

Abstract
The equine herpesvirus 1 (EHV-1) α-trans-inducing factor homologue (ETIF; VP16-E) is a 60-kDa virion component encoded by gene 12 (ORF12) that transactivates the immediate-early gene promoter. Here we report on the function of EHV-1 ETIF in the context of viral infection. An ETIF-null mutant from EHV-1 strain RacL11 (vL11ΔETIF) was constructed and analyzed. After transfection of vL11ΔETIF DNA into RK13 cells, no infectious virus could be reconstituted, and only single infected cells or small foci containing up to eight infected cells were detected. In contrast, after transfection of vL11ΔETIF DNA into a complementing cell line, infectious virus could be recovered, indicating the requirement of ETIF for productive virus infection. The growth defect of vL11ΔETIF could largely be restored by propagation on the complementing cell line, and growth on the complementing cell line resulted in incorporation of ETIF in mature and secreted virions. Low- and high-multiplicity infections of RK13 cells with phenotypically complemented vL11ΔETIF virus resulted in titers of virus progeny similar to those used for infection, suggesting that input ETIF from infection was recycled. Ultrastructural studies of vL11ΔETIF-infected cells demonstrated a marked defect in secondary envelopment at cytoplasmic membranes, resulting in very few enveloped virions in transport vesicles or extracellular space. Taken together, our results demonstrate that ETIF has an essential function in the replication cycle of EHV-1, and its main role appears to be in secondary envelopment.
Equine herpesvirus type 1 (EHV-1) is classified as a member of the Varicellovirus genus within the Alphaherpesvirinae subfamily. In analogy to the situation seen in other alphaherpesviruses, expression of EHV-1 genes is regulated in a cascade-like fashion. An immediate-early (IE), early, and late phase of gene expression are recognized, depending on the order of the appearance of transcripts and proteins during lytic infection (6, 21, 21, 26). From the approximately 76 EHV-1 genes, a single immediate-early (ie- and α-) gene, 49 early (e- and β-) genes, and 26 late (l-, γ1-, and γ2-) genes have been identified (21, 24, 24, 57, 57, 67, 67). The sole IE gene of EHV-1, a homologue of the herpes simplex virus type 1 (HSV-1) ICP4 gene, is transcribed independently of de novo protein synthesis from gene 62 and is absolutely essential for virus replication (18). The EHV-1 IE protein is a multifunctional, regulatory protein capable of modulating early and late promoters independently of or synergistically with early regulatory proteins (4, 25, 27, 55, 56, 67).
IE gene transcription becomes augmented and strongly stimulated by the virion-associated transcriptional regulator of EHV-1 gene expression, variably referred to as ETIF or VP16-E, expressed from the true late (γ2) EHV-1 gene 12 (33, 34, 44, 44, 45, 45). ETIF is the EHV-1 homologue of HSV-1 VP16 also known as α-trans-inducing factor (α-TIF) (5). Homologues of HSV-1 α-TIF have been identified in several other alphaherpesviruses, including varicella-zoster virus (VZV) (39), bovine herpesvirus 1 (BHV-1) (38), pseudorabies virus (PRV) (17), and HSV-2 (22). Although all known α-TIFs share the ability to transactivate IE gene promoters, they differ in their structure, transcriptional activation domains, and properties. HSV-1 VP16 requires a highly acidic C-terminal sequence of about 80 amino acids for transactivation of immediate-early promoters. The transcriptional activation domain consists of two clusters of hydrophobic amino acids with adjacent acidic residues (1, 9, 61, 63). In contrast, the α-TIF of VZV, the open reading frame 10 product, does not contain such a highly acidic C-terminal domain (39). Comparison of other known VP16 homologues revealed that the BHV-1 UL48 product and EHV-1 ETIF lack the majority of the acidic C terminus and contain only a short acidic C-terminal domain (14, 38). Mutational analysis of ETIF showed that the last seven C-terminal amino acids are necessary but not sufficient for ETIF transactivation of the IE promoter (14).
Transcriptional activation by HSV-1 VP16 requires the assembly of a complex consisting of VP16 and the two cellular proteins Oct-1 and host cell factor. That complex is formed on regulatory octamer sites (TAATGARAT) located within the IE promoter, which are required for binding of Oct-1 and VP16 and thus transactivation (reviewed in reference 65). EHV-1 ETIF also forms a higher-order complex with Oct-1 and host cell factor but presumably utilizes octamer sequences other than TAATGARAT within the IE promoter (15, 20).
ETIF is a major tegument component and was identified as a 60-kDa virion protein (34). It is delivered into the host cell by incoming virus particles (33). HSV-1 VP16 and the α-TIF homologues of VZV and PRV are also incorporated into virions and serve as a structural component of the tegument (5, 17, 29). Mutational analysis of HSV-1 VP16 revealed that its function as a transcriptional activator is not essential for productive virus infection since virus unable to transactivate IE gene expression can replicate in noncomplementing cells, although virus growth is drastically reduced (2, 54). In contrast, VP16 is absolutely required for virus assembly. A VP16-null mutant failed to produce infectious progeny virus and DNA packaging appeared to be less efficient (64). More recent work, taking into account that VP16 can interact with vhs (virion host shutoff) and modulate its activity (32, 53), has provided evidence that VP16 is required in HSV-1 virion formation in a step downstream of the primary envelopment at the inner nuclear membrane (41).
Unlike its homologue in HSV-1, the VZV open reading frame 10 and the Marek's disease virus UL48 products are dispensable for virus growth (7, 12). In PRV, a UL48-null mutant is viable and able to replicate in noncomplementing cells, albeit at the cost of reduced virus yield and plaque sizes (17). Similar to the situation in HSV-1, the observed impairment of virus growth in the absence of the PRV UL48 protein appeared to be caused by a severe defect in virion morphogenesis prior to secondary envelopment (17).
The aim of the study presented here was to investigate the role of ETIF in EHV-1 infection. To this end, a recombinant virus lacking gene 12 encoding ETIF was constructed. Our results demonstrate that ETIF is essential for productive virus infection upon DNA transfection in cultured cells that do not express ETIF. Moreover, we provide evidence that the defect in virus replication in absence of ETIF correlates with a defect in virion assembly, likely during secondary envelopment. In addition, the observation that infections at low multiplicities resulted in self-limiting replication of phenotypically complemented ETIF-negative EHV-1 suggest recycling of ETIF delivered via incoming virus particles.
MATERIALS AND METHODS
Plasmids and bacteria.
Plasmids were constructed and maintained in Escherichia coli DH10B by using standard methods (49). For construction of plasmid pc-ETIF a 1.6-kb HindIII-XbaI fragment of pCETIF (28) containing the entire EHV-1 gene 12 coding sequence was cloned into the pcDNA3.1(+) vector (Invitrogen). Recombinant plasmid pKD46 and bacterial strain BW25141 (10), a derivate of E. coli K-12 strain BD792, were kindly provided by Barry L. Wanner, Purdue University, West Lafayette, Ind., and used to perform Red mutagenesis using the EHV-1 bacterial artificial chromosome (BAC) clone of strain RacL11, termed pRacL11 (46). Plasmid p71L11 was described earlier (62). Plasmid pG12 used for generation of the ETIF revertant virus was obtained by TOPO cloning of a 2.6-kbp PCR-product into cloning vector pCR2.1-TOPO (Invitrogen). The 2.6-kbp sequence from EHV-1 strain RacL11 was amplified by standard PCR using primers 12rev-1 (5′-CTGAACACATATACGAAACG-3′) and 12rev-2 (5′-TCTCTATAGCTGAGTCTCGG-3′) and comprises nucleotides 12803 to 15454 based on the published Ab4 sequence (58). The nucleotide sequence of plasmid pG12 encompassing EHV-1 gene 12 and adjoining sequences was determined to verify the correctness of the cloned sequence (Cornell Biotechnology Center).
Mutagenesis of pRacL11.
