

Abstract
The role of Rac family proteins in platelet spreading on matrix proteins under static and flow conditions has been investigated by using Rac-deficient platelets. Murine platelets form filopodia and undergo limited spreading on fibrinogen independent of Rac1 and Rac2. In the presence of thrombin, marked lamellipodia formation is observed on fibrinogen, which is abrogated in the absence of Rac1. However, Rac1 is not required for thrombin-induced aggregation or elevation of F-actin levels. Formation of lamellipodia on collagen and laminin is also Rac1-dependent. Analysis of platelet adhesion dynamics on collagen under flow conditions in vitro revealed that Rac1 is required for platelet aggregate stability at arterial rates of shear, as evidenced by a dramatic increase in platelet embolization. Furthermore, studies employing intravital microscopy demonstrated that Rac1 plays a critical role in the development of stable thrombi at sites of vascular injury in vivo. Thus, our data demonstrated that Rac1 is essential for lamellipodia formation in platelets and indicated that Rac1 is required for aggregate integrity leading to thrombus formation under physiologically relevant levels of shear both in vitro and in vivo.
The Rho family of small GTPases, which includes Rac, Rho, and Cdc42 proteins, plays distinct roles in regulating actin assembly and motility. These proteins cycle between an inactive (GDP-bound) and an active (GTP-bound) conformation that can subsequently interact with specific effector proteins. There are three members of the Rac family in mammals, all of which share a common structural arrangement. Rac1 is ubiquitously expressed and is the most extensively studied isoform. Rac2 is specifically expressed in hematopoietic cells, whereas Rac3 is expressed primarily in the brain during development. These three iso-forms of Rac share between 89 and 93% identity in their amino acid sequence (1).
Initial studies in Swiss 3T3 fibroblasts demonstrated that Rac promotes polymerization of actin at the cell membrane, producing lamellipodia and membrane ruffles (2). This role has since been confirmed in a wide variety of cell types, including platelets, by using constitutively active and dominant negative mutants of Rac (3-5) and through the generation of mice deficient in Rac1 or Rac2 (6, 7), although the role of individual isoforms has not been investigated in platelets. Rac is believed to stimulate actin polymerization through the SCAR (suppressor of cyclic AMP receptor)/WAVE (WASP family verprolin-homologous protein) family of Rac-binding proteins. The SCAR/WAVE family activates the Arp2/3 complex, which nucleates new barbed ends by forming branches on actin filaments near protruding edges of the cell (8-10). The activation of the Rac-SCAR/WAVE-Arp2/3 pathway at the leading edge of a cell triggers actin polymerization and growth of lamellipodia (11). Rac is also implicated in the regulation of a number of other pathways, including the oxidative burst in neutrophils, regulation of synthesis of phosphatidylinositol 4,5-bisphosphate, and gene expression essential for cellular proliferation and hypertrophy (12).
Quiescent platelets circulate in the bloodstream as biconcave discs. The activation of platelets at sites of vascular injury plays a key role in hemostasis. Upon activation on an immobilized matrix, they round up, extend filopodia, and form lamellipodia. This change in morphology serves to increase the platelet surface area and thereby strengthens the contact with the immobilized surface as well as with other platelets. This change in morphology is mediated by the rapid reorganization of the cortical actin cytoskeleton through a combination of uncapping, severing, and nucleation. Previous work has indicated that gelsolin-mediated uncapping and severing of existing F-actin filaments play a critical early role in stimulating polymerization of actin monomers to form new filaments (13). Rac was originally proposed to promote exposure of actin ends through the synthesis of polyphosphoinositides (5), although it now seems more likely that it primarily mediates this event via the Arp2/3 complex. The gelsolin and Rac pathways have been proposed to act in tandem, with gelsolin promoting actin filament uncapping and severing, and Rac activating the Arp2/3 complex, thereby leading to rapid actin polymerization of new filaments at the leading edge of lamellipodia (14). A role for SCAR/WAVE in the development of platelet lamellipodia downstream of Rac and D3-(PI3,4P2,PI3,4,5P3) containing polyphosphoinositides has been proposed (15), although this has not yet been supported by functional evidence. Several groups have reported activation of Rac in platelets by G protein-coupled receptor agonists, including thrombin, although the nature of the active isoform is not established (5, 16-19). Rac is also activated downstream of the tyrosine kinase-linked collagen receptor, GPVI, which signals through sequential activation of Src and Syk tyrosine kinases, through a pathway that this is dependent on release of the G protein-coupled secondary mediators, ADP and thromboxane A2(18-20).
In the present study, we have investigated the presence of all three isoforms of Rac in platelets and have used platelets from Rac1-, Rac2-, and Rac1/Rac2-deficient mice to investigate the role of these two iso-forms in supporting spreading on fibrinogen in the absence and presence of G protein-coupled receptor and GPVI-specific agonists. The results demonstrate a central role for Rac1 in supporting formation of lamellipodia but not filopodia in thrombin- or ADP-stimulated platelets on a fibrinogen surface. Furthermore, Rac1 is shown to be essential for GPVI-regulated platelet spreading as well as sustaining shear-resistant platelet aggregates under dynamic flow conditions both in vitro and in vivo. Together this study demonstrates a cooperative role of integrin and G protein-coupled receptors in regulating Rac activation and lamellipodia formation in platelets that enables them to withstand the high shear forces to which they are exposed in the vasculature.
EXPERIMENTAL PROCEDURES
Reagents—The αIIbβ3 antagonist lotrafiban was supplied by GlaxoSmithKline (King of Prussia, PA). The anti-Rac (23A8) monoclonal antibody was purchased from Upstate Biotechnology (TCS Biologicals, Bucks, UK). Anti-Rac2 polyclonal antibody and anti-Rac3 polyclonal antibody were generously provided from Gary Bokoch (Scripps Institute, La Jolla, CA) and Ivan de Curtis (San Rafaele Scientific Institute, Milan, Italy), respectively. The cDNA for the GST-CRIB domain of PAK1 prepared as described previously (21) and the active form of Rac (L61Rac) were the kind gifts from Dr. Doreen Cantrell (Imperial Cancer Research Fund, London, UK). D-Phenyl-alanyl-1-prolyl-1 arginine chloromethyl ketone was purchased from Calbiochem. Fibrinogen depleted of plasminogen, VWF, and fibronectin was from Kordia Laboratory Supplies, Leiden, Netherlands. VWF was a generous gift from Michael C. Berndt (Monash University, Clayton, Australia). All other reagents were from Sigma or previously named sources (22, 23).
