

Abstract
The lytic switch protein RTA of Kaposi's sarcoma-associated herpesvirus (KSHV) can be targeted to DNA by either direct sequence-specific recognition or via protein-protein interactions with host transcription factors. We have searched for sequences capable of direct RTA binding by screening synthetic oligonucleotide pools and KSHV genomic libraries for RTA-interacting elements, using repeated cycles of in vitro binding followed by amplification of the bound sequences. Multiple low-affinity sequences were recovered from the random pools, with generation of only a weak consensus sequence. The genomic library, by contrast, yielded many biologically relevant fragments, most of which could be shown to interact with RTA in vitro and some of which likely play important regulatory roles in vivo. Surprisingly, the most highly selected fragment came from the promoter of a late gene (gB) and contained at least two direct RTA binding sites, as well as one RBP-Jκ binding site. This raises the possibility that some late KSHV genes may also be subject to direct RTA regulation, though indirect models are not excluded.
Kaposi's sarcoma (KS) is a common neoplasm in immunocompromised humans, and infection by the KS-associated herpesvirus (KSHV, also called human herpesvirus 8) is required for KS development (5, 27, 29). Like other herpesviruses, KSHV exhibits two patterns of gene expression, one responsible for latency and one underlying lytic replication. During the latent cycle, very few viral genes are expressed and no new virions are produced. Upon the switch to lytic infection, the majority of the genome is expressed, new viral progeny are produced, and the host cell is destroyed. Although KSHV-infected tumor cells are predominantly in the latency program (32), lytic KSHV replication is also necessary for disease progression, as viral DNA replication inhibitors like ganciclovir can rapidly block the emergence of KS even in profoundly immunocompromised AIDS patients (26). Therefore, an understanding of the switch from latency to lytic replication is important to a complete understanding of KS pathogenesis.
The latent-to-lytic switch is controlled by a single KSHV transcription factor known as RTA (for replication and transcription activator). Ectopic expression of RTA can induce the lytic program in latently infected cells (13, 25, 33); latent viruses bearing null mutants of RTA are uninducible (37), and dominant-negative mutants of RTA block lytic progression (24). During an infection cycle, RTA is expressed as an immediate-early gene (33) and induces transcription of delayed-early genes; this is then followed by viral DNA replication and subsequent late gene expression. Many delayed-early genes have been shown to be directly controlled by RTA (9, 10, 12, 16, 17, 24, 31, 38); by regulating DNA replication, these genes (and therefore RTA) indirectly regulate the expression of late genes. It is not known, however, whether RTA protein can also directly regulate late gene expression.
RTA is a leucine zipper protein with an N-terminal DNA binding domain. Target genes of RTA identified to date include TK, K2 (v-IL-6), K5 (MIR 2), K8 (RAP), kaposin, ORF57 (MTA), ORF74 (v-GPCR), polyadenylated nuclear RNA (PAN), and RTA itself. A strong interaction between RTA and a specific sequence in the promoter of the PAN and kaposin genes has been demonstrated; the binding sequences in these two promoters are almost identical (7). However, these sequence motifs are not found in most RTA-responsive genes. For example, the ORF57 promoter does not contain a similar sequence, and the activation of this gene (and many other viral genes) by RTA requires a cellular transcription factor, RBP-Jκ (or CSL) (20). Therefore, RTA appears to function in at least two modes (8). It can directly interact with a target gene promoter sequence or be recruited to a target gene promoter by another (usually cellular) cofactor. In addition to interactions with RBP-Jκ, RTA has been found to interact with other transcription factors, including C/EBP (35), c-Jun (34), and Oct-1 (28), which can target RTA to alternative sequences on the genome. RTA can also interact with transcription cofactors like CBP/p300, HDAC1, SWI/SNF chromatin remodeling complex, and TRAP/Mediator coactivator (14, 15).
Previous experiments have searched for RTA target genes genetically by using reporter genes fused with KSHV promoters to test for activation by RTA overexpression. These studies focused on early genes and used deletion analysis to identify the RTA responsive elements (RREs). Sequence analysis of a few of these RREs has not yielded a strong consensus sequence (though one study proposed a tandem array of phased A/T-trinucleotide motifs in RREs) (21). However, difficulties in identifying a conserved motif in this fashion could have been compounded by the inability of such approaches to differentiate target promoters with direct RTA binding sequences from those to which RTA is indirectly recruited by a transcription cofactor.
We sought to simplify the analysis of the DNA binding specificities of RTA by only searching for direct DNA binding sequences. We used an unbiased, genome-wide approach to search for DNA binding sequences that can directly bind to RTA in vitro. Using a tiled KSHV genomic microarray, we identified numerous viral sequences that can bind RTA; interestingly, one of these is located upstream of the ORF8 (glycoprotein B [gB]) gene. Since gB is a late gene, our results suggest that RTA may have roles beyond controlling only delayed-early gene expression.
MATERIALS AND METHODS
Constructs and cell lines.
SLK cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. pGST-50ΔSTAD was generated by cloning a fragment of pcDNA3-Flc50 into pGEX-4T-1 (20). Full-length RTA (pcDNA3-Flc50) used in transfection and in vitro translation assays was described previously (25). pGL3-gB contains 1216 bases upstream of the gB open reading frame (ORF) and was constructed previously (25). Deletion mutations of the gB promoter were generated using digestion with KpnI and BglII followed by exonuclease III and S1 nuclease treatments. Other deletions were generated using specific PCR primers. Four base pair mutations were introduced with the QuikChange mutagenesis kit (Stratagene). Oligonucleotide sequences can be quickly provided upon request.
Protein expression.
Glutathione S-transferase (GST)-50ΔSTAD (amino acids 1 to 530) was bacterially expressed, and lysates were generated in NETN buffer (20 mM Tris, pH 8, 200 mM NaCl, 1 mM EDTA, 0.5% NP-40, and protease inhibitor cocktail). This material was fractionated over a heparin column using an AKTA system (Amersham Pharmacia). The sample was eluted using a NaCl step gradient, with the majority of RTA eluting around 750 mM NaCl. Eluted fractions from the heparin column were further purified over glutathione-Sepharose 4B beads and eluted with glutathione. Rabbit reticulocyte lysate-expressed proteins are called IVT in figures. They were generated with the TNT T7 quick-coupled transcription-translation system (Promega). Reaction mixtures contained 2 μg of plasmid DNA.