For modification of pRacL11, Red mutagenesis was used (10) and adapted for BACs (60). Briefly, BW25141 harboring pRacL11 and the Red helper plasmid pKD46 were grown in Luria-Bertani broth (LB) with chloramphenicol (30 μg/ml), ampicillin (100 μg/ml), and l-arabinose (0.1% final concentration) at 30°C to an optical density at 600 nm of 0.6 and then made electrocompetent exactly as previously described (10). To delete gene 12 encoding ETIF in pRacL11, resulting in recombinant BAC termed pRacL11ΔETIF, the aminoglycoside phosphotransferase (aphAI) gene of plasmid pACYC177 (MBI Fermentas) conferring kanamycin resistance was amplified by PCR. The designed primers 48kan-1 (5′-TCCGGTTAACGCTTATTTGCATAAATTCATAACACTGTGCCCTCAATAAAGCCAGTGTTACAACCAATTAACC-3′) and 48kan-2 (5′-CGACGGAAAATACGGCCAACATGTTAAGTCTGGAAACATACTTTATTGTACGATTTATTCAACAAAGCCACG-3′) contained 50-nucleotide homology arms bordering the desired deletion from position 13505 to 14945 of gene 12 and 20 nucleotides (in boldface) for amplification of the aphAI sequences. The resulting 1-kb PCR fragment was purified from an agarose gel (QIAquick gel extraction kit; QIAGEN) and electroporated into pRacL11- and pKD46-containing electrocompetent BW25141 using 0.1-cm cuvettes (Bio-Rad Laboratories) under standard electroporation conditions (1.25 kV/cm, 200 Ω, 25 μF). After electroporation, cells were grown in 1 ml of SOC for 60 min at 37°C and plated onto LB agar plates containing 30 μg of chloramphenicol/ml and 50 μg of kanamycin/ml. Double-resistant colonies were picked into liquid LB medium, grown at 37°C, and small-scale preparations of mutant pRacL11 DNA were obtained by alkaline lysis of E. coli (49), digested with various restriction enzymes, and analyzed for changes in their restriction patterns.
Viruses and cells.
EHV-1 strain RacL11 was grown on rabbit kidney cells (RK13), which were propagated in Dulbecco minimal essential medium supplemented with 10% fetal bovine serum. A complementing cell line, RK-ETIF, was generated by transfection of 3 μg of recombinant plasmid pc-ETIF using Superfect (QIAGEN) into RK13 cells. Clones developing under Dulbecco modified Eagle medium containing 500 μg of G418 (Calbiochem)/ml were isolated and expanded for further analysis by indirect immunofluorescence (IIF) using anti-EHV-1 monoclonal antibody (MAb) L3ab directed against ETIF (34), as well as in a trans-complementation assay. One positive cell clone was selected, named RK-ETIF, and used for all further experiments. The recombinant ETIF-gp2 double-negative virus vL11ΔETIF/gp2 was reconstituted by calcium phosphate transfection of pRacL11ΔETIF BAC DNA into RK-ETIF cells. To generate the gene 12 single deletion mutant vL11ΔETIF and restore expression of gp2, pRacL11ΔETIF DNA was cotransfected with plasmid p71L11 (62) into RK-ETIF cells exactly as described before (46). Five days after cotransfection, supernatant was harvested and transferred to fresh RK-ETIF cells, and recombinant nonfluorescing virus plaques were picked and purified to homogeneity by two rounds of plaque purification (47). An ETIF revertant virus, vL11ΔETIF-R, was isolated after cotransfection of 5 μg of vL11ΔETIF virus DNA with 3 μg of plasmid pG12 into RK13 cells and subsequent plaque purification of virus progeny.
DNA analysis.
Viral DNA was isolated from eukaryotic cells by using standard phenol-chloroform extraction (49), cleaved with restriction enzymes, and separated by 0.8% agarose gel electrophoresis. In addition, viral DNA was analyzed with sequencing primers Kan1 (5′-CACGTTCGCCGGCTTTCCCC-3′) and Kan2 (5′-GGGATCTCATGCTGGAGTTC-3′) to verify the insertion of the aphAI sequence into the genome of EHV-1 instead of EHV-1 gene 12.
Antibodies.
To detect EHV-1 ETIF by IIF and Western blot analysis, MAb L3ab was used (34). MAb E2 directed against EHV-1 gM was kindly provided by Lindsey Day and Richard Killington, University of Leeds, Leeds, United Kingdom (11, 48, 51). A monospecific polyclonal rabbit antiserum recognizing the EHV-1 US3 protein (US3p) was used exactly as described earlier (8). A β-actin antibody (ab3280) cross-reacting with the rabbit protein was purchased from Abcam Inc, Cambridge, Mass., and used as a control antibody.
IIF and confocal microscopy.
RK13 or RK-ETIF cells were grown on six-well plates or coverslips for confocal laser-scanning microscopy (CLSM) and infected at a multiplicity of infection (MOI) of 0.01. Alternatively, cells were transfected with viral DNA or the pc-ETIF expression plasmid. Cells were fixed at 48 to 72 h postinfection (p.i.) or transfection with 90% ice-cold acetone for 10 min, and IIF was performed exactly as described previously (46, 47). For CLSM, Alexa 568-conjugated secondary antibody (Molecular Probes) was used, and fluorescence was preserved with Fluoromount-G (Southern Biotechnology Associates, Inc.). Coverslips were analyzed by using the Olympus IX70 microscope.
Western blot analysis.
Cell lysates or extracellular virions purified by sucrose gradient centrifugation were examined by Western blotting (62). Lysates were adjusted to equal protein concentrations determined by the method of Bradford (Bio-Rad Protein Assay; Bio-Rad Laboratories). Samples were separated by sodium dodecyl sulfate-12% polyacrylamide gel electrophoresis (SDS-12% PAGE) and transferred to nitrocellulose membranes (Schleicher & Schuell) or polyvinylidene difluoride membranes (Millipore) by the semidry method (30). Free binding sites on the sheets were blocked by addition of 5% skim milk in phosphate-buffered saline containing 0.3% Tween (PBST) before the antibodies (suspended in PBST) were added. Bound antibodies were detected with anti-mouse immunoglobulin G (IgG)-peroxidase conjugates (Jackson Immunoresearch Laboratories) or anti-rabbit IgG-peroxidase conjugates (Pharmacia-Amersham) and visualized by enhanced chemiluminescence (ECL Kit; Pharmacia-Amersham).
Plaque size determination and virus titer.
Plaque sizes were measured after plating of the viruses on RK13 or RK-ETIF cells and 3 days of incubation at 37°C under a 0.6 to 0.7% methylcellulose overlay (48). Cells were fixed with acetone and stained by IIF with anti-gM MAb E2. Plaque areas were determined exactly as described recently (62), and virus growth kinetics were examined after infection of 105 RK13 or RK-ETIF cells with various MOIs of the different viruses. Virus infection was synchronized by incubating cells with viruses for 1 h at 4°C and subsequently shifting of the temperature to 37°C for another hour to allow penetration. Residual input infectivity was neutralized by addition of ice-cold citrate buffer (pH 3.0) for 3 min (48). Finally, prewarmed medium was added, and the cells were incubated at 37°C. At various time points after infection, extracellular and cell-associated virus titers were determined by culturing 10-fold virus dilutions on RK13 or RK-ETIF cells overlaid with 0.6 to 0.7% methylcellulose and final staining with crystal violet after 3 days of incubation.
Stability of ETIF.
RK13 cells (105) were infected at an MOI of 20 of the different viruses. The infection was synchronized by incubating cells with virus for 1.5 h on ice and subsequent incubation at 37°C for another 1.5 h to allow penetration. Cells were washed intensively with medium to remove residual input virus. After the addition of prewarmed medium, the cells were incubated at 37°C. At various time points after infection, cells were separated from the supernatant, lysed, and examined by Western blot analysis.
Electron microscopy.
RK13 or RK-ETIF cells were infected at an MOI of 3 with wild-type or phenotypically complemented vL11ΔETIF, which had been propagated on RK-ETIF. At 16 h p.i., virus-infected cells were fixed and embedded for electron microscopy exactly as described previously (50). Ultrathin sections of embedded material were counterstained with uranyl acetate and lead salts and examined in an electron microscope (Philips EM 400Tecnai). For determination of total virus particle numbers, a diluted mixture of purified virions with a defined number of latex beads (0.3 μm; Ladd Research Industries, Williston, VT) was adsorbed to Formvar-carbon coated 300 mesh copper grids, stained for 1 min with uranyl acetate, and examined by electron microscopy.
RESULTS
ETIF is stably expressed in RK-ETIF cells.