Preparation of Human Washed Platelets—Human venous blood was drawn by venipuncture from healthy volunteers into sodium citrate and acid/citrate/dextrose as described previously (23). Platelet-rich plasma (PRP) was prepared by centrifugation of whole blood at 200 × g for 20 min. The platelets were then isolated from PRP by centrifugation at 1000 × g for 10 min in the presence of prostacyclin (0.1 μg/ml). The pellet was resuspended in modified HEPES/Tyrodes buffer (in mM: 129 NaCl, 0.34 Na2HPO4, 2.9 KCl, 12 NaHCO3, 20 HEPES, 5 glucose, 1 MgCl2; pH 7.3) containing 0.1 μg/ml prostacyclin. The platelets were washed once via centrifugation (1000 × g for 10 min) and resuspended at the desired concentration with HEPES/Tyrode buffer.
Preparation of Murine Washed Platelets—The generation of mice bearing a conditional loxP-flanked allele of Rac1, Rac1flox, has been described previously (6). To induce expression of the Mx1-Cre trans-gene, the mice were given a 150-μl intraperitoneal injection of synthetic double-stranded RNA polyinosinic-polycytidylic acid (2 mg/ml) every other day for a total of three injections, and blood was taken at least 14 days after the last injection to ensure a complete turnover of platelets. The protein expression of Rac1 and Rac2 was verified for both control and Rac-deficient mice for each experiment (data not shown). The number of platelets in whole blood from Rac1-/-, Rac2-/-, or Rac1-/-Rac2-/- mice was no different from wild type (data not shown). Bleeding problems, such as the intraperitoneal hemorrhage seen in Syk- and SLP-76-deficient mice, were not observed for these mice.
Murine blood was drawn from CO2terminally anesthetized mice by cardiac puncture and taken into 100 μl of acid/citrate/dextrose. PRP was obtained by centrifugation at 200 χ g for 6 min. Washed platelets were prepared via centrifugation of PRP at 1000 × g in the presence of prostacyclin (0.1 μg/ml) for 6 min. The pellet was resuspended in modified HEPES/Tyrode buffer to the desired platelet level. All animals were maintained using housing and husbandry in accordance with local and national legal regulations.
In separate experiments, human or murine platelet suspensions were treated with 10 μM cytochalasin D, 0.1-10 μg/ml CRP, 1-10 μM ADP, 0.04-1 units/ml thrombin, 10 μM lotrafiban or 1 mM adenosine 3′,5′-diphosphate (A3P5P), and 1 μM AR-C67085 for 10-30 min before use in the assays. Unless otherwise stated, all experiments were performed in the presence of 2 units/ml apyrase and 10 μM indomethacin and in the absence of exogenously added Ca2+.
Measurement of Rac Activity—Rac activity was measured essentially as described in Pearce et al. (18) using the CRIB domain of PAK1 (amino acids 67-150), which binds the GTP-bound form of Rac. Following stimulation of platelet suspensions (3 × 108/ml), reactions were stopped with an equal volume of 2× lysis buffer (2% (v/v) Nonidet P-40, 2% (w/v) N-octyl glucoside, 300 mM NaCl, 20 mM Tris/HCl, 2 mM EGTA, 20 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 μg/ml pepstatin A, pH 7.4, and 2 mM orthovana-date). Alternatively, platelets (3 × 108/ml) were incubated for 10-45 min in dishes coated with fibrinogen, collagen, laminin, or BSA2in the absence or presence of thrombin (1 units/ml) and apyrase (2 units/ml). Unbound platelets were removed by two washes with phosphate-buffered saline followed by aspiration, and adherent cells were solubilized with 1× lysis buffer. A sample of the suspension over BSA was taken and used as a control. Insoluble material was then removed by centrifugation (5 min, 13,000 rpm), and GST-PAK1, previously incubated with glutathione agarose beads, stored in glycerol at -80 °C, and washed with 1× lysis buffer, was added to the lysates and incubated for 1 h at 4 °C. Beads were then washed with 1× lysis buffer, and the bound protein was taken up into Laemmli buffer. The resulting samples were separated by 12% SDS-PAGE, transferred to polyvinylidene difluoride membranes, and immunoblotted with a Rac-specific antibody and horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences). Protein was detected using ECL (Amersham Biosciences).
Measurement of Filamentous Actin Content—Filamentous actin content of washed platelets was measured using a modification of the method of Machesky and Hall (24). Basal or activated platelets (2 × 108/ml) were fixed with an equal volume of 3.7% formaldehyde containing a saturating amount of FITC-phalloidin (20 mM KH2PO4, 10 mM Pipes, 5 mM EGTA, 2 mM MgCl2, 0.1% Triton X-100, 3.7% formalde-hyde, 2 μM FITC-phalloidin) and incubated for1hat room temperature on a nutator. The platelets were then pelleted for 2 min in a microcentrifuge, and pellets were washed in 0.1% saponin, 20 mM KH2PO4, 10 mM Pipes, 5 mM EGTA, 2 mM MgCl2. Pellets were then resuspended in methanol to extract the FITC-phalloidin and incubated for 1 h on a nutator at room temperature. FITC-phalloidin binding was measured for each sample with the fluorescence emission at 520 nm and excitation at 488 nm. Alternatively, F-actin levels were assessed via flow cytometry after fixation, permeabilization, and staining of platelets with FITCphalloidin as described previously (18). Filamentous actin content was expressed as a comparison with values obtained for untreated cells processed in parallel on the same day.
