res than those treated with scrapie microsomes in suspension. However, data from our previous cell-free conversion studies have shown that PrP-res/TSE infectivity immobilized onto solid surfaces can efficiently induce conversion of PrPC (38). Alternatively, perhaps this is evidence of a polar effect on infection, as has been observed in epithelial cell models of infection (44). In contrast to Weissmann et al. (57), we observed a positive correlation between infection efficiency and the quantity of input brain material (Fig. 7C, lanes 8 to 12), suggesting that in our preparations the presence of other brain proteins was not inhibitory, at least for the quantities of total protein we added to the wells (from 0.2 to 25 μg). It is possible that the inhibitory factors present in brain homogenates are removed during the preparation of the microsomes. We have noted that large quantities of microsomes can be added to cells without significant effects on cell viability (data not shown). In any case, our observations demonstrate the use of microsome preparations as a highly efficient means of initiating infection in cultured cell lines.
Acknowledgments
We thank Andrew Hughson and Gregory and Lynne Raymond for technical assistance. We thank Bruce Chesebro, Derek Dimcheff, and Suzette Priola for critical reading of the manuscript. We also thank Bruce Wainer for providing the SN56 cell line.
A.C.M. received a predoctoral exchange fellowship from CAPES. M.A.M.P is a fellow of the John Simon Guggenheim Memorial Foundation (2004-2005) and receives research support from CNPq, PRONEX-MG, and the American Health Assistance Foundation. This research was supported in part by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases (NIAID).
All animals were treated in accordance with the regulations and guidelines of the Animal Care and Use Committee of the Rocky Mountain Laboratories and the National Institutes of Health.
REFERENCES
- 1.Aguzzi, A., F. L. Heppner, M. Heikenwalder, M. Prinz, K. Mertz, H. Seeger, and M. Glatzel. 2003. Immune system and peripheral nerves in propagation of prions to CNS. Br. Med. Bull. 66:141-159. [DOI] [PubMed] [Google Scholar]
- 2.Angrand, M., A. Briolay, F. Ronzon, and B. Roux. 1997. Detergent-mediated reconstitution of a glycosyl-phosphatidylinositol-protein into liposomes. Eur. J. Biochem. 250:168-176. [DOI] [PubMed] [Google Scholar]
- 3.Archer, F., C. Bachelin, O. Andreoletti, N. Besnard, G. Perrot, C. Langevin, A. Le Dur, D. Vilette, A. Baron-Van Evercooren, J. L. Vilotte, and H. Laude. 2004. Cultured peripheral neuroglial cells are highly permissive to sheep prion infection. J. Virol. 78:482-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbosa, J., Jr., A. R. Massensini, M. S. Santos, S. I. Meireles, R. S. Gomez, M. V. Gomez, M. A. Romano-Silva, V. F. Prado, and M. A. Prado. 1999. Expression of the vesicular acetylcholine transporter, proteins involved in exocytosis, and functional calcium signaling in varicosities and soma of a murine septal cell line. J. Neurochem. 73:1881-1893. [PubMed] [Google Scholar]
- 5.Baron, G. S., and B. Caughey. 2003. Effect of glycosylphosphatidylinositol anchor-dependent and -independent prion protein association with model raft membranes on conversion to the protease-resistant isoform. J. Biol. Chem. 278:14883-14892. [DOI] [PubMed] [Google Scholar]
- 6.Baron, G. S., K. Wehrly, D. W. Dorward, B. Chesebro, and B. Caughey. 2002. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J. 21:1031-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batista, F. D., D. Iber, and M. S. Neuberger. 2001. B cells acquire antigen from target cells after synapse formation. Nature 411:489-494. [DOI] [PubMed] [Google Scholar]