We anticipated that an EHV-1 mutant virus lacking the ETIF expressing gene 12 might grow very inefficiently in RK13 cells. In order to be able to construct and investigate such a mutant virus, generation of a cell line constitutively expressing ETIF seemed necessary. After transfection of RK13 cells with ETIF expression plasmid pc-ETIF, G418-resistant cell clones were isolated. Expression of ETIF by individual cell clones was constantly monitored by IIF with anti-ETIF MAb L3ab. Notable was that expression levels of ETIF in all tested cell clones decreased rapidly within the few passages that were necessary for isolation and purification of the cell line and only very low levels of ETIF expression were detected in final clones (Fig. 1). All tested cell clones were able to complement the deletion of gene 12. One clone, termed RK-ETIF, was chosen and used for all further studies. In the majority of the RK-ETIF cells, the VP16 homologue predominantly localized to the nucleus, but weak cytoplasmic staining with the anti-ETIF antibody was detected (Fig. 1). Localization of ETIF in RK-ETIF cells upon virus infection was mainly cytoplasmic at later times p.i. and indistinguishable from that in virus-infected RK13 cells (data not shown).
FIG. 1.

CLSM of RK13 cells transfected with ETIF expression plasmid pc-ETIF (A), RK-ETIF cells (B), and RK13 cells (C). ETIF was detected with MAb L3ab and visualized with anti-mouse IgG-Alexa Fluor 568 conjugate.
Deletion of gene 12 from the EHV-1 genome and virus reconstitution.
Using Red mutagenesis in E. coli, gene 12 was deleted in the infectious full-length clone of EHV-1 strain RacL11 (pRacL11) (46). gene 12 of pRacL11 was replaced with an aminoglycoside phospho-transferase gene (aphAI), which confers kanamycin resistance (Fig. 2). DNA from several kanamycin-resistant bacterial clones was isolated and then analyzed by DNA restriction enzyme analysis and Southern blot hybridization. One clone exhibiting the correct restriction enzyme fragment pattern was chosen and termed pRacL11ΔETIF (data not shown). The correct insertion of the aphAI gene was verified by sequencing using aphAI specific primers. The DNA analyses confirmed that the deletion of gene 12 in pRacL11-ETIF was introduced as planned.
FIG. 2.

Generation of mutant virus vL11ΔETIF and plasmid pG12. (A) Shown is the overall organization of the EHV-1 genome consisting of a unique long (UL) and a unique short (US) region, which is bracketed by inverted internal repeat (IR) and terminal repeat (TR) sequences, and the BamHI map. (B) Depiction of the unique long region of RacL11 from gene 11 (UL49 homologue) through gene 14 (UL46 homologue), with arrowheads indicating the transcriptional directions of the various genes. Arrows show the site of the aphAI gene insertion using Red mutagenesis using infectious clone pRacL11. (C) Organization of mutant virus vL11ΔETIF in which the aphAI gene is inserted in lieu of gene 12. (D) Plasmid pG12 contains a 2.6-kbp PCR product comprising gene 12 and flanking sequences and was used to generate ETIF-revertant virus vL11ΔETIF-R.
Transfection of pRacL11ΔETIF BAC DNA into RK-ETIF cells generated a virus mutant, designated vL11ΔETIF/gp2, which in addition to gene 12 also lacks gene 71 encoding gp2 (46). To restore expression of gene 71, RK-ETIF cells were cotransfected with pRacL11ΔETIF DNA and plasmid p71L11 to reinsert gene 71 by homologous recombination. Nonfluorescent plaques as the result of the loss of the egfp gene and reinsertion of gp2-encoding sequences were purified until a homogenous population of nonfluorescing virus progeny was obtained (62). The resulting mutant virus lacking the ETIF encoding sequences only was termed vL11ΔETIF and propagated on RK-ETIF cells.
ETIF is essential for EHV-1 growth.
The effect of the deletion of gene 12 on virus growth was initially analyzed by transfection of pRacL11ΔETIF BAC-DNA, prepared from E. coli, into RK13 cells. Transfection efficiencies and plaque formation could easily be monitored after transfection of pRacL11ΔETIF DNA by using fluorescence microscopy because EGFP is expressed from pRacL11 (46). Although green fluorescing cells could be detected as early as 1 day after transfection, no infectious virus in the supernatant of transfected cells or developing virus plaques were detected after transfection for the entire observation period. Only a few foci comprising up to eight cells could be found, but we were unable to passage or expand the number of infected cells after reseeding. Single infected cells and foci of infected cells could also be stained with an anti-gM MAb by IIF. At no time point were we able to propagate virus after transfection of pRacL11ΔETIF DNA into RK13 cells.
In contrast, cotransfection of pRacL11ΔETIF DNA with the ETIF expression plasmid pc-ETIF or transfection of pRacL11ΔETIF DNA into the ETIF-expressing cell line resulted in plaque formation and infectious virus that could readily be passaged onto new RK13 cells. To verify that the lack of ETIF alone was responsible for the phenotype of vL11ΔETIF/gp2, transfection experiments with vL11ΔETIF DNA isolated from RK-ETIF cells were performed. As described above for pRacL11ΔETIF BAC DNA, only single infected cells or small foci that could not be expanded were detected after transfection of vL11ΔETIF DNA into noncomplementing RK13 cells by IIF with an anti-gM MAb, while transfection of vL11ΔETIF DNA together with plasmid pc-ETIF or transfection of vL11ΔETIF DNA into RK-ETIF cells resulted in plaque formation (Fig. 3). Parallel transfections of identical amounts of wild-type RacL11 DNA into RK13 cells resulted in complete infection of cells and high virus titers by 3 days p.i. (data not shown). Based on these results, we concluded that ETIF, the HSV-1 VP16 homologue encoded by EHV-1, is essential for virus growth.
FIG. 3.

IIF analysis of RK13 cells transfected with vL11ΔETIF DNA, RK-ETIF cells transfected with vL11ΔETIF DNA, and RK13 cells. Cells were fixed with acetone on day 3 after transfection and incubated with anti-gM MAb E2, which was visualized by using anti-mouse IgG Alexa 488 antibody (Molecular Probes). Individual panels represent views 1,000 by 1,000 μm in size.
Reversion of the vL11ΔETIF mutation with a wild-type allele of ETIF.
To exclude the possibility that adventitious second site mutations occurred during the process of generating vL11ΔETIF, a genetic revertant of vL11ΔETIF was constructed in which the ETIF-encoding sequence was restored. The revertant was constructed by cotransfection of vL11ΔETIF viral DNA and plasmid pG12 containing wild-type gene 12 into RK13 cells (Fig. 2). It was expected that the ability to produce virus progeny resulting from this cotransfection was restored and the revertant virus would be able to grow in RK13 cells, which proved to be the case (Fig. 4). Virus progeny isolated from the cotransfection was plaque purified and analyzed by IIF with the anti-ETIF MAb L3ab. All tested virus clones were positive for ETIF, and one virus clone was chosen and termed vL11ΔETIF-R.
FIG. 4.

Plaque areas of RacL11, vL11ΔETIF, and vL11ΔETIF-R on RK13 cells (A) and RK-ETIF cells (B). Shown are means and standard deviations (error bars) of plaque areas of 100 plaques measured for each virus at 3 days p.i. in two independent experiments. Plaque areas of RacL11 were set at 100% for each cell line.
Growth defect of vL11ΔETIF: efficiency of plaque formation.
The ETIF-negative mutant of EHV-1 can be propagated on RK-ETIF cells, as demonstrated by the isolation of vL11ΔETIF but not in noncomplementing RK13 cells after transfection of vL11ΔETIF DNA. To further investigate the growth properties of the ETIF-negative virus, plaque formation and direct cell-to-cell spread abilities of parental wild-type RacL11 virus, vL11ΔETIF virus propagated on RK-ETIF cells, and the vL11ΔETIF-R revertant virus were measured on noncomplementing RK13 or RK-ETIF cells (Fig. 4).
Plaque areas of RacL11 and vL11ΔETIF-R were virtually identical on RK13 and RK-ETIF cells, respectively. However, the ratio of plaque sizes formed on RK-ETIF cells versus RK13 cells was 0.85 for RacL11 and 0.84 for vL11ΔETIF-R (data not shown). This indicated a slight impairment of direct cell-to-cell spread on cells constitutively expressing ETIF compared to plaque formation on RK13 cells.