Platelet Aggregation and Shape Change—A quantity of 300 μl of PRP or washed platelets (2 × 108/ml) was used for aggregation. Stimulation of platelets was performed in a PAP-4 aggregometer (Bio/Data Corp., Horsham, PA) with continuous stirring at 1200 rpm at 37 °C for the times shown. Aggregation of platelets was monitored by measuring changes in light transmission.
Static Adhesion Assays—Coverslips were incubated with a suspension of fibrinogen (100 μg/ml), collagen (100 μg/ml), or laminin (50 μg/ml) overnight at 4 °C. Surfaces were then blocked with denatured BSA (5 mg/ml) for 1 h at room temperature followed by subsequent washing with phosphate-buffered saline before use in spreading assays. Quiescent platelets failed to bind to surfaces coated with denatured BSA (data not shown).
Platelet spreading (2 × 107/ml) was imaged in real time using Köhler illuminated Nomarski differential interference contrast optics with a Zeiss 63× oil immersion 1.40 NA plan-apochromat lens on a Zeiss Axiovert 200M microscope. Time-lapse events were captured by a Hamamatsu Orca 285 cooled digital camera (Cairn Research, Kent, UK) using Slidebook 4.0 (Intelligent Imaging Innovations, Inc., Denver, CO). To compute the length and thickness of filopodia and surface area of spreading platelets, images were manually outlined and quantitated by determining the number of pixels within each outline using a Java plugin for the Image J software package as described previously (23). Imaging a graticule under the same conditions allowed the conversion of pixels size to microns.
Flow Adhesion Studies—For flow adhesion studies using collagen, mouse blood was drawn into sodium heparin (10 IU/ml) and D-phenylalanyl-1-prolyl-1 arginine chloromethyl ketone (40 μM). Alternatively, mouse blood was drawn into sodium citrate (0.38% w/v) for immobilized VWF adhesion studies. Glass capillary tubes (Camlab, Cambridge, UK) were coated with 100 μg/ml type I collagen from equine tendon (Horm, Nycomed, Munich, Germany) or 100 μg/ml VWF, 100 units/ml thrombin for1hat room temperature. The capillaries were washed and blocked with phosphate-buffered saline containing 5 mg/ml BSA for 1 h at room temperature before being mounted on the stage of an inverted microscope (DM IRB; Leica, Milton Keynes, UK). Anticoagulated whole blood was perfused through the chamber for 4 min at a wall shear rate of 1000 s-1, followed by washing for 3 min at the same shear rate with modified Tyrodes buffer before being fixed with 3.7% paraformaldehyde for 30 min and imaged using DIC microscopy. In selected experiments, fixed samples were incubated overnight with the fluorescent dye DiOC6(2 μM; Molecular Probes Inc., Eugene, Oregon) before being imaged using confocal microscopy (DM IRE2; Leica, Milton Keynes, UK). In separate experiments, whole blood was fluorescently labeled with DiOC6(2 μM, 10 min at 37 °C), and the accumulation of DiOC6-labeled platelets was monitored in real time using fluorescence microscopy (CoolSnap ES, Photometrics, Huntington Beach, CA).
Image analysis was performed off-line using ImageJ. Platelet adhesion results are expressed as the percentage of surface area covered by platelets.
Laser-induced Vessel Wall Injury—All procedures were undertaken with approval from the United Kingdom Home Office in accordance with the Animals (Scientific Procedures) Act of 1986. Male mice were anesthetized with ketamine (100 mg/kg Vetalar; Amersham Biosciences and Upjohn Ltd., UK) and 2% xylazine (20 mg/kg; Millpledge Pharmaceuticals, UK). The cremaster, a transparent muscle surrounding the testicle, was exteriorized and continuously superfused with a bicarbonate-buffered saline (36 °C) aerated with 5% CO2, 95% N2. High speed intravital microscopy experiments were performed as described previously by Falati et al. (25). Arterioles with a diameter of 25-35 μm were selected for study, and endothelial injury was induced using a pulsed nitrogen dye laser (coumarin 440 nm) focused on the luminal surface. To label platelets fluorescently, 20 μl of Alexa Fluor 488 conjugated to goat anti-rat antibody (Molecular Probes, Eugene, OR) and 5 μl of rat anti-mouse CD41 antibody (Pharmingen) were added to 70 μl of saline and infused via the carotid cannula. Multiple thrombi in each cremaster preparation were generated upstream to previous injuries in the same or similar sized arterioles. The background fluorescence intensity, predominantly because of freely circulating platelets, was determined and subtracted from the fluorescence intensity of the developing thrombus. The resulting value was multiplied by the sum of all pixels above background to give a value for integrated intensity at each time point. This integrated intensity value was directly proportional to the size of the developing thrombus and when plotted against time provided a graph that illustrated the dynamic kinetics of platelet accumulation.
Analysis of Data—Experiments were carried out on at least three occasions, and images shown are representative data from one experiment. Where applicable, results are shown as mean ± S.E. Statistical significance of differences between the means was determined by analysis of variance. If the means were shown to be significantly different, multiple comparisons were performed by the Tukey test. Probability values of p < 0.01 were selected to be statistically significant.
RESULTS
Identification of Rac Isoforms Present in Platelets—To study the role of Rac proteins in regulating the organization of the platelet actin cytoskeleton, we first examined which Rac proteins are expressed in murine and human platelets. Previous reports have shown that Rac1 is ubiquitously expressed, whereas Rac2 is a hematopoietic cell-specific GTPase (26). Rac3 is thought to play an important role in neural development and has been shown to have a specific localization in the developing mouse brain (27). In an attempt to identify which Rac isoforms are present in platelets, we first examined a serial analysis of gene expression (SAGE) library that had been prepared from primary murine megakaryocytes, the platelet precursor cell.3This revealed the presence of 57 Rac1, 4 Rac2, and 0 Rac3 sequence tags out of a total of 53,046, indicating that Rac1 was likely to be the predominant isoform and that Rac3 was very unlikely to be present, consistent with its restricted distribution.