- 8.Birkett, C. R., R. M. Hennion, D. A. Bembridge, M. C. Clarke, A. Chree, M. E. Bruce, and C. J. Bostock. 2001. Scrapie strains maintain biological phenotypes on propagation in a cell line in culture. EMBO J. 20:3351-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blusztajn, J. K., A. Venturini, D. A. Jackson, H. J. Lee, and B. H. Wainer. 1992. Acetylcholine synthesis and release is enhanced by dibutyryl cyclic AMP in a neuronal cell line derived from mouse septum. J. Neurosci. 12:793-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bolton, D. C., R. D. Rudelli, J. R. Currie, and P. E. Bendheim. 1991. Copurification of Sp33-37 and scrapie agent from hamster brain prior to detectable histopathology and clinical disease. J. Gen. Virol. 72:2905-2913. [DOI] [PubMed] [Google Scholar]
- 11.Bosque, P. J., and S. B. Prusiner. 2000. Cultured cell sublines highly susceptible to prion infection. J. Virol. 74:4377-4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caughey, B., G. J. Raymond, M. A. Callahan, C. Wong, G. S. Baron, and L. Xiong. 2001. Interactions and conversions of prion protein isoforms. Adv. Prot. Chem. 57:139-169. [DOI] [PubMed] [Google Scholar]
- 13.Caughey, B., G. J. Raymond, D. Ernst, and R. E. Race. 1991. N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J. Virol. 65:6597-6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caughey, W. S., L. D. Raymond, M. Horiuchi, and B. Caughey. 1998. Inhibition of protease-resistant prion protein formation by porphyrins and phthalocyanines. Proc. Natl. Acad. Sci. USA 95:12117-12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denzer, K., M. J. Kleijmeer, H. F. Heijnen, W. Stoorvogel, and H. J. Geuze. 2000. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 113:3365-3374. [DOI] [PubMed] [Google Scholar]
- 16.Enari, M., E. Flechsig, and C. Weissmann. 2001. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc. Natl. Acad. Sci. USA 98:9295-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fevrier, B., and G. Raposo. 2004. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr. Opin. Cell Biol. 16:415-421. [DOI] [PubMed] [Google Scholar]
- 18.Fevrier, B., D. Vilette, F. Archer, D. Loew, W. Faigle, M. Vidal, H. Laude, and G. Raposo. 2004. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 101:9683-9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flechsig, E., I. Hegyi, M. Enari, P. Schwarz, J. Collinge, and C. Weissmann. 2001. Transmission of scrapie by steel-surface-bound prions. Mol. Med. 7:679-684. [PMC free article] [PubMed] [Google Scholar]
- 20.Follet, J., C. Lemaire-Vieille, F. Blanquet-Grossard, V. Podevin-Dimster, S. Lehmann, J. P. Chauvin, J. P. Decavel, R. Varea, J. Grassi, M. Fontes, and J. Y. Cesbron. 2002. PrP expression and replication by Schwann cells: implications in prion spreading. J. Virol. 76:2434-2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gabizon, R., M. P. McKinley, D. F. Groth, L. Kenaga, and S. B. Prusiner. 1988. Properties of scrapie prion protein liposomes. J. Biol. Chem. 263:4950-4955. [PubMed] [Google Scholar]
- 22.Gabizon, R., M. P. McKinley, and S. B. Prusiner. 1987. Purified prion proteins and scrapie infectivity copartition into liposomes. Proc. Natl. Acad. Sci. USA 84:4017-4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammond, D. N., H. J. Lee, J. H. Tonsgard, and B. H. Wainer. 1990. Development and characterization of clonal cell lines derived from septal cholinergic neurons. Brain Res. 512:190-200. [DOI] [PubMed] [Google Scholar]
- 24.Hetz, C., M. Russelakis-Carneiro, K. Maundrell, J. Castilla, and C. Soto. 2003. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 22:5435-5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horiuchi, M., J. Chabry, and B. Caughey. 1999. Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO J. 18:3193-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ilangumaran, S., P. J. Robinson, and D. C. Hoessli. 1996. Transfer of exogenous glycosylphosphatidylinositol (GPI)-linked molecules to plasma membranes. Trends Cell Biol. 6:163-167. [DOI] [PubMed] [Google Scholar]
- 27.Jeffrey, M., C. M. Goodsir, M. Bruce, P. A. McBride, J. R. Scott, and W. G. Halliday. 1994. Correlative light and electron microscopy studies of PrP localisation in 87V scrapie. Brain Res. 656:329-343. [DOI] [PubMed] [Google Scholar]
- 28.Jeffrey, M., C. M. Goodsir, M. E. Bruce, P. A. McBride, and J. R. Fraser. 1997. In vivo toxicity of prion protein in murine scrapie: ultrastructural and immunogold studies. Neuropathol. Appl. Neurobiol. 23:93-101. [PubMed] [Google Scholar]