In contrast, infection of RK13 cells with vL11ΔETIF resulted in barely measurable plaque areas that reached less than 3% of the plaque areas of wild-type or vL11ΔETIF-R virus. This defect of vL11ΔETIF on RK13 could be complemented on the ETIF-expressing cell line even though plaque areas did not quite reach plaque sizes of RacL11 or vL11ΔETIF-R. The results of the plaque assay indicated that the cell-to-cell spread of EHV-1 is dramatically diminished in RK13 cells in the absence of ETIF (Fig. 4).
Growth defect of vL11ΔETIF: efficiency of replication.
In a second set of experiments, replication of RacL11, the ETIF-negative, and the revertant virus (vL11ΔETIF-R) was examined by single-step growth kinetics in both RK13 and RK-ETIF. Cells were infected at an MOI of 3, and extracellular (Fig. 5) and cell-associated virus titers (data not shown) were determined at various time points after infection by titration on RK-ETIF cells. All three viruses replicated efficiently in RK-ETIF cells, although reduced extracellular titers of vL11ΔETIF were observed compared to those of wild-type and the ETIF revertant virus. However, the time course of virus replication was similar for each virus as reflected by production of cell-associated infectivity (data not shown). In RK13 cells, extracellular virus titers and the time course of replication of RacL11 and vL11ΔETIF-R were virtually identical (Fig. 5). In contrast and as expected from the transfection experiments, replication of the ETIF deletion mutant was severely impaired on RK13 cells, and extracellular titers were reduced up to 63-fold compared to that of wild-type virus and ETIF revertant virus (Fig. 5).
FIG. 5.

Single-step growth kinetics of RacL11 (▴), vL11ΔETIF (○), and vL11ΔETIF-R (▪) on RK13 or RK-ETIF cells. At the indicated times p.i., extracellular virus titers were determined on RK13 or RK-ETIF cells as described in Materials and Methods. Shown are means and standard deviations (error bars) for each virus and cell type.
Taken together, data from the plaque assay and single-step growth studies indicate that the ETIF deletion mutant of EHV-1 produced on the complementing cell line has a severe growth defect on noncomplementing cells. This defect could partially, but not completely, be overcome in RK-ETIF cells that constitutively express low levels of EHV-1 ETIF. Moreover, since the revertant virus possesses growth properties that are virtually identical to those of parental virus, we concluded that vL11ΔETIF does not harbor fortuitous mutations that affect viral replication in vitro.
Infection with vL11ΔETIF is self-limiting.
The results of the single-step growth kinetics described above indicated that the ETIF-null mutant produced on complementing cells is still able to grow on noncomplementing cells to a certain extent, while ETIF proved to be absolutely essential for growth after transfection of vL11ΔETIF DNA into RK13 cells. In order to investigate the role of ETIF in virus replication in greater detail, low-MOI experiments were undertaken. RK13 cells and RK-ETIF cells were infected with various MOIs of wild-type or vL11ΔETIF. At 3 days p.i., endpoint virus titers were determined. In low-MOI infections, wild-type virus grew to similar virus titers of up to 1.3 × 107 in both RK13 and RK-ETIF cells (Fig. 6). In contrast, infection of RK13 cells with the complemented ETIF-null virus mutant resulted in MOI-dependent endpoint titers, which were drastically lower than those observed for wild-type RacL11 and were not more than 10-fold higher than the titers used for infection. Reinfection of RK13 cells with vL11ΔETIF obtained from the first round of a low-MOI infection resulted in drastically reduced end-point titers compared to the titers used for infection. In the case of the reinfection of the vL11ΔETIF virus obtained from the lowest-MOI infection, no infectious virus could be detected at day 3 p.i. In contrast, vL11ΔETIF grew to similar titers on complementing RK-ETIF cells, which were slightly lower compared to those of wild-type virus. From these data we concluded that infection of RK13 cells with the complemented ETIF-negative virus is progressively self-limiting, such that a state is reached in which no infectivity, neither cell associated nor extracellular, can be detected.
FIG. 6.

RK13 (A) or RK-ETIF cells (B) were infected with the indicated MOIs of RacL11 (▪) and vL11ΔETIF (□) corresponding to 104, 103, 102, and 101 PFU. At 3 days p.i., virus titers were determined on RK13 or RK-ETIF cells as described in Materials and Methods. vL11ΔETIF virus obtained from the first round of infection was used to reinfect RK13 or RK-ETIF cells (░⃞). Shown are means and standard deviation of each virus and cell type of three independent experiments.
Incorporation of ETIF in vL11ΔETIF virions.
The results described above showed that the ETIF expressing cell line is complementing for the absence of gene 12 in vL11ΔETIF. Besides the well-studied function of ETIF as a potent transactivator of IE gene expression, ETIF is also incorporated into virions as a structural component of the virus tegument. The possibility was considered that ETIF expressed by RK-ETIF cells was incorporated into virions during propagation of vL11ΔETIF and might be responsible for the differences in growth observed after infection or transfection of vL11ΔETIF. Western blots of purified virions were carried out to investigate whether ETIF was incorporated into virus particles. In lysates of virus-infected cells (Fig. 7A), as well as in purified virions (Fig. 7B) of both wild-type and vL11ΔETIF-R, the previously reported 60-kDa ETIF protein was detected by using anti-ETIF MAb L3ab. Furthermore, the antibody recognized two additional ETIF species of lower molecular masses of 50 and 46 kDa, which were also incorporated into virions (Fig. 7A and B). In lysates of RK-ETIF cells that had been infected with vL11ΔETIF, as well as in RK-ETIF cells, at least the two larger ETIF moieties of 60 and 50 kDa were detected. Notable were the reduced expression levels of ETIF in complementing RK-ETIF cells compared to RK13 and RK-ETIF cells infected with parental or revertant virus vL11ΔETIF-R (Fig. 7A). All three ETIF species were also present in purified virions of vL11ΔETIF propagated on RK-ETIF cells (data not shown), whereas no ETIF protein could be detected in vL11ΔETIF virions passaged once on RK13 cells (Fig. 7B). Incorporation of the 60-kDa and the 50-kDa moieties of ETIF into virions of vL11ΔETIF was less efficient compared to RacL11 and vL11ΔETIF-R virions, which could be a result of the overall lower expression levels of ETIF in the complementing cell line. To confirm that absence of ETIF in vL11ΔETIF virions propagated on RK13 cells was not the result of an absence of virions altogether, an identical nitrocellulose membrane containing separated virion proteins of RacL11, the ETIF rescuant virus, and vL11ΔETIF propagated on RK-ETIF or RK13, was incubated with a polyclonal antibody specific for the US3 protein (US3p) (Fig. 7B). US3p was found to be present in comparable amounts in each of the virion preparations. The detection of US3p in purified virions confirmed that ETIF protein is specifically absent from vL11ΔETIF virions purified from RK13 cells. We therefore concluded that, as a result of propagation in RK-ETIF cells, ETIF was incorporated in vL11ΔETIF virions to serve as a structural component. Furthermore, the slight increase in virus titers in the first passage in noncomplementing cells and the following massive decrease from the second passage onward indicated that ETIF might be recycled and be responsible for the gradual loss of infectivity and the self-limiting nature of vL11ΔETIF infection in noncomplementing cells, which, in turn, could be a direct effect of a gradual decrease of ETIF incorporation.
FIG. 7.

(A) Detection of ETIF in lysates of RK13 and RK-ETIF cells infected with the indicated viruses 18 h p.i. Lysates were separated by SDS-12% PAGE, transferred to a polyvinylidene difluoride membrane, and incubated with either anti-ETIF MAb L3ab or a control anti-β-actin MAb. (B) Purified virions of RacL11, vL11ΔETIF propagated on RK-ETIF cells, vL11ΔETIF-R, and vL11ΔETIF grown on RK13 cells were incubated with either anti-ETIF MAb L3ab or control anti-US3p polyclonal antibody. Only the full-length 60-kDa form of ETIF is indicated by a solid arrowhead. The sizes of the Precision Plus Protein (Bio-Rad) marker are given in thousands.
In addition to the incorporation of ETIF into vL11ΔETIF particles when the mutant virus was propagated on complementing cells, the particle/PFU ratio of phenotypically complemented vL11ΔETIF is an important parameter in experiments in which different MOIs are used for different viruses. Therefore, the particle/PFU ratio of purified virions of parental and rescuant virus (both produced on RK13 cells), as well as vL11ΔETIF (produced on RK-ETIF cells), was determined. Virus progeny of the ETIF-negative mutant grown on the complementing cell line exhibited an ∼7-fold increase in the particle/PFU ratio compared to that of parental RacL11 (Table 1). Considering this difference in the particle/PFU ratio, the high input of ETIF protein via infecting particles in the previously described infection experiments could be responsible for the growth differences of vL11ΔETIF observed after infection or transfection of RK13 cells.