SELEX.
Oligonucleotides mentioned below in Fig. 1 were synthesized (Invitrogen) for the random library, gel purified, and amplified using PCR. The KSHV genomic library was created using the same oligonucleotides and method described in reference 30. PCR products were gel purified and included an average of 200 to 300 bases of the genome. The initial binding reaction mixtures contained 5 μg of the random library and 0.5 μg of the genomic library with 5 μg and 0.5 μg of recombinant RTA, respectively. Recombinant RTA used in the binding reactions was purified by using a heparin column followed by a glutathione column (see above). Library DNA and RTA were incubated for 1 h at 25°C in 50 mM Tris, pH 7.5, 300 mM KCl, 10 mM MgCl2. The binding reaction mixture was passed through a filter (HAWP-025-00; Millipore) and washed with binding buffer, and membrane-associated protein and DNA were eluted with 8 M urea, 1× Tris-borate-EDTA at 65°C for 10 min. DNA was purified using phenol-chloroform-isoamyl alcohol extraction and ethanol precipitation. A 75% portion of purified material was amplified for the next round of amplification using standard PCR amplification.
FIG. 1.

Selection strategy for RTA aptamers. (A) Design of the starting material for selection with the random library. Forty bases of random sequence were flanked by conserved sequences to allow for PCR amplification. (B) Construction scheme for the KSHV genomic library. Starting with fragments of the KSHV genome, the library was generated using a series of PCR amplification reactions with primers containing conserved and random sequences. A gel purification step purified fragments between 200 and 300 bp. (C) RTA was bound to each pool, and bound elements were purified, amplified by PCR, and resubmitted for additional rounds of binding and selection. Following eight cycles of enrichment, the enriched DNAs were cloned and characterized.
Electrophoresis mobility shift assays (EMSAs).
Probes were labeled with 32P using T4 polynucleotide kinase (NEB) and purified using G25 microspin columns (Amersham Pharmacia). Typical reaction mixtures contained 0.4 pmol of probe, 350 ng of GST-50ΔSTAD or 3 μl of in vitro-translated sample, 25 ng poly(dI-dC), and 19 ng of herring sperm DNA in 20 mM HEPES, pH 7.9, 50 mM KCl, 10% glycerol, 1 mM EDTA, 1 mM MgCl2, 1 mM dithiothreitol. Reaction mixtures were incubated at 25°C for 30 min and resolved on 3.5% nondenaturing polyacrylamide gels in 0.5× Tris-borate-EDTA. Gels were dried and analyzed using a PhosphorImager (Amersham Pharmacia) and autoradiography.
Microarray analysis.
Equal amounts of the unenriched and enriched libraries were labeled with amino-allyl UTP using PCR. Purified PCR products were labeled with either Cy3 or Cy5 (Amersham Pharmacia) in 0.1 M sodium bicarbonate. Equal amounts of labeled DNA from each library were hybridized at 65°C to the KSHV genomic array, which contained 500-bp nonoverlapping fragments of the KSHV genome (3). After washing, arrays were scanned with a GenePix 4000B scanner and analyzed using GenePix Pro version 4 (Molecular Devices). The enrichment ratio represents the average of the ratio of medians from multiple independent assays. Data were normalized so that the average enrichment ratio of all included spots was 1.0.
Luciferase assays.
SLK cells were plated in 24-well dishes and transfected at 60% confluence using Fugene 6 (Roche) according to the manufacturer's instructions. For each well, 0.15 μg of reporter plasmid (in pGL3-Basic parental vector) and expression plasmid (in pcDNA3.1) was transfected. Lysates were prepared using the dual-luciferase reporter assay system (Promega). Each assay was performed in triplicate and performed multiple independent times.
RESULTS
Generation of aptamer libraries for RTA binding.
To begin our investigation into the DNA binding properties of RTA, we constructed two distinct libraries of DNA fragments. The first library contained synthetic oligonucleotides of 40 random bases flanked by PCR primer annealing sequences (Fig. 1A). PCR amplification was used to generate a random library of double-stranded oligonucleotides. Given the synthesis scale and random region length, this library does not completely cover the sequence space, but calculations suggest that over 1 × 1015 sequences were represented in this library. We believed this was large enough to support the search for RTA aptamers from random sequences. This library will be referred to subsequently as the random library or pool. This library could serve for investigations of the best binding sequences among a diverse population of sequences.
The other library was designed to allow us to identify RTA binding sites within the KSHV genome. The KSHV genomic library construction is outlined in Fig. 1B and is based on reference 30. Starting with the KSHV genome represented in a bacterial artificial chromosome (BAC), a series of PCR amplification steps were performed using PCR primers containing random bases at the 3′ end and conserved sequences at the 5′ end. In the first step, the first primer should anneal randomly throughout the genome and generate random genomic fragments with a conserved 5′ end after amplification. The second step generates random fragments with a different conserved 5′ end, and some of these double-stranded fragments will contain the different conserved 5′ sequences at each end. PCR products around 200 to 300 bp were size selected using gel purification. This material was then PCR amplified to generate the starting population of genomic fragments. To assess the coverage of the genomic library, we used a portion of the library as a 32P-labeled probe in a Southern blot analysis of the KSHV BAC. The banding pattern of the Southern blot generated with this probe was remarkably similar to the ethidium bromide staining pattern of the digested BAC DNA, indicating that the pool was broadly representative of the KSHV genome (data not shown).