- 29.Jeffrey, M., C. M. Goodsir, M. E. Bruce, P. A. McBride, J. R. Scott, and W. G. Halliday. 1992. Infection specific prion protein (PrP) accumulates on neuronal plasmalemma in scrapie infected mice. Neurosci. Lett. 147:106-109. [DOI] [PubMed] [Google Scholar]
- 30.Kanu, N., Y. Imokawa, D. N. Drechsel, R. A. Williamson, C. R. Birkett, C. J. Bostock, and J. P. Brockes. 2002. Transfer of scrapie prion infectivity by cell contact in culture. Curr. Biol. 12:523-530. [DOI] [PubMed] [Google Scholar]
- 31.Klohn, P. C., L. Stoltze, E. Flechsig, M. Enari, and C. Weissmann. 2003. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc. Natl. Acad. Sci. USA 100:11666-11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kocisko, D. A., G. S. Baron, R. Rubenstein, J. Chen, S. Kuizon, and B. Caughey. 2003. New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J. Virol. 77:10288-10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kushmerick, C., M. A. Romano-Silva, M. V. Gomez, and M. A. Prado. 2001. Changes in Ca2+ channel expression upon differentiation of SN56 cholinergic cells. Brain Res. 916:199-210. [DOI] [PubMed] [Google Scholar]
- 34.Lee, K. S., A. C. Magalhaes, S. M. Zanata, R. R. Brentani, V. R. Martins, and M. A. Prado. 2001. Internalization of mammalian fluorescent cellular prion protein and N-terminal deletion mutants in living cells. J. Neurochem. 79:79-87. [DOI] [PubMed] [Google Scholar]
- 35.Liu, T., R. Li, T. Pan, D. Liu, R. B. Petersen, B. S. Wong, P. Gambetti, and M. S. Sy. 2002. Intercellular transfer of the cellular prion protein. J. Biol. Chem. 277:47671-47678. [DOI] [PubMed] [Google Scholar]
- 36.Mack, M., A. Kleinschmidt, H. Bruhl, C. Klier, P. J. Nelson, J. Cihak, J. Plachy, M. Stangassinger, V. Erfle, and D. Schlondorff. 2000. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat. Med. 6:769-775. [DOI] [PubMed] [Google Scholar]
- 37.Magalhaes, A. C., G. S. Baron, K. S. Lee, O. Steele-Mortimer, D. Dorward, M. A. Prado, and B. Caughey. 2005. Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J. Neurosci. 25:5207-5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maxson, L., C. Wong, L. M. Herrmann, B. Caughey, and G. S. Baron. 2003. A solid-phase assay for identification of modulators of prion protein interactions. Anal. Biochem. 323:54-64. [DOI] [PubMed] [Google Scholar]
- 39.Mayer, R. J., M. Landon, L. Laszlo, G. Lennox, and J. Lowe. 1992. Protein processing in lysosomes: the new therapeutic target in neurodegenerative disease. Lancet 340:156-159. [DOI] [PubMed] [Google Scholar]
- 40.McKinley, M. P., R. K. Meyer, L. Kenaga, F. Rahbar, R. Cotter, A. Servan, and S. B. Prusiner. 1991. Scrapie prion rod formation in vitro requires both detergent extraction and limited proteolysis. J. Virol. 65:1340-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medof, M. E., S. Nagarajan, and M. L. Tykocinski. 1996. Cell-surface engineering with GPI-anchored proteins. FASEB J. 10:574-586. [DOI] [PubMed] [Google Scholar]
- 42.Monnet, C., V. Marthiens, H. Enslen, Y. Frobert, A. Sobel, and R. M. Mege. 2003. Heterogeneity and regulation of cellular prion protein glycoforms in neuronal cell lines. Eur. J. Neurosci. 18:542-548. [DOI] [PubMed] [Google Scholar]
- 43.Nishida, N., D. A. Harris, D. Vilette, H. Laude, Y. Frobert, J. Grassi, D. Casanova, O. Milhavet, and S. Lehmann. 2000. Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. J. Virol. 74:320-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paquet, S., E. Sabuncu, J. L. Delaunay, H. Laude, and D. Vilette. 2004. Prion infection of epithelial Rov cells is a polarized event. J. Virol. 78:7148-7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raymond, G. J., and J. Chabry. 2004. Purification of the pathological isoform of prion protein (PrPSc or PrP-res) from transmissible spongiform encephalopathy-affected brain tissue., p. 16-26. In S. A. G. J. Lehmann (ed.), Methods and tools in biosciences and medicine: techniques in prion research. Birkhäuser Verlag, Basel, Switzerland.