Specific antibodies were then used to verify which Rac isoforms are present at the protein level in murine and human platelets. By using specific antibodies against all three family members, which do not exhibit cross-reactivity with the other isoforms (data not shown), immunoblot analyses established that murine and human platelets express Rac1 but have no detectable Rac2 expression at the protein level (Fig. 1). Wild-type and Rac2-/-thymocyte lysates were run alongside as a positive control. Furthermore, there were no compensatory changes in the levels of Rac1 or Rac 2 in Rac2-/-and Rac1-/-platelets, respectively. A specific antibody to Rac3, raised against the 15 carboxyl-terminal residues (27), failed to detect expression of Rac3 in human or murine platelets (data not shown). These data confirm that Rac1 is the predominant isoform in human and murine platelets. They further indicate that neither Rac2 nor Rac3 is likely to be present in platelets, although the small number of SAGE tags for Rac2 raises the possibility that it may be expressed at a very low level, and for this reason later experiments were performed on mice deficient in Rac1, Rac2, and both of these Rac isoforms.
FIGURE 1.

Rac isoforms present in murine and human platelets (plt). Equal amounts of human and murine platelet lysates were analyzed for Rac expression using noncross-reactive Rac1 (A) or Rac2 (B) antibodies. Lysates from wild-type (WT) and Rac2-/-thymus were included as a positive control.
Regulation of Rac Activity in Platelets—With a view to studying the role of Rac1, we measured the activation state of the small GTPase in stimulated human and murine platelets by precipitation with a glutathione S-transferase fusion protein encompassing the CRIB domain of PAK, which has the binding region for the GTP-bound but not GDP-bound state. Stimulation of human platelet suspensions with the G protein-coupled agonist thrombin caused a marked increase in GTP-bound Rac1, which was not significantly altered in the presence of the ADP scavenger apyrase, thereby demonstrating that thrombin stimulation can lead to an increase in Rac activation independent of ADP (Fig. 2A). Stimulation with ADP also caused a smaller increase in active Rac1 (1.33 ± 0.06-fold increase as assessed by densitometry; mean ± S.E.; n = 3), which was abrogated in the presence of apyrase (1.02 ± 0.03-fold). Similar trends were observed for murine platelets (Fig. 2B). Thrombin stimulated a robust increase in GTP-bound Rac1 in the absence and presence of apyrase, whereas the smaller, ADP-dependent increase (1.23 ± 0.09-fold) was eliminated in the presence of apyrase (0.99 ± 0.04-fold).
FIGURE 2.

Effects of agonists on Rac1 activation in platelets both in suspension and on a fibrinogen surface. Equal numbers of washed human (A) or murine (B) platelets (5 × 108/ml) were stimulated with thrombin (thr; 1 unit/ml) or ADP (10μM) in the absence or presence of ADP scavenger apyrase (2 units/ml) for 60 s before lysis. Subsequently platelet lysates were incubated with agarose beads coupled with GST-PBD fusion proteins to bind GTP-bound forms of Rac1. Total lysates or material binding to the GST-PBD beads were solubilized, separated by SDS-PAGE, and detected by immunoblotting with an anti-Rac1 monoclonal antibody. In separate experiments, washed human (C) and murine (D) platelets (5 × 108/ml) were added to BSA- or fibrinogen (FG)-coated dishes in the absence or presence of thrombin (1 unit/ml) and incubated for 10 min. Dishes coated with fibrinogen were washed twice to remove nonadherent cells. Platelets adherent to fibrinogen or in suspension over BSA were lysed, and Rac activity was determined as described above. Each panel depicts one experiment representative of three independent experiments.
It is well established that platelets undergo a dramatic change in morphology upon binding to immobilized adhesive proteins. Thus, experiments were designed to determine whether Rac1 was activated during spreading on immobilized surfaces. For these studies, purified platelets were placed on fibrinogen- or BSA-coated dishes in the presence of ADP inhibitors for 10 min before lysis. As shown in Fig. 2, C and D, we were unable to detect an increase in Rac1 activation following spreading of human or mouse platelets on fibrinogen (1.04 ± 0.02- and 1.05 ± 0.04-fold increase in GTP-bound Rac1 relative to BSA sample, respectively). A similar result was observed when spreading was allowed to proceed between 1 and 45 min (data not shown). In contrast, stimulation of fibrinogen-bound platelets with thrombin caused a sustained level of Rac1 activation (Fig. 2, C and D).
The Role of Rac in Platelet Actin Assembly—Agonist-induced platelet activation is associated with an increase in cellular F-actin levels. We measured F-actin levels in human and mouse platelets using a modified version of the FITC-phalloidin binding assay (24) after stimulation with vehicle, thrombin, or ADP. As shown in Fig. 3, thrombin stimulation caused a 1.8- and 2.6-fold increase in F-actin content above resting levels in human and murine platelets, respectively, in good agreement with previous findings (28, 29). A slightly smaller increase in the levels of F-actin was observed in response to ADP stimulation in both species.
FIGURE 3.