Once the random library and the genomic library were constructed, we performed multiple rounds of SELEX (systematic evolution of ligands by exponential enrichment) to enrich for sequences that interact with RTA. As shown in Fig. 1C, this strategy involved selecting sequences that bind directly to RTA from those sequences that do not interact. Recombinant RTA was incubated with the unenriched libraries, and binding sequences were selected using a nitrocellulose filter binding method. DNA molecules that interacted with RTA were retained on the filter, while unbound molecules passed through the membrane. (We also performed some rounds of negative selection to remove sequences that would nonspecifically interact with the membrane in the absence of RTA.) The binding DNA sequences were purified and amplified by PCR using primers directed to the conserved flanking sequences. This amplified material was then used as the starting material for the next generation of selection and amplification. Eight rounds of positive selection were performed with recombinant purified RTA. After these rounds of selection, the enriched DNA fragments were cloned into a plasmid vector.
Analysis of binding sequences.
To assess the ability to bind RTA directly, 16 individual DNA fragments selected from the random pool were radioactively labeled and included in EMSAs with purified recombinant RTA (Fig. 2). Slower-migrating bands suggested a direct interaction between the specific DNA sequence and RTA (Fig. 2A). Compared to the negative control in the left lane, we observed shifted bands in reactions with numerous different selected sequences, affirming that the procedure did indeed enrich for sequences capable of direct binding to RTA. Those nine sequences that bound to RTA in vitro were analyzed using a computer program, MEME (2), to search for common motifs among the selected sequences. The most conserved motif is shown in Fig. 2B. However, the degree of conservation of this motif is low and may suggest a diverse DNA binding sequence specificity of RTA.
FIG. 2.
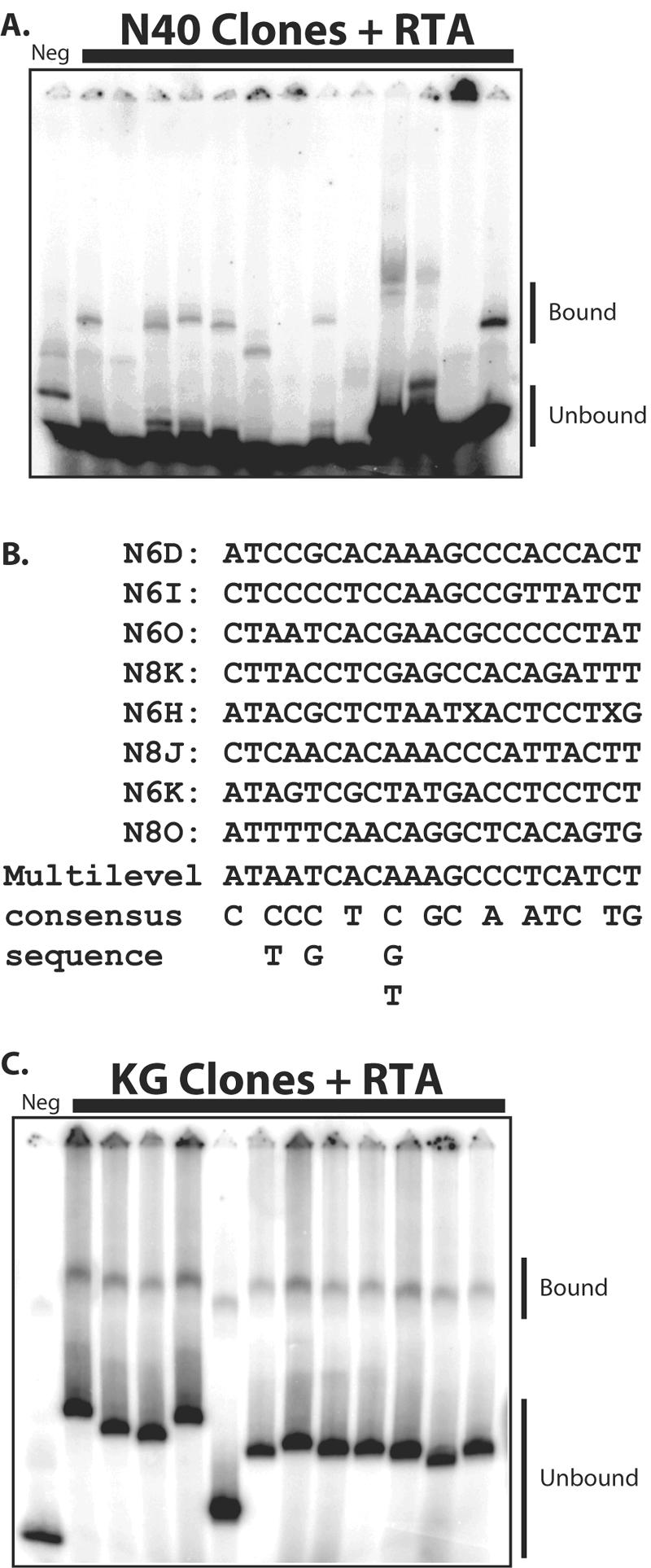
Binding of enriched sequences to RTA. Clones from enriched pools were cloned into plasmid vector and transformed into bacterial hosts, and colonies were randomly selected for individual analysis. (A) Recombinant RTA was incubated with individual sequences from the random pool, and EMSAs were performed. (B) Sequences revealing a shifted band were analyzed using the program MEME to search for a common motif. The most common motif is shown. (C) EMSAs with sequences from the genomic library selection. The negative control (Neg) was a randomly selected sequence obtained from the starting library.
In addition to investigating the selected sequences from the random pool, we also performed EMSAs with 13 sequences that were selected and amplified from the KSHV genomic pool. We observed that most of the sequences randomly chosen from the enriched pool also showed a shifted band (Fig. 2C), again suggesting a direct interaction with RTA. These 13 clones and 10 additional clones from the genomic pool were mapped to the KSHV genome, and their genomic locations are shown in Table 1. Reassuringly, one of these clones (K6P) contains the high-affinity RRE previously described in the kaposin promoter (5).
TABLE 1.