- 46.Safar, J., M. Ceroni, P. Piccardo, P. P. Liberski, M. Miyazaki, D. C. Gajdusek, and C. J. Gibbs, Jr. 1990. Subcellular distribution and physicochemical properties of scrapie-associated precursor protein and relationship with scrapie agent. Neurology 40:503-508. [DOI] [PubMed] [Google Scholar]
- 47.Safar, J., H. Wille, V. Itri, D. Groth, H. Serban, M. Torchia, F. E. Cohen, and S. B. Prusiner. 1998. Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 4:1157-1165. [DOI] [PubMed] [Google Scholar]
- 48.Schatzl, H. M., L. Laszlo, D. M. Holtzman, J. Tatzelt, S. J. DeArmond, R. I. Weiner, W. C. Mobley, and S. B. Prusiner. 1997. A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J. Virol. 71:8821-8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silveira, J. R., G. J. Raymond, A. G. Hughson, R. E. Race, V. L. Sim, S. F. Hayes, and B. Caughey. 2005. The most infectious prion protein particles. Nature 437:257-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stoorvogel, W., M. J. Kleijmeer, H. J. Geuze, and G. Raposo. 2002. The biogenesis and functions of exosomes. Traffic 3:321-330. [DOI] [PubMed] [Google Scholar]
- 51.Thery, C., L. Zitvogel, and S. Amigorena. 2002. Exosomes: composition, biogenesis and function. Nat. Rev. Immunol. 2:569-579. [DOI] [PubMed] [Google Scholar]
- 52.Vilette, D., O. Andreoletti, F. Archer, M. F. Madelaine, J. L. Vilotte, S. Lehmann, and H. Laude. 2001. Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc. Natl. Acad. Sci. USA 98:4055-4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vorberg, I., A. Raines, and S. A. Priola. 2004. Acute formation of protease-resistant prion protein does not always lead to persistent scrapie infection in vitro. J. Biol. Chem. 279:29218-29225. [DOI] [PubMed] [Google Scholar]
- 54.Vorberg, I., A. Raines, B. Story, and S. A. Priola. 2004. Susceptibility of common fibroblast cell lines to transmissible spongiform encephalopathy agents. J. Infect. Dis. 189:431-439. [DOI] [PubMed] [Google Scholar]
- 55.Wadsworth, J. D., S. Joiner, A. F. Hill, T. A. Campbell, M. Desbruslais, P. J. Luthert, and J. Collinge. 2001. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 358:171-180. [DOI] [PubMed] [Google Scholar]
- 56.Walter, E. I., W. D. Ratnoff, K. E. Long, J. W. Kazura, and M. E. Medof. 1992. Effect of glycoinositolphospholipid anchor lipid groups on functional properties of decay-accelerating factor protein in cells. J. Biol. Chem. 267:1245-1252. [PubMed] [Google Scholar]
- 57.Weissmann, C., M. Enari, P. C. Klohn, D. Rossi, and E. Flechsig. 2002. Transmission of prions. J. Infect. Dis. 186(Suppl. 2):S157-S165. [DOI] [PubMed] [Google Scholar]
- 58.Zobeley, E., E. Flechsig, A. Cozzio, M. Enari, and C. Weissmann. 1999. Infectivity of scrapie prions bound to a stainless steel surface. Mol. Med. 5:240-243. [PMC free article] [PubMed] [Google Scholar]