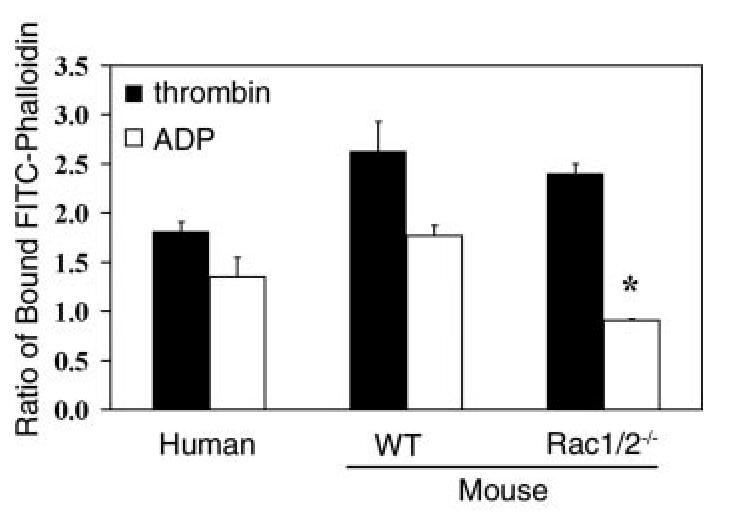
Quantitation of F-actin in human and murine platelets responding to various stimuli. Purified human and wild-type (WT) or Rac1-/-Rac2-/-murine platelets were activated with 1 unit/ml thrombin or 10 μM ADP for 60 s. Samples were then fixed with 3.7% formaldehyde, permeabilized with 0.1% Triton X-100 containing 2 μM FITC-phalloidin, and analyzed in a fluorometer as described. Results are expressed as the ratio between the fluorescence of activated cells versus resting cells. Values are mean ± S.D. (n = 2 performed in triplicate). *,p < 0.01 with respect to wild-type platelets.
The availability of mice lacking Rac1, Rac2, or both isoforms allowed us to examine the functional role of Rac in promoting platelet actin assembly. Thrombin stimulated a similar increase in the level of F-actin in platelets from Rac1-/-Rac2-/-mice to that in controls (Fig. 3). In sharp contrast, the stimulation of actin polymerization by ADP was abrogated in Rac1-/-Rac2-/-platelets. Similar results were obtained when increases in cellular F-actin content was quantified via flow cytometry (thrombin stimulation, 2.04 ± 0.08-fold versus 2.20 ± 0.28-fold increase in F-actin; ADP stimulation, 1.52 ± 0.03-fold versus 0.96 ± 0.05-fold increase in F-actin for wild-type and Rac1-/-Rac2-/-platelets, respectively; mean ± S.E.; n = 3). Similar results to those recorded for Rac1-/-Rac2-/- platelets were also observed in Rac1-deficient platelets, whereas thrombin- and ADP-induced changes in F-actin formation were indistinguishable between control and Rac2-/- platelets (data not shown). Furthermore, we found that collagen induced an increase in F-actin content in a Rac1/Rac2-dependent manner. However, we found that collagen-induced F-actin level increases were largely dependent upon the action of secondary mediators, therefore demonstrating that Rac1/Rac2 is required for the secondary effects of ADP to induced F-actin changes following collagen stimulation (data not shown). Taken together, these data demonstrate that Rac1 plays the major role in ADP-induced actin polymerization in platelets. The similar level of formation of F-actin in response to thrombin in the absence of Rac1 and Rac2 demonstrates the presence of an alternative pathway of assembly of F-actin (see “Discussion?”).
We next aimed to investigate the role of Rac in integrin-mediated F-actin assembly by measuring the temporal exposure of actin filament barbed ends in platelets spreading on fibrinogen. This was achieved by monitoring the incorporation of Cy3-labeled monomeric actin into permeabilized platelets, which directly correlates with the level of barbed-end exposure (24). Our data demonstrate that wild-type and Rac1-/-Rac2-/-platelets incorporate equivalent levels of Cy3-labeled monomeric actin as they spread on immobilized fibrinogen (supplemental Fig. S1). In contrast, incorporation of monomeric Cy3-labeled actin into platelets was abrogated in the presence of cytochalasin D, a potent actin-polymerization inhibitor (data not shown). Taken together, these data demonstrate that αIIbβ3integrin engagement of immobilized fibrinogen induces barbed-end exposure (as measured by the incorporation of monomeric actin) in wild-type platelets and that this process is independent of Rac1/Rac2.
The Role of Rac in Platelet Aggregation—The role of Rac GTPases in the process of agonist-induced platelet shape change and aggregation was examined by using a Born aggregometer. This apparatus measures light transmission through a platelet suspension, with an increase in optical density correlating to platelet spheration (shape change) and a decrease indicative of platelet aggregation. As shown in Fig. 4, the magnitude and time course of both shape change and aggregation in response to intermediate concentrations of thrombin (0.1 units/ml) or ADP (10 μM) was similar for wild-type and Rac1-/-Rac2-/- mouse platelets. We obtained similar results when platelets were treated with submaximal concentrations of thrombin (0.04 units/ml) or ADP (1 μM) (data not shown). It is noteworthy that aggregation to thrombin is not altered in the absence of Rac1/Rac2, even though this response is dependent on hydrolysis of phosphatidylinositol 4,5-bisphosphate by phospholipase C (5). Similar results to those recorded for Rac1-/-Rac2-/- platelets were also observed for Rac1-deficient and Rac2-deficient platelets (data not shown).
FIGURE 4.

Platelets from Rac1/Rac2-deficient mice have normal aggregation responses. A, washed platelets (2 × 108/ml) from wild-type (WT) and Rac1-/-Rac2-/-mice were stimulated with 0.1 unit/ml thrombin and the change in optical density indicative of aggregation recorded. B, heparinized PRP (2 × 108/ml) from wild-type and Rac1/Rac2-deficient mice was stimulated with 10 μM ADP, and the aggregation was recorded as described above. One representative experiment of three separate trials is depicted.
The Role of Rac in Platelet Spreading on Fibrinogen—Ensuing experiments aimed to examine the role of Rac in mediating platelet spreading. A reproducible series of morphological changes occurs when platelets contact a fibrinogen-coated surface in the presence of inhibitors of ADP and thromboxanes. However, there exists a marked difference between the patterns of spreading observed for human versus murine platelets. Human platelets bind and fully spread on immobilized fibrinogen in the absence of external agonists (Fig. 5). Moreover, stimulation with either thrombin or ADP has a minimal effect on the extent of human platelet spreading (TABLE ONE). In contrast, wild-type murine platelets exhibit only partial formation of a lamellae-like structure on fibrinogen under nonstimulated conditions (Fig. 5). However, stimulation of murine platelets with either thrombin or ADP greatly enhances the degree of lamellipodia formation, resulting in a dramatic increase in the final platelet surface area (TABLE ONE). In both species, spreading is completely inhibited in the presence of the actin-polymerization inhibitor cytochalasin D (TABLE ONE), consistent with previous work (22, 30).