Clones obtained after multiple rounds of selection and amplification for RTA binding sequences
| Clone | Map position | KSHV sequence | Expression |
|---|---|---|---|
| K6G | 8181-8431 | In ORF7 | Early |
| K8K | 16945-17126 | Downstream of ORF11 and ORF K2 | Unknown, early |
| K6O | 26292-26574 | Upstream of ORF K5 | IE/early |
| K8N | 26752-26939 | Upstream of ORF K5 | IE/early |
| K8J | 29094-29203 | In ORF K7 | Unknown |
| K8L | 29242-29413 | Upstream of ORF16 | Early |
| K6Q | 31140-31303 | Downstream of ORF16 and ORF17 | Early |
| K8F | 31176-31391 | In ORF17 | Early |
| K8A | 32616-32724 | Upstream of ORF17 and ORF18 | Early, unknown |
| K8T | 42751-42994 | In ORF25 | Late |
| K6R | 60750-60969 | In ORF40 | Early |
| K8B | 84807-84998 | In ORFK9 | Early |
| K8S | 88487-88717 | Upstream of K10 | Unknown |
| K6E | 101657-101957 | Upstream of ORF62 and ORF63 | Late |
| K8H | 104278-104495 | In ORF64 | Early |
| K6B | 104394-104747 | In ORF64 | Early |
| K8D | 104398-104606 | In ORF64 | Early |
| K6A | 104396-104605 | In ORF64 | Early |
| K6P | 118622-118989 | Upstream of K12 | Latent |
| K6H | 125940-126013 | In ORF73 | Latent |
| K6K | 125955-126014 | In ORF73 | Latent |
| K6M | 125977-126017 | In ORF73 | Latent |
| K8R | 125998-126043 | In ORF73 | Latent |
Microarray analysis of binding sequences.
Since sequencing individual clones yields only a limited perspective of the enriched binding sequences, we sought to analyze the entire enriched population of binding elements. For this purpose we employed a KSHV genomic array. The array consists of 500-bp nonoverlapping genomic fragments tiled across the genome and covers the majority of the KSHV genome, including previously identified RREs (see Fig. 4C, below). To detect the enrichment of certain regions of the genome in the selected pool, we labeled the unenriched genomic starting population with Cy3 and the enriched (selected) population with Cy5. We observed Cy3 intensity signals above background for the vast majority of the array, which again affirmed that the starting genomic pool adequately covered the KSHV genome. We then hybridized this mixture to the genomic array and measured the ratio of Cy5 to Cy3 (Fig. 3A). Higher ratios represent positive selection for binding to RTA and amplification of a given binding sequence. We selected those microarray spots with a ratio of more than 2.0 and mapped the corresponding sequences to the genome (Fig. 3B); the exact nucleotides spanned are shown in Table 2. Presumably, the array spots with high enrichment ratios contain sequences that RTA preferentially bound in vitro during the SELEX rounds of selection and amplification. By using comparative hybridization, we normalized for under- or overrepresented sequences in the starting unenriched pool.
FIG. 4.

Analysis of the glycoprotein B promoter. (A) The map shows initial promoter deletion mutants and the corresponding response to RTA expression in luciferase assays using SLK cells. Fold activation was calculated by comparing the luciferase levels in SLK cells transfected with an RTA expression plasmid or a control empty vector. The region responsible for RTA activation is shown in the map. (B) Sequence comparison using MEME for the region of the glycoprotein B promoter to other RREs suggests a common sequence motif. (C) Diagram of the gB region of interest with identified sequence elements. GBS1, -2, -3, and -4 refer to probes used in Fig. 6, below.
FIG. 3.

Microarray analysis of enriched sequences from the genomic selection. (A) The average ratio of enrichment was plotted by comparing the signal intensity from unenriched versus enriched libraries. A ratio of 1 represents no enrichment by the SELEX rounds of selection. Sequence ID refers to the name given to each spot of the array. (B) A diagram of the KSHV genome displays the ORFs as arrows and the microarray hits as rectangles above the ORFs. Hits shown had an enrichment ratio above 2.
TABLE 2.
Microarray hits from enriched SELEX population
| SELEX hit(s) | PCR ID | Enrichment Ratio | Map positiona |
|---|---|---|---|
| ORF7 | KS017 | 3.92 | 8073-8564 |
| DR2/K5 | KS047 | 2.56 | 24941-25428 |
| ORF K5 | KS048 | 2.27 | 25634-25939 |
| ORF29, ORF29a | KS105 | 2.98 | 54005-54505 |
| ORF40 | KS118 | 2.31 | 60505-61009 |
| ORF43, ORF44 | KS126 | 2.60 | 64505-65000 |
| ORF48 | KS280 | 3.34 | 70822-71221 |
| ORF50 | KS145 | 2.07 | 74010-74504 |
| ORF56 | KS159 | 2.68 | 81009-81516 |
| ORF K9 | KS165 | 2.17 | 84004-84503 |
| ORF K10.5 | KS177 | 2.07 | 90004-90503 |
| ORF K10.6 | KS179 | 2.23 | 91004-91503 |
| ORF K10.6 | KS282 | 2.18 | 91302-91711 |
| ORF59 | KS189 | 2.53 | 95986-96493 |
| ORF60, ORF61 | KS192 | 2.21 | 97476-98022 |
| ORF61 | KS196 | 2.64 | 99500-99999 |
| LIR1′/ORF71 | KS234 | 2.19 | 119879-120378 |
| ORF71/K13, ORF72 | KS239 | 3.08 | 122372-122877 |
Map position refers to accession number NC_003409.
As shown in Table 2 and Fig. 3B, many of these SELEX hits fell within coding regions (ORFs). Given the compactness of the viral genome, most of these are likely to represent promoter elements of genes for adjacent (downstream) ORFs; however, we do not bar the possibility that in some cases RTA may also be able to regulate promoters from an intragenic position. We also note that very strong DNA binding sites were localized to two distinct intergenic (noncoding) regions, one between ORFs K4.2 and K5 and a second between ORFs K12 and K13. These are regions of multiple, tandem repeats; work from several groups indicates that these likely define origins of lytic cycle DNA replication (1, 22). Interestingly, a recent study documents that the first of these loci (between K4.2 and K5) encodes a noncoding transcript that is strongly inducible by RTA (36). The region we identified in the second lytic origin (nucleotides [nt] 119,879 to 120,378) (Table 2) maps to the right of the already-recognized RRE in the K12/kaposin promoter. Thus, there are at least two RTA binding sites in the vicinity of this lytic origin.