TABLE 1.
Particle/PFU ratio
Data are expressed as the mean particle/PFU ratio, with the standard deviation given.
vL11ΔETIF was propagated on RK-ETIF cells.
Stability of incoming ETIF.
The fate of ETIF that is delivered into the cells with the infecting particle is poorly understood. The stability of incoming ETIF was examined by Western blot analyses after infection of RK13 cells with vL11ΔETIF using an MOI of 20. ETIF was detected for up to 6.5 h p.i., the end of the sampling period, with no detectable degradation of ETIF (Fig. 8). Stability of ETIF was also observed in lysates of cells infected with RacL11 or the ETIF-revertant virus until de novo synthesis of ETIF started (data not shown). From these observations we concluded that ETIF delivered by incoming EHV-1 particles is stable for extended periods of time and could theoretically be recycled for at least one virus passage.
FIG. 8.
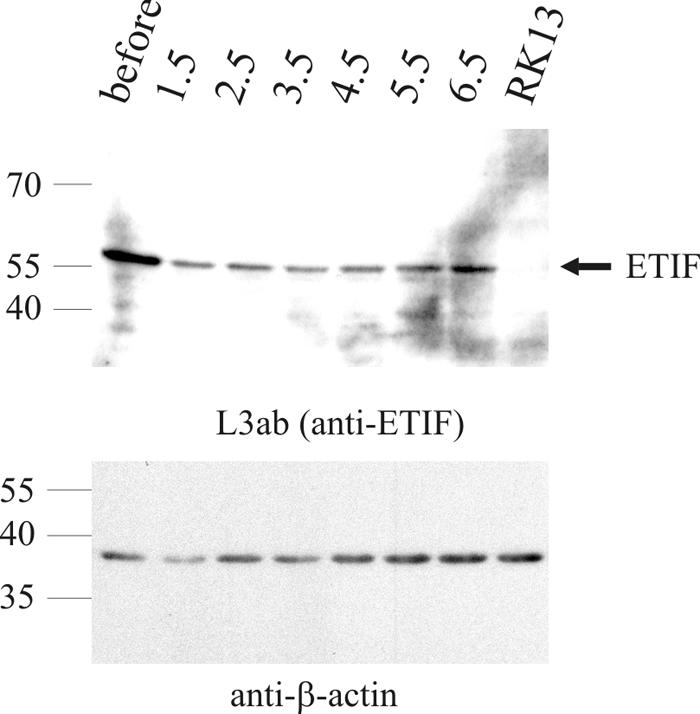
Stability of ETIF in RK13 cells infected with complemented vL11ΔETIF using an MOI of 20. ETIF was detected with anti-ETIF MAb L3ab at the indicated times p.i. in infected-cell lysates before and after intensive washing steps. Full-length ETIF is indicated by a solid arrow. The membrane was stripped and incubated with a control anti-β-actin MAb in a second reaction. The sizes of the PageRuler Prestained Protein Ladder (Fermentas) are given in thousands.
vL11ΔETIF exhibits a defect in virus assembly in the cytoplasm.
To determine whether the replication defect of the ETIF-null mutant was associated with a defect in virus assembly, ultrastructural analyses were performed. These studies indicated that vL11ΔETIF exhibited a defect in secondary envelopment, inasmuch as budding into cytoplasmic vesicles appeared to be initiated, but remained incomplete. Early stages of virus assembly such as nucleocapsid formation in infected-cell nuclei, primary budding at the inner lamella of the nuclear membrane, and de-envelopment at the outer lamella of the nuclear membrane, however, did not appear to be different from those observed in wild-type-infected cells (Fig. 9A). De-enveloped nucleocapsids accumulated in the vicinity of cytoplasmic membranes (Fig. 9B), and incomplete budding processes of nucleocapsids at vesicle membranes were observed more frequently (Fig. 9C and D). Furthermore, assembly stages after secondary envelopment such as fully enveloped virions in the cytoplasm or virions released into extracellular space were found only very rarely. In contrast, replication of vL11ΔETIF on complementing RK-ETIF cells was morphologically indistinguishable from that of wild-type RacL11 virus, and large numbers of virions released into extracellular space and finally enveloped virions in transport vesicles were found (Fig. 9E and F) (51). We concluded from the electron microscopic studies that the absence of ETIF during EHV-1 infection results in a profound defect in the assembly of infectious virions at the site of secondary envelopment at cytoplasmic vesicles.
FIG. 9.

Electron micrographs of RK13 (A to D) or RK-ETIF (E and F) cells infected with vL11ΔETIF at 16 h p.i. (A) Overview of a section of an infected cell containing nucleocapsids in the nucleus and cytoplasmic nucleocapsids in the cytoplasm. No completely enveloped virions could be detected in the cytoplasm or extracellular space. (B) Nonenveloped cytoplasmic nucleocapsids released from infected-cell nuclei accumulated in the neighborhood of stacks of cytoplasmic membranes. (C) A cytoplasmic particle is budding into a presumably TGN originated membrane as indicated by an arrow. (D) Higher magnification demonstrated fuzzy material around cytoplasmic nucleocapids while budding into TGN membranes (arrows). (E) Infection of RK-ETIF with vL11ΔETIF at 16 h p.i. revealed nucleocapsids in the nucleus of infected cells, some which are adjacent to the nuclear envelope (arrowhead) and many fully enveloped particles in the extracellular space (arrows). (F) Secondary enveloped particles in the cytoplasm and extracellular virions are indicated by arrows. Bars, 500 nm (A to F).
DISCUSSION
In this study, experiments were conducted to elucidate the role of ETIF, the EHV-1 homologue of HSV-1 VP16 (α-TIF), in virus replication. An ETIF deletion mutant of EHV-1 was constructed in E. coli using an infectious EHV-1 BAC clone and progeny vL11ΔETIF virus was propagated on an ETIF-expressing cell line. The failure to propagate an ETIF-negative virus on noncomplementing cells and the abortive nature of an infection with complemented virus of an ETIF mutant virus suggests an absolute requirement of ETIF for productive virus replication. It could further be demonstrated that absence of ETIF resulted in a massive defect in virus morphogenesis that may be caused by a defect in tegumentation and/or secondary envelopment.
The part of the EHV-1 genome where gene 12 encoding ETIF is located is transcriptionally very active and surrounded by genes expressing homologues of other tegument proteins, e.g., VP13/14 (UL46), VP17/18 (UL47), and VP22 (UL49) (58). Therefore, the specificity of the essential phenotype of the vL11ΔETIF virus was addressed in two ways. First, a site-specific revertant based on vL11ΔETIF was generated in which the ETIF-encoding sequence was restored. The revertant virus regained wild-type growth properties, indicating that no secondary mutations occurred during isolation of vL11ΔETIF and that the observed lethal phenotype of vL11ΔETIF mapped within the DNA fragment used to rescue the mutation. Second, specificity of the deletion was shown by trans-complementing the replication defect of vL11ΔETIF using RK-ETIF cells, which constitutively express ETIF. The RK-ETIF cell line efficiently complemented the growth defect in vL11ΔETIF, although titers and plaque sizes were slightly reduced. The reduction in virus growth properties compared to those of parental or revertant virus could be caused by the significantly lower expression levels of ETIF in this cell line compared to virus infection (Fig. 7A), which also results in relatively less efficient incorporation of ETIF into virus particles (Fig. 7B). We view it as unlikely that the cellular distribution of ETIF in RK-ETIF cells was altered, because ETIF compartmentalization appeared to be unaffected and consistent with earlier reports showing ETIF in both the cytoplasm and the nucleus (20). In addition, distribution of ETIF after infection in both RK13 cells and RK-ETIF cells was indistinguishable (data not shown). Localization of ETIF into vesicular compartments in the cytoplasm as described for HSV-1 VP16 could not be observed (31) but will be investigated in future studies in greater detail.