FIGURE 5.

Spreading of human and murine platelets on fibrinogen. Purified human and wild-type (WT) or Rac1-/-Rac2-/-murine platelets (2 × 107/ml) were placed on coverslips coated with fibrinogen for 45 min and imaged using DIC microscopy. Platelets were treated with apyrase (2 units/ml) and indomethacin (10 μM) in the absence or presence of 1 unit/ml thrombin. In separate experiments, platelets were treated with 10 μM ADP in the absence of inhibitors of secondary mediators. Results are representative of at least three experiments.
TABLE ONE.
Effects of external stimulation on platelet surface area following adhesion to fibrinogen Purified human and wild-type (WT) and Rac1-/-Rac2-/- murine platelets (2 × 107/ml) were placed on coverslips coated with fibrinogen for 45 min. Platelets were treated with apyrase (2 units/ml) and indomethacin (10 μM) in the absence or presence of 1 unit/ml thrombin. In separate experiments, platelets were treated with 10 μM ADP or 10 μM cytochalasin D (Cyto D) in the absence of inhibitors of secondary mediators. The mean surface area of the adherent platelets was determined as described under “Experimental Procedures.” Values are reported as follows: platelet surface area = mean ± S.E. of at least 250 cells.
| Analysis of platelet surface area on fibrinogen (μm2) | |||
|---|---|---|---|
| Treatment | Human | WT | Rac1-/-Rac2-/- |
| 47.9 ± 0.27 | 14.3 ± 0.12 | 13.5 ± 0.11 | |
| ADP | 49.5 ± 0.23 | 19.0 ± 0.18a | 14.0 ± 0.19 |
| Thrombin | 51.5 ± 0.34 | 25.1 ± 0.10a | 19.2 ± 0.13a |
| Cyto D | 10.9 ± 0.10a | 8.1 ± 0.13a | 8.0 ± 0.14a |
p < 0.01 with respect to no treatment control for each genotype.
Lamellipodial actin assembly has been reported to be regulated by Rac in permeabilized platelets using a dominant negative form of the GTPase (5). The mutant mouse platelets were used to address the roles of Rac1 and Rac2 in mediating spreading in platelets. Upon exposure to immobilized fibrinogen, Rac1-/-Rac2-/- platelets underwent a similar level of adhesion and changes in morphology, extending filopodia and forming partial lamellae-like structures, to that observed for wild-type mouse platelets (Fig. 5). More specifically, quantitative image analysis revealed that Rac1-/-Rac2-/- platelets formed equivalent number of filopodia as observed for wild-type platelets, and these filopodia were of similar thickness and length (TABLE TWO). However, upon thrombin stimulation and exposure to immobilized fibrinogen a dramatic inhibition of lamellipodia formation was observed in Rac1-/-Rac2-/-platelets. More importantly, these cells retain the ability to form pronounced filopodia, which were significantly thicker and longer than those observed in the absence of thrombin (Fig. 5 and TABLE TWO). In sharp contrast, ADP-induced morphological changes were absent in Rac1-/-Rac2-/- platelets, with the platelets being indistinguishable from non-stimulated levels of spreading on fibrinogen. Similar results to those recorded for Rac1-/-Rac2-/-platelets were also observed in Rac1-deficient platelets, although there was no apparent difference in the spreading of Rac2-deficient platelets compared with wild-type cells in the presence of thrombin or ADP stimulation (data not shown).
TABLE TWO.
Quantitative analysis of platelet morphology following adhesion to fibrinogen Purified wild-type (WT) and Rac1-/-Rac2-/- murine platelets (2 × 107/ml) were placed on coverslips coated with fibrinogen for 45 min. Platelets were treated with apyrase (2 units/ml) and indomethacin (10 μM) in the absence or presence of 1 unit/ml thrombin. The length and thickness of filopodial protrusions were determined as described under “Experimental Procedures.” Values are reported as follows: mean ± S.E. of at least 100 cells.
| Genotype | Treatment No. | filopodia per cell | Length of filopodia | Thickness of filopodia |
|---|---|---|---|---|
| μm | μm | |||
| WT | 9.6 ± 0.20 | 0.92 ± 0.02 | 0.50 ± 0.01 | |
| Rac1-/-Rac2-/- | 9.8 ± 0.19 | 0.89 ± 0.02 | 0.48 ± 0.01 | |
| Rac1-/-Rac2-/- | Thrombin | 9.7 ± 0.09 | 2.05 ± 0.04a | 0.74 ± 0.01a |
p < 0.01 with respect to no treatment control.
To investigate further the kinetics of filopodia and lamellipodia formation in the absence of the two Rac isoforms, we monitored spreading of wild-type and Rac1-/-Rac2-/-platelets on fibrinogen using time-lapse video microscopy (Fig. 6). Upon initial contact with the surface, both control and Rac1/Rac2-deficient platelets undergo rounding, before generating short, dynamic filopodia and limited formation of lamellae-like structures. However, in the presence of thrombin, platelets rapidly generate sheet-like lamellipodial membrane that proceed to fill in the gaps between the filopodia (supplemental video 1), resulting in a 60-70% increase in surface area, as shown in Fig. 6C. In contrast, thrombin-stimulated Rac1/Rac2-deficient platelets extend elongated filopodia, but fail to generate lamellipodia (supplemental video 2), and so have a corresponding reduction in surface area (Fig. 6C). Furthermore, the ADP-dependent increase in surface area observed in wild-type platelets is eliminated in the absence of Rac1 and Rac2 (Fig. 6C). Similar results were observed for Rac1-/-platelets, whereas Rac2-/-platelet spreading was indistinguishable from control platelets (data not shown). Taken together, these studies demonstrate an important role for Rac1, but not Rac2, in G protein receptor-coupled agonist-induced lamellipodia formation in platelets. However, Rac1 and Rac2 are not required for the limited spreading on fibrinogen that is observed in the presence of inhibitors of thromboxanes and ADP.