Identification of a novel RTA target in a late gene.
Inspection of the regions selected from the genomic pool (Table 2) showed that a site in ORF7 (just upstream of the glycoprotein B open reading frame, ORF8) showed the highest enrichment value in the selection experiment (Fig. 3A). To determine if this site was functional for RTA activation, we fused 1.2 kb of KSHV DNA from upstream of ORF8 (positions 7483 to 8699) to a luciferase reporter and transfected the chimera into 293 cells in the presence or absence of an RTA expression vector. This revealed a strong (400-fold) response to RTA expression (Fig. 4A). Initial deletion analysis of this region mapped the RTA-responsive element to a 120-bp region (Fig. 4A). To further examine this region, we searched this region for a possible motif represented in previously mapped RTA-responsive elements using the MEME program. This revealed an 8-bp CT-rich motif (which we term the A site) shared among several RTA-responsive elements (Fig. 4B). This region also contains two elements (here denoted as sites B and C) that are closely similar to the canonical RBP-Jκ binding sequence in this 120-bp region. Site B, interestingly, is immediately adjacent to site A (Fig. 4C).
Following the identification of these potentially important sequences for RTA response, we made further 5′ deletions in this region upstream of the glycoprotein B promoter. Figure 5A shows that all information upstream of sites A, B, and C is dispensable for strong RTA induction (Fig. 4C, deletion Δ33, which terminates at nt 33). A 5′ deletion spanning sites A and B (Fig. 4C, deletion Δ66, which ends at nt 66) results in a 75% (4-fold) reduction in inducibility (Fig. 5A), while deletion Δ89, which extends to nt 89 (Fig. 4C), virtually abolished the remaining RTA responsiveness. This suggests that an additional element important for RTA responsiveness maps from nt 66 to 89; we have denoted this element as site D in Fig. 4C. Since deletion Δ89 removes sites A, B, and D but leaves site C intact, the isolated C site must be insufficient for RTA inducibility.
FIG. 5.
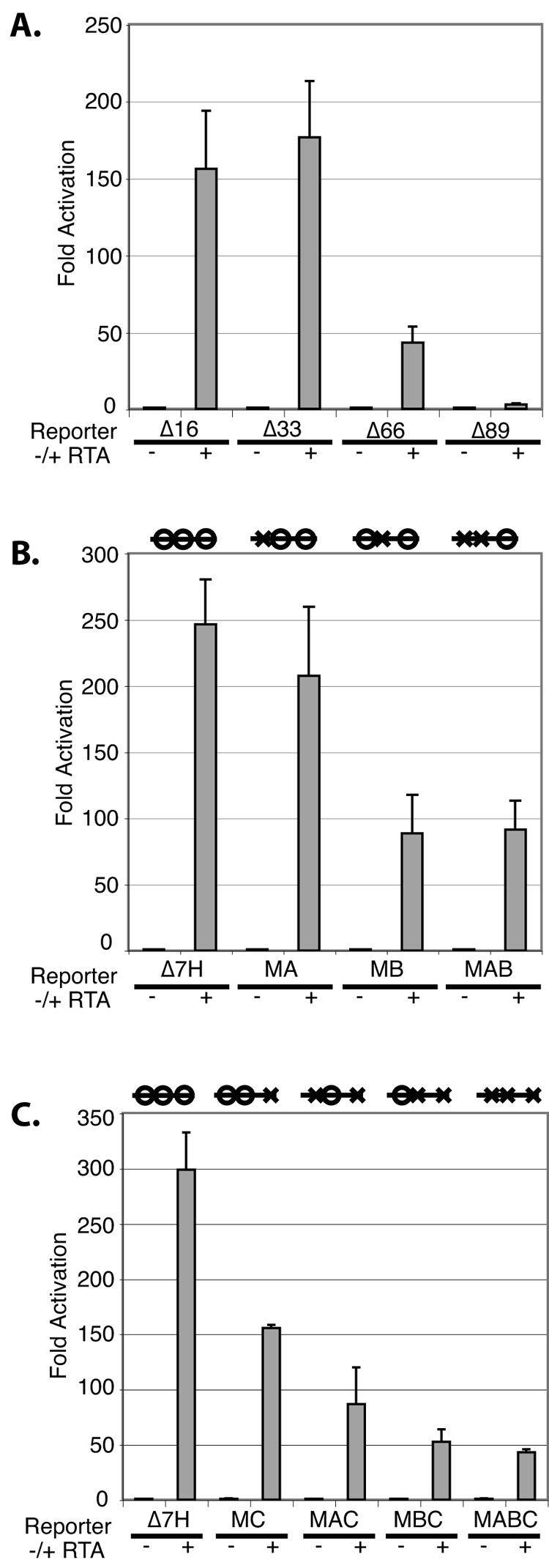
(A) Smaller promoter deletion reporters were tested in luciferase assays similar to those described in the legend for Fig. 4A. The reporter numbering refers to the sequence in Fig. 4C. (B and C) Similar luciferase assays were performed with a promoter mutant with four-base deletions in the A, B, and/or C site. The diagram above each reporter set denotes whether the above-mentioned sequence elements were wild type (open circle) or mutant (X).
To probe the importance of these elements with more precision, we constructed 4-bp mutations engineered into the individual A, B, and C sites. Lesions in the A site alone had minimal impact on RTA induction (Fig. 5B), while lesions in the B site (or in the A and B sites together) had larger effects. Solitary lesions in site C reduced induction only twofold (Fig. 5C). However, inactivating any two of the three sites, or all three sites, reduced activity by ca. 85% (Fig. 5B and C). This suggests that B and C sites are the most important for responsiveness to RTA expression but the A site is also important in the absence of these other sites. The source of the residual activity in the absence of all three elements most likely resides in the region between sites B and C; the deletion analysis of Fig. 5A maps the responsible region to site D (Fig. 5A and 4C).