Previous studies have shown that ETIF accomplishes at least two functions during EHV-1 infection. ETIF serves as a structural component of the tegument (3, 34) and can stimulate the transcription of the sole EHV-1 immediate-early gene (33). It shares these functional properties with α-TIFs of HSV-1, BHV-1, PRV, and VZV (5, 17, 38, 39). In EHV-1, the sole and essential IE gene, transcribed independently of de novo EHV-1 proteins, functions as a transcriptional transactivator of the early genes and initiates the transcriptional cascade of EHV-1 genes. However, EHV-1 DNA is infectious, and virus progeny can be produced after transfection of virus DNA or of plasmids carrying the EHV-1 genome into permissive cells, suggesting that ETIF delivered by incoming virions is not absolutely required for the onset of viral replication (46; the present study). This is further supported by results obtained from experiments using HSV-1 expressing mutated VP16 in which the transactivation domain was deleted. Such mutants were viable, although both the level and timing of viral gene expression were severely disrupted (66). The defect in viral gene expression caused by the absence of the transactivation domain of VP16 resulted in significantly reduced virus titers, which could be increased by a high-MOI infection or induction of cellular stress (2, 35, 66).
In the case of EHV-1, foci or single infected cells upon transfection of mutant virus DNA lacking the entire coding sequence for ETIF could be identified by an antibody directed against gM. Expression of gM starts in the late phase of virus replication (43), indicating that virus genome replication and onset of viral gene expression is not dependent on the presence of ETIF, which is consistent with the situation seen in HSV-1 (2, 42, 54). Production of late viral proteins in the absence of ETIF suggests that the transactivator function of ETIF is dispensable for initiation of the cascade-like transcription pattern of EHV-1 genes. However, the exact role of the ETIF function as transactivator of the IE gene expression and its requirement in the early stages of infection remains to be determined and will be the object of future studies.
The second function of ETIF is of a structural nature. ETIF is a virion component (34), and we could detect at least three different sizes of ETIF-specific bands in both virus-infected cell lysates and purified virions by Western blot analysis using an MAb directed against ETIF. These various forms of ETIF are also found in lysates of RK-ETIF cells and of RK13 cells transfected with pc-ETIF. The nature of the multiple bands of ETIF in Western blot analysis is not completely understood and could reflect different phosphorylation states or be the result of proteolytic cleavage. The most likely explanation, however, is the use of alternative start codons that can be found in the sequence of ETIF and would result in the expression of shorter forms with predicted molecular masses corresponding to the observed smaller bands. In fact, recent data indicate that mutation of the first start methionine leads to absence of the largest form (60 kDa) of ETIF, while the two lower ETIF species were still expressed (59).
The presence of ETIF in phenotypically complemented vL11ΔETIF, which carry the risk of “phenotypical leakage,” may explain seemingly contradictory results of the transfection and infection experiments. High-MOI infection of RK13 cells with vL11ΔETIF propagated on RK-ETIF cells resulted in extracellular titers that were significantly reduced compared to those of wild-type or rescuant virus but were still measurable and reached 3 × 105 PFU/ml. This result was unexpected and was in stark contrast to the essential requirement of ETIF for virus egress as observed in transfection experiments (the present study) and previous reports on a VP16-null mutant of HSV-1 showing loss of infectivity after infection in one-step growth kinetics (64). One explanation for this phenomenon could be that delivery of functional ETIF by incoming complemented virions results in “recycling” for at least another round of virion assembly. However, it cannot be entirely ruled out that the requirement of ETIF for virus growth can be overcome by infections at high MOI. Recycling of ETIF would require that the incoming protein is stable and remains functional so that it can be sequestered and serve as a structural component of newly synthesized virions. In fact, in immunoblot analyses, incoming ETIF was stable beyond 6 h p.i. (Fig. 8) and is therefore available long enough to be recycled for virion assembly. In addition, the higher particle/PFU ratio of vL11ΔETIF propagated on RK-ETIF cells compared to wild-type and ETIF-rescuant virus will result in larger amounts of ETIF delivered to RK13 cells by incoming virus particles. The fate, however, of incoming ETIF is not completely understood. Dissociation of HSV-1 VP16 from the incoming particle and localization to the nucleus early in infection was reported (31, 40). It was also shown that HSV-1 VP16 is relatively stable upon entry into cells, does not undergo any apparent changes, and can be detected up to 4 h after infection (40). Similar results, i.e., dissociation of the UL48 protein from the incoming virion after entry, were recently obtained by immunoelectron analyses of the early stages of infection of RK13 cells with PRV (19). Although there are no supporting data available, dissociation of incoming ETIF from the EHV-1 particle and subsequent localization to the nucleus is likely. Considering that ETIF can be recycled, it is conceivable that ETIF is added to progeny virus capsids within the nucleus or relocated to the cytoplasm, where it is added to newly formed nucleocapsids. This latter hypothesis will be addressed in future experiments.
Our hypothesis of recycling of ETIF after infection with phenotypically complemented ETIF-negative EHV-1 is also supported by the results of the low-MOI infections, which minimized the amount of incoming ETIF. Infection of noncomplementing RK13 cells with low MOIs of complemented ETIF-null virus resulted in strictly MOI-dependent titers of virus progeny that were up to 10-fold higher than input titers. In contrast to infection with wild-type virus or infection of complementing cell line RK-ETIF, complete infection of all cells was never observed after infection of RK13 cells with vL11ΔETIF. Upon serial propagation of vL11ΔETIF in RK13 cells, virus titers diminished until no infectious virus could be recovered, usually around the fourth passage. These data indicate that input ETIF protein derived from infecting virions is not sufficient to serve for more than few rounds of replication. Recycling of ETIF for production of finally enveloped particles would likely require incorporation into newly formed particles. In purified virions of ETIF-negative virus propagated once on noncomplementing cells, no ETIF, however, could ever be detected by Western blotting. This likely indicates that the amounts of ETIF that need to be incorporated into virions for successful secondary envelopment are minute. Taken together, our results suggested that infection of RK13 cells with complemented vL11ΔETIF and subsequent passage on RK13 cells is a self-limiting infection, which support data from transfection experiments that ETIF is required for productive virus assembly and egress.
The assembly and the egress properties of ETIF mutant viruses were addressed by electron microscopy. In the late phase of infection of the ETIF-null mutant on RK13 cells, a clear defect in virus morphogenesis was detected, similar to the observations of VP16-negative HSV mutants, which were severely impaired in virus egress, presumably caused by a defect in an assembly step after primary envelopment (41, 64). Characteristic for this mutant was that capsid formation, DNA encapsidation, and nuclear egress seemed to be identical to the situation in cells infected with wild-type virus. However, many naked capsids in the cytoplasm but not enveloped particles in the cytoplasm or the extracellular space could be detected (41). Similarly, a UL48-negative mutant of PRV exhibiting a severe growth defect but able to grow on noncomplementing cells showed a massive defect in virus morphogenesis that resulted in accumulation of nucleocapsids in the cytoplasm (17). The molecular and mechanistic details of herpesvirus egress are still not completely understood. In a proposed model comprising the sequential envelopment/de-envelopment/re-envelopment process for herpesvirus maturation (36, 52), tegumentation of herpesvirus capsids occurs at two different sites (reviewed in reference 37). One site is the nucleocapsid in the cytoplasm after de-envelopment where UL36 and UL37 are added, and the other is the future budding site involving other tegument proteins and their interaction with cytoplasmic domains of viral glycoproteins located at membranes of cytoplasmic vesicles. Previous studies and the present study indicate that HSV-1 VP16, the PRV UL48 protein, and EHV-1 ETIF are apparently not required for the initiation of both processes (17, 41, 64). It is therefore plausible that this tegument protein plays an important structural role in linking tegument proteins to each other, tegument proteins with envelope proteins, or may be required for efficient connection of the tegumented capsid with the glycoprotein-containing vesicle during budding. Multiple interactions of HSV-1 VP16 with tegument (e.g., VP22, Vhs [the UL41 product]) and envelope glycoproteins (e.g., gH) have been reported and emphasize its important structural role (13, 23, 53, 68). The interaction of VP16 with Vhs, which precludes degradation of viral mRNA mediated by Vhs, is an important posttranscriptional regulatory function of VP16 (32). In EHV-1, a similar interaction between ETIF and Vhs might be possible and could contribute to some extent to the phenotype of vL11ΔETIF. Studies on Vhs in EHV-1 have shown that no host protein shutoff was mediated by the UL41 homologous protein, although the shutoff function is conserved as shown by transient-transfection experiments of vhs (16). It is not known, however, whether ETIF interacts with Vhs and thereby modulates its function. Further studies will be conducted to identify possible interaction partners of ETIF and the exact role of EHV-1 ETIF in virus maturation, with particular emphasis on the nature and role of the three different ETIF protein species detected both in infected cells and purified EHV-1 virions.