FIGURE 6.

Real time imaging of platelet spreading on fibrinogen. Purified murine platelets (2 × 107/ml) were exposed to a fibrinogen-coated surface in the presence of apyrase (2 units/ml) and indomethacin (10 μM) and observed in real time using DIC microscopy. Representative morphology of a single wild-type (WT)(A) and (B) Rac1-/-Rac2-/- murine platelet spreading on fibrinogen in the absence or presence of thrombin (1 units/ ml). Corresponding videos 1 and 2 can be found in the supplemental material. C, the mean surface area of wild-type and Rac1-/-Rac2-/- murine platelets in the absence (filled boxes) or presence of thrombin (filled circles) or ADP (open triangles) was quantitated at the indicated time points using ImageJ as described under “Experimental Procedures.” ADP stimulation was performed in the absence of inhibitors of secondary mediators. One experiment representative of 3-5 is depicted, and values are mean ± S.E. of at least 15 cells.
The Role of Rac in Arp2/3 Complex Redistribution in Spread Murine Platelets—Recent reports have identified the Arp2/3 complex as a major regulator of platelet actin dynamics, being incorporated into the actin cytoskeleton and redistributed to the edge of actin-rich lamellae in platelets following spreading on glass (31, 32). To explore whether Rac plays a role in this process, we used double-label immunofluorescence to compare the location of Arp2/3 with that of filamentous actin in murine platelet spreading on fibrinogen. Under nonstimulated conditions, a speckled cytoplasmic staining of Arp2/3 was observed in both wild-type and Rac1-/-Rac2-/-platelets (Fig. 7). However, upon thrombin stimulation, the Arp2/3 complex localizes uniformly around the rim of spread wild-type platelets and in the center of activated platelets, in agreement with previous observations (14, 31, 32). A similar distribution was observed in human platelets on fibrinogen (data not shown). In the absence of Rac1 (not shown) or Rac1 and Rac2, however, the Arp2/3 complex was localized to the tip and to the base of the filopodial protrusions, whereas in some cases, it was also present on the shaft of the filopodia. Higher magnification of the filopodia from the Rac1/Rac2-deficient platelets, as denoted by arrows in Fig. 7A, emphasize the relationship between Arp2/3 and actin filaments (Fig. 7B).
FIGURE 7.

Location of Arp2/3 in spread platelets. A, purified wild-type (WT) and Rac1/ 2-/-murine platelets (2 × 107/ml) were exposed to a fibrinogen-coated surface in the presence of apyrase (2 units/ml) and indomethacin (10 μM) with or without 1 unit/ml thrombin for 45 min. Cells were fixed with 3.7% paraformaldehyde containing 0.2% Triton and stained for actin filaments (FITC-phalloidin; column 1) and Arp2/3 (rhodaminp34; column 2) before examination by confocal microscopy. B, higher magnification of the two filopodia indicated by an arrow in A. Images are representative pictures from three experiments.
Role of Rac in Platelet Spreading on Collagen and Laminin—Recent reports have demonstrated that Rac is activated during platelet spreading on the extracellular matrix protein collagen in the absence of inhibitors of the secondary mediators, ADP and thromboxanes (20). In platelet suspensions, activation of Rac by collagen has been shown previously to be entirely dependent on the release of ADP and thromboxanes, thereby revealing that engagement of GPVI or integrin α2β1 by collagen is unable to promote detectable activation of the small G protein Rac (18). This observation was confirmed on murine platelets that had been allowed to spread on immobilized collagen in the presence of inhibitors of the actions of thromboxanes and ADP (Fig. 8A), whereas in the absence of inhibitors, an increase in Rac activation by collagen was observed (data not shown). Furthermore, pre-stimulation of collagen-bound platelets with thrombin caused a sustained level of Rac activation (Fig. 8A). Thrombin was also observed to stimulate activation of Rac on a laminin surface, which supports adhesion through integrin α6β1, although the extracellular matrix protein was unable to activate Rac on its own (Fig. 8A).
FIGURE 8.

Spreading of murine platelets on collagen and laminin. A, effects of platelet spreading on collagen and laminin on Rac activation. Washed murine platelets (5 × 108/ml) were added to BSA-, collagen (Coll)-, or laminin (LM)-coated dishes in the presence of apyrase (2 units/ml) and indomethacin (10 μM) and incubated for 10 min at 37 °C. Where indicated, platelets were pretreated with thrombin (thr; 1 units/ml). Dishes were washed twice to remove nonadherent cells. Platelets adherent to either collagen or laminin or in suspension over BSA were lysed, and Rac activity was determined as described in Fig. 2. B, purified wild-type (WT), and Rac1-/-Rac2-/-, murine platelets (2 × 107/ml) were placed on coverslips coated with either collagen or laminin in the presence of apyrase (2 units/ml) and indomethacin (10 μM) for 45 min before being fixed and imaged using DIC microscopy. C, aggregation of platelets from wild-type (WT) and Rac1/Rac2-deficient mice was measured as described in the legend to Fig. 4. One experiment representative of three is depicted.
We next examined whether Rac is required for spreading on collagen and laminin. In contrast to observations on fibrinogen, murine platelets bind and fully spread on immobilized collagen or laminin in the absence of external agonists (Fig. 8B). Further stimulation with either thrombin or ADP had minimal effect on the extent of wild-type murine platelet spreading on collagen or laminin (TABLE THREE). Most strikingly, lamellipodia formation was abolished for both Rac1- and Rac1/Rac2-deficient platelets on collagen or laminin, although a small degree of filopodia formation remained. Filopodia elongation and thickening was markedly enhanced when Rac-deficient platelets were treated with thrombin, in a similar way to that seen on fibrinogen, whereas ADP treatment had no effect upon the platelet morphology (TABLE THREE). Taken together, our data demonstrate that Rac1 plays a critical role in cytoskeletal reorganization downstream of engagement of receptors for collagen and laminin.