To better understand the role of these sequences in activation, we also performed in vitro binding assays with fragments of the glycoprotein B promoter. For this we synthesized three oligonucleotides (Fig. 4C): GBS2 (nt 30 to 60), GBS3 (nt 60 to 90), and GBS4 (nt 90 to 120). Each fragment was end labeled and then used in EMSAs with purified, recombinant RTA expressed in Escherichia coli (Fig. 6A). We observed direct binding of RTA to fragments GBS2 (which contains sites A and B) and GBS3 (which contains site D), but only very weak binding to GBS4 (which contains only element C). It is noteworthy that the efficiency of complex formation with the gB promoter sites in GBS2 and GBS3 was much less than that observed with the known high-affinity PAN site (Fig. 6A, lane 7), suggesting that all these elements are of lower affinity that the PAN site. In fact, competition experiments have revealed that the GBS2 fragment competes for RTA binding 200- to 400-fold less efficiently than does the PAN site (data not shown).
FIG. 6.

RTA binding assays with fragments of the gB promoter. (A) Recombinant GST-RTA (R) or GST alone (G) was incubated with fragments of the gB promoter shown in Fig. 4C. (B) Recombinant RTA (R), in vitro-translated sample with empty control vector (NT), in vitro-translated RBP-Jκ (RB), and the combination of recombinant RTA and in vitro-translated RBP-Jκ (RR) protein samples were incubated with GBS4 and GBS2. The positive control probe, PAN RRE, was also included. (C) Two forms of RTA, full-length in vitro-translated RTA (IVT RTA), recombinant GST-RTAΔSTAD, or in vitro-translated RBP-Jκ (IVT RBP) were incubated with the probe GBS2. (D) Identical protein combinations as in panel B were incubated with wild-type GBS2 or a probe with a mutation in the A site (2MA), a mutation in the B site (2MB), or mutation in both the A and B sites (2MAB).
Since GBS2 and GBS4 both contain potential RBP-Jκ sites, we also examined the impact of RBP-Jκ on RTA interactions with these fragments (Fig. 6B). With the GBS2 probe (which contains sites A and B), we observe a strong shifted band in the presence of RBP-Jκ, affirming that site B is a high-affinity RBP-Jκ site (lane 7). The GBS4 probe, which spans site C, also binds RBP-Jκ, but considerably less efficiently than does the site B-containing GBS2 probe (compare lanes 3 and 7). This suggests that site C is a weaker RBP-Jκ site than site B, a result consistent with the fact that its sequence (TTGGAA) differs more from the established consensus sequence (TGGGAA) for RBP-Jκ binding. We again detected direct RTA binding to the GBS2 probe (sites A and B) (Fig. 6B, lane 5). Importantly, this band is supershifted in the presence of RBP-Jκ protein (lane 8). Evidence that this supershifted band in fact contains RTA is derived from two sources: (i) its formation is blocked by anti-RTA antibody (not shown), and (ii) its mobility is altered when RTA deletion mutants of differing sizes are employed in the EMSA reaction (Fig. 6C, lanes 1 and 2). Although the same amount of full-length in vitro-translated RTA was included in reactions 1 and 4 of Fig. 6C, we observed the lower-mobility (RTA-containing) complex only in the presence of RBP-Jκ. This indicates that virtually all of the DNA molecules in this complex contain both RTA and RBP-Jκ and is consistent with a cooperative interaction between RTA and RBP-Jκ on DNA. Control EMSA experiments with the PAN site again revealed much more efficient binding of RTA to this site and, as expected, no impact of RBP-Jκ on the interaction (Fig. 6B, lanes 9 to 12).
Next, we examined the effects of mutations in sites A or B on the binding of RTA to the GBS2 fragment in vitro; for this purpose we employed the same mutants we had earlier examined in vivo (Fig. 5B). Figure 6D shows the results of this experiment. Mutation of the A site reduced the direct RTA binding below the detection limit of this assay (compare lanes 1 and 5), again affirming the direct interaction of RTA with site A and suggesting that this site may well have been responsible for the recovery of this region in the SELEX experiment. As expected, lesions in site A have no impact on RBP-Jκ binding (lane 7). However, when the site A mutant fragment was incubated with both RTA and RBP-Jκ, a lower-mobility complex containing RTA was again observed (lane 8). This indicates that RTA can be recruited by RBP-Jκ in the absence of site A, either via protein-protein interactions or by cooperative binding to the mutant site. As expected, mutations in the B site dramatically inhibited RBP-Jκ binding (lanes 11 and 12), but weak binding of RTA to this probe was still detectable (lane 9). The combination of mutations in the A and B sites abolishes all binding by RTA or RBP-Jκ (lanes 13 to 15).
Taken together with the luciferase data, we conclude that site A is a low-affinity site for direct RTA binding and site B is a high-affinity RBP-Jκ recognition site that can also recruit RTA to the template even in the absence of a functional site A. An additional RTA binding site (site D) is located at nt 66 to 89 of the GBS3 fragment; together these three elements are responsible for the majority of the RTA responsiveness of the gB promoter. A lower-affinity, noncanonical RBP-Jκ site (site C) is present downstream of these sites; it cannot mediate RTA responses on its own, but it can augment responsiveness to RTA in the presence of the upstream elements.
DISCUSSION
In this work we have employed an unbiased selection procedure to identify and characterize sequences that can be directly bound by KSHV RTA. Using a pool of random sequence oligonucleotides, we derived a loose consensus sequence among the amplified elements. However, the resulting consensus is not highly conserved, reflecting a considerable degree of divergence among the recovered sequences. Although most of the sequences we tested were indeed capable of binding to RTA in vitro, their affinities appeared low, and several of the sequences we tested by cloning into reporter constructs did not confer appreciable RTA responsiveness in transfected cells (J. Ziegelbauer and Y. Xu, unpublished results). Perhaps a more robust consensus element could be arrived at by repeating the SELEX protocol under different binding conditions.