Acknowledgments
We thank Barry Wanner for providing recombinant plasmids and bacteria and Lindsey Day and Richard Killington for supplying MAbs.
This study was supported by grant DEQ-73 from the Morris Animal Foundation and the Harry M. Zweig Memorial Fund for Equine Research to N.O. and by NIH grants AI22001 P20-RR018724 to D.J.O.
REFERENCES
- 1.Ace, C. I., M. A. Dalrymple, F. H. Ramsay, V. G. Preston, and C. M. Preston. 1988. Mutational analysis of the herpes simplex virus type 1 trans-inducing factor Vmw65. J. Gen. Virol. 69:2595-2605. [DOI] [PubMed] [Google Scholar]
- 2.Ace, C. I., T. A. McKee, J. M. Ryan, J. M. Cameron, and C. M. Preston. 1989. Construction and characterization of a herpes simplex virus type 1 mutant unable to transinduce immediate-early gene expression. J. Virol. 63:2260-2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bowles, D. E., V. R. Holden, Y. Zhao, and D. J. O'Callaghan. 1997. The ICP0 protein of equine herpesvirus 1 is an early protein that independently transactivates expression of all classes of viral promoters. J. Virol. 71:4904-4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowles, D. E., S. K. Kim, and D. J. O'Callaghan. 2000. Characterization of the transactivation properties of equine herpesvirus 1 EICP0 protein. J. Virol. 74:1200-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell, M. E., J. W. Palfreyman, and C. M. Preston. 1984. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate-early transcription. J. Mol. Biol. 180:1-19. [DOI] [PubMed] [Google Scholar]
- 6.Caughman, G. B., J. Staczek, and D. J. O'Callaghan. 1985. Equine herpesvirus type 1-infected cell polypeptides: evidence for immediate early/early/late regulation of viral gene expression. Virology 145:49-61. [DOI] [PubMed] [Google Scholar]
- 7.Cohen, J. I., and K. Seidel. 1994. Varicella-zoster virus (VZV) open reading frame 10 protein, the homolog of the essential herpes simplex virus protein VP16, is dispensable for VZV replication in vitro. J. Virol. 68:7850-7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colle, C. F., and D. J. O'Callaghan. 1996. Localization of the Us protein kinase of equine herpesvirus type 1 is affected by the cytoplasmic structures formed by the noval IR6 protein. Virology 220:424-435. [DOI] [PubMed] [Google Scholar]
- 9.Cousens, D. J., R. Greaves, C. R. Goding, and P. O'Hare. 1989. The C-terminal 79 amino acids of the herpes simplex virus regulatory protein, Vmw65, efficiently activate transcription in yeast and mammalian cells in chimeric DNA-binding proteins. EMBO J. 8:2337-2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Day, L. 1999. Ph.D. thesis. University of Leeds, United Kingdom. Characterisation of selected glycoproteins of equine herpesvirus-1: immune in the murine model.
- 12.Dorange, F., B. K. Tischer, J. F. Vautherot, and N. Osterrieder. 2002. Characterization of Marek's disease virus serotype 1 (MDV-1) deletion mutants that lack UL46 to UL49 genes: MDV-1 UL49, encoding VP22, is indispensable for virus growth. J. Virol. 76:1959-1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliott, G., G. Mouzakitis, and P. O'Hare. 1995. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J. Virol. 69:7932-7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elliott, G. D. 1994. The extreme carboxyl terminus of the equine herpesvirus 1 homolog of herpes simplex virus VP16 is essential for immediate-early gene activation. J. Virol. 68:4890-4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elliott, G., and P. O'Hare. 1995. Equine herpesvirus 1 Gone 12, the functional homologue of herpes simplex virus VP16, transactivates via octamer sequences in the equine herpesvirus IE Gone promoter. Virology 213:258-262. [DOI] [PubMed] [Google Scholar]
- 16.Feng, X., Y. G. Thompson, J. B. Lewis, and G. B. Caughman. 1996. Expression and function of the equine herpesvirus 1 virion-associated host shutoff homolog. J. Virol. 70:8710-8718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuchs, W., H. Granzow, B. G. Klupp, M. Kopp, and T. C. Mettenleiter. 2002. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J. Virol. 76:6729-6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garko-Buczynski, K. A., R. H. Smith, S. K. Kim, and D. J. O'Callaghan. 1998. Complementation of a replication-defective mutant of equine herpesvirus type 1 by a cell line expressing the immediate-early protein. Virology 248:83-94. [DOI] [PubMed] [Google Scholar]
- 19.Granzow, H., B. G. Klupp, and T. C. Mettenleiter. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J. Virol. 79:3200-3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grapes, M., and P. O'Hare. 2000. Differences in determinants required for complex formation and transactivation in related VP16 proteins. J. Virol. 74:10112-10121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gray, W. L., R. P. Baumann, A. T. Robertson, G. B. Caughman, D. J. O'Callaghan, and J. Staczek. 1987. Regulation of equine herpesvirus type 1 gene expression: characterization of immediate early, early, and late transcription. Virology 158:79-87. [DOI] [PubMed] [Google Scholar]
- 22.Greaves, R. F., and P. O'Hare. 1991. Sequence, function, and regulation of the Vmw65 gene of herpes simplex virus type 2. J. Virol. 65:6705-6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gross, S. T., C. A. Harley, and D. W. Wilson. 2003. The cytoplasmic tail of herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology 317:1-12. [DOI] [PubMed] [Google Scholar]
- 24.Harty, R. N., and D. J. O'Callaghan. 1991. An early gene maps within and is 3′ coterminal with the immediate-early gene of equine herpesvirus 1. J. Virol. 65:3829-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holden, V. R., Y. Zhao, Y. Thompson, G. B. Caughman, R. H. Smith, and D. J. O'Callaghan. 1995. Characterization of the regulatory function of the ICP22 protein of equine herpesvirus type 1. Virology 210:273-282. [DOI] [PubMed] [Google Scholar]
- 26.Honess, R. W., and B. Roizman. 1974. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 14:8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim, S. K., V. R. Holden, and D. J. O'Callaghan. 1997. The ICP22 protein of equine herpesvirus 1 cooperates with the IE protein to regulate viral gene expression. J. Virol. 71:1004-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim, S. K., and D. J. O'Callaghan. 2001. Molecular characterizations of the equine herpesvirus 1 ETIF promoter region and translation initiation site. Virology 286:237-247. [DOI] [PubMed] [Google Scholar]
- 29.Kinchington, P. R., J. K. Hougland, A. M. Arvin, W. T. Ruyechan, and J. Hay. 1992. The varicella-zoster virus immediate-early protein IE62 is a major component of virus particles. J. Virol. 66:359-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kyhse-Andersen, J. 1984. Electroblotting of multiple gels: a simple apparatus without buffer tank for rapid transfer of proteins from polyacrylamide to nitrocellulose. J. Biochem. Biophys. Methods 10:203-209. [DOI] [PubMed] [Google Scholar]
- 31.La Boissiere, S., A. Izeta, S. Malcomber, and P. O'Hare. 2004. Compartmentalization of VP16 in cells infected with recombinant herpes simplex virus expressing VP16-green fluorescent protein fusion proteins. J. Virol. 78:8002-8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lam, Q., C. A. Smibert, K. E. Koop, C. Lavery, J. P. Capone, S. P. Weinheimer, and J. R. Smiley. 1996. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J. 15:2575-2581. [PMC free article] [PubMed] [Google Scholar]
- 33.Lewis, J. B., Y. G. Thompson, and G. B. Caughman. 1993. Transcriptional control of the equine herpesvirus 1 immediate-early gene. Virology 197:788-792. [DOI] [PubMed] [Google Scholar]
- 34.Lewis, J. B., Y. G. Thompson, X. Feng, V. R. Holden, D. O'Callaghan, and G. B. Caughman. 1997. Structural and antigenic identification of the ORF12 protein ([alpha]TIF) of equine herpesvirus 1. Virology 230:369-375. [DOI] [PubMed] [Google Scholar]