By contrast, the biological relevance of the sequences selected and amplified from the genomic library was more readily apparent. First, among the sequences most preferentially amplified were two regions that are known to contain RTA-responsive elements. One of these is the canonical RRE from the K12/kaposin lytic cycle promoter, and the second is derived from the lytic origin between ORFs K4.2 and K5, which is known to express a noncoding RNA that is strongly responsive to RTA (36). Sequences upstream of vIL6, a known RTA-responsive gene, were also identified. In addition, sequences from upstream of RTA itself were recovered in the genomic SELEX protocol, and many groups have observed autoinduction of RTA transcription (11). We were puzzled not to have recovered the PAN element itself in our screen. However, it is important to note the different selection pressures involved in the SELEX technology. For example, the ability to be amplified in PCRs may limit the amplification of some sequences that may bind to RTA. In addition, the binding ability of sequences can be influenced by the binding conditions, and the binding conditions used in the SELEX method are not identical to standard conditions used in electrophoresis mobility shift assays. So, the absence of a specific sequence in the enriched pools could be the result of multiple factors, and the interpretation must be limited as a result. These limitations are noteworthy and prevent an estimation of the total number of RTA target genes exhibited in vivo.
Surprisingly, the most highly represented clone from the genomic SELEX experiment was one corresponding to the gB promoter. This was quite unanticipated, since an earlier experiment using a gB reporter had led us to conclude that this gene was not induced by RTA (25). However, we have been unable to confirm that negative result, even using the original plasmid vector; instead, multiple assays (Fig. 4 and 5) have shown that gB reporters are indeed strongly activated by RTA expression. EMSA experiments confirm that RTA indeed binds to at least two sequences genetically linked to RTA responsiveness (site A in GBS2 and site D in GBS3), though the affinity of these interactions, which was evidently sufficient to allow their recovery in a SELEX protocol, is much lower than that observed with the canonical PAN site in EMSAs. Moreover, much of the activation appears to be mediated by nearby RBP-Jκ sites, ablation of both of which reduces induction by 85% (Fig. 5C). Further work will be required to determine if cooperativity exists between RBP-Jκ bound at site B and RTA bound to site A, a model rendered attractive by the proximity of these sites.
We believe that gB is the first KSHV late gene for which a role for RTA has been proposed. Validation that RTA is involved in vivo in gB regulation will not be simple, however, since gB expression follows and may be influenced by both delayed-early gene expression and DNA replication, both of which are directly or indirectly controlled by RTA. We have recently examined the effects of DNA synthesis inhibitors on gB mRNA induction during lytic replication induced by exogenous RTA expression. These studies showed that gB gene induction, although potent (1,000-fold over the uninduced culture as judged by quantitative reverse transcription-PCR), was only modestly inhibited by ganciclovir (similar, in fact, to the DE gene ORF59 and much less than the classical late gene ORF25 [unpublished data]), a result that agrees well with studies by others (6, 23). This indicates that most of the upregulation of gB transcription at late times is not due to copy number amplification and is consistent with the idea that gB transcription is upregulated by transacting factors. In view of the present results, RTA is an obvious candidate for such a factor; however, we cannot exclude that a delayed-early protein induced by RTA might also be responsible.
Be that as it may, our results may also shed light on another regulatory issue in gB expression. A recent study of gene expression in cultured cells shortly after de novo infection revealed expression of both RTA and gB within a few hours of infection, before the onset of most delayed-early gene expression. Expression of gB was selective—most other glycoproteins were not upregulated at this time point (18). This burst of aberrant lytic gene expression was transient, giving way to latency by 24 h. We speculate that perhaps the RTA site we have identified in gB could have been responsible for ectopic gB expression in the context of deregulated RTA expression. We and others have recently found that RTA is also encapsidated in KSHV virions (4, 19), and so this pool of newly introduced, preformed RTA might have contributed to the transient induction of gB observed immediately post-viral entry.
In conclusion, the present findings illustrate the power of sequence amplification approaches to detect potential regulatory interactions in large viral genomes. We anticipate that careful analysis of additional sequences from the amplified genomic library should yield further insights into KSHV lytic replication and its regulation by RTA.
Acknowledgments
We thank members of the Ganem and DeRisi laboratories for advice and support. We also thank Yuying Liang for reagents and protocols.
J.Z. is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-1793). D.G. is an investigator of the Howard Hughes Medical Institute.
REFERENCES
- 1.AuCoin, D. P., K. S. Colletti, Y. Xu, S. A. Cei, and G. S. Pari. 2002. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) contains two functional lytic origins of DNA replication. J. Virol. 76:7890-7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey, T. L., and M. Gribskov. 1998. Combining evidence using p-values: application to sequence homology searches. Bioinformatics 14:48-54. [DOI] [PubMed] [Google Scholar]
- 3.Bechtel, J., A. Grundhoff, and D. Ganem. 2005. RNAs in the virion of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:10138-10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bechtel, J. T., R. C. Winant, and D. Ganem. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:4952-4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boshoff, C., and R. A. Weiss. 1998. Kaposi's sarcoma-associated herpesvirus. Adv. Cancer Res. 75:57-86. [DOI] [PubMed] [Google Scholar]
- 6.Chang, J., and D. Ganem. 2000. On the control of late gene expression in Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8). J. Gen. Virol. 81:2039-2047. [DOI] [PubMed] [Google Scholar]
- 7.Chang, P. J., D. Shedd, L. Gradoville, M. S. Cho, L. W. Chen, J. Chang, and G. Miller. 2002. Open reading frame 50 protein of Kaposi's sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J. Virol. 76:3168-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang, P. J., D. Shedd, and G. Miller. 2005. Two subclasses of Kaposi's sarcoma-associated herpesvirus lytic cycle promoters distinguished by open reading frame 50 mutant proteins that are deficient in binding to DNA. J. Virol. 79:8750-8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, J., K. Ueda, S. Sakakibara, T. Okuno, and K. Yamanishi. 2000. Transcriptional regulation of the Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor gene. J. Virol. 74:8623-8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng, H., M. J. Song, J. T. Chu, and R. Sun. 2002. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 76:8252-8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng, H., A. Young, and R. Sun. 2000. Auto-activation of the rta gene of human herpesvirus-8/Kaposi's sarcoma-associated herpesvirus. J. Gen. Virol. 81:3043-3048. [DOI] [PubMed] [Google Scholar]
- 12.Duan, W., S. Wang, S. Liu, and C. Wood. 2001. Characterization of Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF57 promoter. Arch. Virol. 146:403-413. [DOI] [PubMed] [Google Scholar]
- 13.Gradoville, L., J. Gerlach, E. Grogan, D. Shedd, S. Nikiforow, C. Metroka, and G. Miller. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74:6207-6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gwack, Y., H. J. Baek, H. Nakamura, S. H. Lee, M. Meisterernst, R. G. Roeder, and J. U. Jung. 2003. Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi's sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol. Cell. Biol. 23:2055-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gwack, Y., H. Byun, S. Hwang, C. Lim, and J. Choe. 2001. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi's sarcoma-associated herpesvirus open reading frame 50. J. Virol. 75:1909-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haque, M., J. Chen, K. Ueda, Y. Mori, K. Nakano, Y. Hirata, S. Kanamori, Y. Uchiyama, R. Inagi, T. Okuno, and K. Yamanishi. 2000. Identification and analysis of the K5 gene of Kaposi's sarcoma-associated herpesvirus. J. Virol. 74:2867-2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeong, J., J. Papin, and D. Dittmer. 2001. Differential regulation of the overlapping Kaposi's sarcoma-associated herpesvirus vGCR (orf74) and LANA (orf73) promoters. J. Virol. 75:1798-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishnan, H. H., P. P. Naranatt, M. S. Smith, L. Zeng, C. Bloomer, and B. Chandran. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lan, K., D. A. Kuppers, S. C. Verma, N. Sharma, M. Murakami, and E. S. Robertson. 2005. Induction of Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J. Virol. 79:7453-7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang, Y., J. Chang, S. J. Lynch, D. M. Lukac, and D. Ganem. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jκ (CSL), the target of the Notch signaling pathway. Genes Dev. 16:1977-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao, W., Y. Tang, Y. L. Kuo, B. Y. Liu, C. J. Xu, and C. Z. Giam. 2003. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 transcriptional activator Rta is an oligomeric DNA-binding protein that interacts with tandem arrays of phased A/T-trinucleotide motifs. J. Virol. 77:9399-9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin, C. L., H. Li, Y. Wang, F. X. Zhu, S. Kudchodkar, and Y. Yuan. 2003. Kaposi's sarcoma-associated herpesvirus lytic origin (ori-Lyt)-dependent DNA replication: identification of the ori-Lyt and association of K8 bZip protein with the origin. J. Virol. 77:5578-5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu, M., J. Suen, C. Frias, R. Pfeiffer, M. H. Tsai, E. Chuang, and S. L. Zeichner. 2004. Dissection of the Kaposi's sarcoma-associated herpesvirus gene expression program by using the viral DNA replication inhibitor cidofovir. J. Virol. 78:13637-13652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lukac, D. M., J. R. Kirshner, and D. Ganem. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lukac, D. M., R. Renne, J. R. Kirshner, and D. Ganem. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304-312. [DOI] [PubMed] [Google Scholar]
- 26.Martin, D. F., B. D. Kuppermann, R. A. Wolitz, A. G. Palestine, H. Li, C. A. Robinson, et al. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N. Engl. J. Med. 340:1063-1070. [DOI] [PubMed] [Google Scholar]
- 27.Ojala, P. M., M. Tiainen, P. Salven, T. Veikkola, E. Castanos-Velez, R. Sarid, P. Biberfeld, and T. P. Makela. 1999. Kaposi's sarcoma-associated herpesvirus-encoded v-cyclin triggers apoptosis in cells with high levels of cyclin-dependent kinase 6. Cancer Res. 59:4984-4989. [PubMed] [Google Scholar]
- 28.Sakakibara, S., K. Ueda, J. Chen, T. Okuno, and K. Yamanishi. 2001. Octamer-binding sequence is a key element for the autoregulation of Kaposi's sarcoma-associated herpesvirus ORF50/Lyta gene expression. J. Virol. 75:6894-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulz, T. F. 1999. Epidemiology of Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. Adv. Cancer Res. 76:121-160. [DOI] [PubMed] [Google Scholar]
- 30.Singer, B. S., T. Shtatland, D. Brown, and L. Gold. 1997. Libraries for genomic SELEX. Nucleic Acids Res. 25:781-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song, M. J., H. J. Brown, T. T. Wu, and R. Sun. 2001. Transcription activation of polyadenylated nuclear RNA by RTA in human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:3129-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Staskus, K. A., W. Zhong, K. Gebhard, B. Herndier, H. Wang, R. Renne, J. Beneke, J. Pudney, D. J. Anderson, D. Ganem, and A. T. Haase. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71:715-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang, S. E., F. Y. Wu, H. Chen, M. Shamay, Q. Zheng, and G. S. Hayward. 2004. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 78:4248-4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang, S. E., F. Y. Wu, M. Fujimuro, J. Zong, S. D. Hayward, and G. S. Hayward. 2003. Role of CCAAT/enhancer-binding protein alpha (C/EBPα) in activation of the Kaposi's sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J. Virol. 77:600-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang, Y., H. Li, M. Y. Chan, F. X. Zhu, D. M. Lukac, and Y. Yuan. 2004. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J. Virol. 78:8615-8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu, Y., D. P. AuCoin, A. R. Huete, S. A. Cei, L. J. Hanson, and G. S. Pari. 2005. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 79:3479-3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang, L., J. Chiu, and J. C. Lin. 1998. Activation of human herpesvirus 8 (HHV-8) thymidine kinase (TK) TATAA-less promoter by HHV-8 ORF50 gene product is SP1 dependent. DNA Cell Biol. 17:735-742. [DOI] [PubMed] [Google Scholar]