- 35.McFarlane, M., J. I. Daksis, and C. M. Preston. 1992. Hexamethylene bisacetamide stimulates herpes simplex virus immediate-early gene expression in the absence of trans-induction by Vmw65. J. Gen. Virol. 73:285-292. [DOI] [PubMed] [Google Scholar]
- 36.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mettenleiter, T. C. 2004. Budding events in herpesvirus morphogenesis. Virus Res. 106:167-180. [DOI] [PubMed] [Google Scholar]
- 38.Misra, V., A. C. Bratanich, D. Carpenter, and P. O'Hare. 1994. Protein and DNA elements involved in transactivation of the promoter of the bovine herpesvirus (BHV) 1 IE-1 transcription unit by the BHV alpha gene trans-inducing factor. J. Virol. 68:4898-4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moriuchi, H., M. Moriuchi, S. E. Straus, and J. I. Cohen. 1993. Varicella-zoster virus open reading frame 10 protein, the herpes simplex virus VP16 homolog, transactivates herpesvirus immediate-early gene promoters. J. Virol. 67:2739-2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morrison, E. E., A. J. Stevenson, Y. F. Wang, and D. M. Meredith. 1998. Differences in the intracellular localization and fate of herpes simplex virus tegument proteins early in the infection of Vero cells. J. Gen. Virol. 79:2517-2528. [DOI] [PubMed] [Google Scholar]
- 41.Mossman, K. L., R. Sherburne, C. Lavery, J. Duncan, and J. R. Smiley. 2000. Evidence that herpes simplex virus VP16 is required for viral egress downstream of the initial envelopment event. J. Virol. 74:6287-6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mossman, K. L., and J. R. Smiley. 1999. Truncation of the C-terminal acidic transcriptional activation domain of herpes simplex virus VP16 renders expression of the immediate-early genes almost entirely dependent on ICP0. J. Virol. 73:9726-9733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Osterrieder, N., A. Neubauer, B. Fakler, C. Brandmuller, C. Seyboldt, O. R. Kaaden, and J. D. Baines. 1997. Synthesis and processing of the equine herpesvirus 1 glycoprotein M. Virology 232:230-239. [DOI] [PubMed] [Google Scholar]
- 44.Purewal, A. S., R. Allsopp, M. Riggio, E. A. Telford, S. Azam, A. J. Davison, and N. Edington. 1994. Equid herpesviruses 1 and 4 encode functional homologs of the herpes simplex virus type 1 virion transactivator protein, VP16. Virology 198:385-389. [DOI] [PubMed] [Google Scholar]
- 45.Purewal, A. S., A. V. Smallwood, A. Kaushal, D. Adegboye, and N. Edington. 1992. Identification and control of the cis-acting elements of the immediate-early gene of equid herpesvirus type 1. J. Gen. Virol. 73:513-519. [DOI] [PubMed] [Google Scholar]
- 46.Rudolph, J., D. J. O'Callaghan, and N. Osterrieder. 2002. Cloning of the genomes of equine herpesvirus type 1 (EHV-1) strains KyA and racL11 as bacterial artificial chromosomes (BAC). J. Vet. Med. B Infect. Dis. Vet. Public Health 49:31-36. [DOI] [PubMed] [Google Scholar]
- 47.Rudolph, J., and N. Osterrieder. 2002. Equine herpesvirus type 1 devoid of gM and gp2 is severely impaired in virus egress but not direct cell-to-cell spread. Virology 293:356-367. [DOI] [PubMed] [Google Scholar]
- 48.Rudolph, J., C. Seyboldt, H. Granzow, and N. Osterrieder. 2002. The gene 10 (UL49.5) product of equine herpesvirus 1 is necessary and sufficient for functional processing of glycoprotein M. J. Virol. 76:2952-2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sambrook, J., and D. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 50.Schumacher, D., B. K. Tischer, S. Trapp, and N. Osterrieder. 2005. The protein encoded by the US3 orthologue of Marek's disease virus is required for efficient de-envelopment of perinuclear virions and involved in actin stress fiber breakdown. J. Virol. 79:3987-3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seyboldt, C., H. Granzow, and N. Osterrieder. 2000. Equine herpesvirus 1 (EHV-1) glycoprotein M: effect of deletions of transmembrane domains. Virology 278:477-489. [DOI] [PubMed] [Google Scholar]
- 52.Skepper, J. N., A. Whiteley, H. Browne, and A. Minson. 2001. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment → deenvelopment → reenvelopment pathway. J. Virol. 75:5697-5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smibert, C. A., B. Popova, P. Xiao, J. P. Capone, and J. R. Smiley. 1994. Herpes simplex virus VP16 forms a complex with the virion host shutoff protein vhs. J. Virol. 68:2339-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smiley, J. R., and J. Duncan. 1997. Truncation of the C-terminal acidic transcriptional activation domain of herpes simplex virus VP16 produces a phenotype similar to that of the in1814 linker insertion mutation. J. Virol. 71:6191-6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith, R. H., G. B. Caughman, and D. J. O'Callaghan. 1992. Characterization of the regulatory functions of the equine herpesvirus 1 immediate-early gene product. J. Virol. 66:936-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith, R. H., V. R. Holden, and D. J. O'Callaghan. 1995. Nuclear localization and transcriptional activation activities of truncated versions of the immediate-early gene product of equine herpesvirus 1. J. Virol. 69:3857-3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith, R. H., Y. Zhao, and D. J. O'Callaghan. 1993. The equine herpesvirus 1 (EHV-1) UL3 gene, an ICP27 homolog, is necessary for full activation of gene expression directed by an EHV-1 late promoter. J. Virol. 67:1105-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Telford, E. A., M. S. Watson, K. McBride, and A. J. Davison. 1992. The DNA sequence of equine herpesvirus-1. Virology 189:304-316. [DOI] [PubMed] [Google Scholar]
- 59.Tischer, B. K., J. von Einem, B. Kaufer, and K. Osterrieder. Two-step Red-mediated recombination for versatile, high-efficiency markerless DNA manipulation in Escherichia coli. BioTechniques, in press. [DOI] [PubMed]
- 60.Trapp, S., N. Osterrieder, G. M. Keil, and M. Beer. 2003. Mutagenesis of a bovine herpesvirus type 1 genome cloned as an infectious bacterial artificial chromosome: analysis of glycoprotein E and G double deletion mutants. J. Gen. Virol. 84:301-306. [DOI] [PubMed] [Google Scholar]
- 61.Triezenberg, S. J., R. C. Kingsbury, and S. L. McKnight. 1988. Functional dissection of VP16, the transactivator of herpes simplex virus immediate-early gene expression. Genes Dev. 2:718-729. [DOI] [PubMed] [Google Scholar]
- 62.von Einem, J., J. Wellington, J. M. Whalley, K. Osterrieder, D. J. O'Callaghan, and N. Osterrieder. 2004. The truncated form of glycoprotein gp2 of equine herpesvirus 1 (EHV-1) vaccine strain KyA is not functionally equivalent to full-length gp2 encoded by EHV-1 wild-type strain RacL11. J. Virol. 78:3003-3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walker, S., R. Greaves, and P. O'Hare. 1993. Transcriptional activation by the acidic domain of Vmw65 requires the integrity of the domain and involves additional determinants distinct from those necessary for TFIIB binding. Mol. Cell. Biol. 13:5233-5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weinheimer, S. P., B. A. Boyd, S. K. Durham, J. L. Resnick, and D. R. O'Boyle. 1992. Deletion of the VP16 open reading frame of herpes simplex virus type 1. J. Virol. 66:258-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wysocka, J., and W. Herr. 2003. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem. Sci. 28:294-304. [DOI] [PubMed] [Google Scholar]
- 66.Yang, W. C., G. V. Devi-Rao, P. Ghazal, E. K. Wagner, and S. J. Triezenberg. 2002. General and specific alterations in programming of global viral gene expression during infection by VP16 activation-deficient mutants of herpes simplex virus type 1. J. Virol. 76:12758-12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao, Y., V. R. Holden, R. H. Smith, and D. J. O'Callaghan. 1995. Regulatory function of the equine herpesvirus 1 ICP27 gene product. J. Virol. 69:2786-2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu, Q., and R. J. Courtney. 1994. Chemical cross-linking of virion envelope and tegument proteins of herpes simplex virus type 1. Virology 204:590-599. [DOI] [PubMed] [Google Scholar]