TABLE THREE.
Effects of external stimulation on platelet surface area following adhesion to collagen and laminin Purified human and murine platelets (2 × 107/ml) were placed on coverslips coated with collagen or laminin for 45 minutes. Platelets were treated with apyrase (2 units/ml) and indomethacin (10 μM) in the absence or presence of 1 unit/ml thrombin. In separate experiments, platelets were treated with 10 μM ADP in the absence of inhibitors of secondary mediators. Values are reported as follows: platelet surface area = mean ± S.E. of at least 250 cells.
| Murine platelet spreading on collagen (SA; μm2) | ||
|---|---|---|
| Treatment | WT | Rac1-/-Rac2-/- |
| 18.1 ± 0.18 | 7.8 ± 0.07a | |
| ADP | 18.4 ± 0.09 | 7.5 ± 0.18a |
| Thrombin | 18.3 ± 0.08 | 12.1 ± 0.12a |
| Murine platelet spreading on laminin (SA; μm2) | ||
| Treatment | WT | Rac1-/-Rac2-/- |
| 23.9 ± 0.27 | 7.4 ± 0.06a | |
| ADP | 22.9 ± 0.26 | 7.0 ± 0.09a |
| Thrombin | 23.5 ± 0.18 | 17.3 ± 0.10a |
p < 0.01 with respect to no treatment control for wild-type platelets.
Most interestingly, the weak aggregation response to a low concentration of collagen (0.6 μg/ml) was significantly reduced in the absence of Rac1/Rac2, whereas the response to higher concentrations (3-10 μg/ml) was not altered (Fig. 8C). More importantly, Rac1/Rac2-deficient platelets failed to aggregate in response to low concentrations of the GPVI-specific agonist collagen-related peptide (Fig. 8C). Therefore, this prompted us to probe the possible role of Rac in mediating the ability of GPVI to activate platelets. Our data demonstrate that tyrosine phosphorylation of Syk and phospholipase Cλ2, two proteins that play a central role in GPVI signaling, was not altered in response to CRP in Rac1/Rac2-deficient platelets (not shown). Furthermore, equivalent increases in intracellular calcium, as measured using the calcium reporter Oregon Green 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid 1-AM, was observed for both Rac1/Rac2-deficient and wild-type platelets in response to CRP stimulation (not shown). Similar results were observed in the absence of Rac1, although there was no defect in response in the absence of Rac2 (not shown). It is noteworthy that all experiments examining the effects of CRP stimulation were performed in the presence of inhibitors of the positive feedback signals arising from ADP and thromboxanes. These results therefore demonstrate an important role for Rac in mediating aggregation downstream of GPVI, although this does not appear to be due to an impairment in activation of phospholipase C.
Investigation of the Role of Rac in Platelet Adhesion and Thrombus Formation under Flow in Vitro—We sought to investigate further the functional role of Rac on platelet adhesion to and platelet aggregation on collagen and VWF/thrombin under shear conditions using an in vitro flow-based assay. We perfused whole blood from wild-type, Rac1-/-, Rac2-/-, and Rac1-/-Rac2-/-mice over either a collagen-coated or VWF/thrombin-coated surface at 1000 s-1for 4 min. Subsequently, samples were fixed, and adherent platelets were imaged by either DIC or confocal icroscopy. Thrombin was co-immobilized with VWF to enhance thrombus formation, whereas immobilized thrombin alone did not support platelet recruitment (data not shown). Blood from control mice exhibited robust formation of densely packed platelets on both collagen and VWF/thrombin (Fig. 9A). In marked contrast, Rac1-/-Rac2-/-platelets attached along the length of the collagen fibers, but for the most part did not support platelet aggregate formation (Fig. 9A). Similarly, thrombus formation of Rac1-/-Rac2-/-platelets on VWF/thrombin was markedly reduced (Fig. 9A). Quantitation of surface area coverage by platelets revealed a significant decrease for Rac1-/-Rac2-/-platelets as compared with wild-type on both surfaces (TABLE FOUR). Furthermore, the analysis of the three-dimensional structure of the aggregate using confocal microscopy revealed that the mean height of the aggregate was reduced from 7.87 ± 0.41 μm2in control platelets to 2.34 ± 0.12 μm2for Rac1-/-Rac2-/- platelets on collagen (n = 20). These results are consistent with the notion that Rac1-/-Rac2-/- platelets formed a single layer on the collagen fibers in contrast to the multilayer platelet thrombi observed in control blood. In support of these findings, platelet treatment with an αIIbβ3antagonist, which has been shown previously to produce a single layer of platelets bound to collagen (33), resulted in a 2.07 ± 0.42 μm2 mean thrombus height. Similar results to those recorded for Rac1-/-Rac2-/- platelets were also observed in Rac1-deficient platelets, whereas Rac2-/-platelet aggregate formation was indistinguishable from that of control platelets (data not shown).
FIGURE 9.

Role of Rac in platelet adhesion and aggregate stability on collagen under flow. A, mouse blood anticoagulated with D-phenyl-alanyl-1-prolyl-1 arginine chloromethyl ketone and heparin was perfused through a collagen-coated microslide at a shear rate of 1000 s-1 for 4 min followed by modified Tyrode buffer for 3 min to remove nonadherent cells. In separate experiments, mouse blood was anticoagulated with sodium citrate and perfused over VWF/thrombin-coated microslides as described above. Subsequently, slides were fixed and visualized using DIC microscopy. Representative images of platelet adhesion from wild-type (left panel) and Rac1-/-Rac2-/- (right panel) mice are shown. B, alternatively, mouse blood was fluorescently labeled with DiOC6 prior to perfusion over collagen as described above. A representative time course for both wild-type (WT) (top panel) and Rac1-/-Rac2-/- (bottom panel) platelet accumulation on collagen is shown. Note the Rac1-/-Rac2-/- platelet embolization as indicated by the arrowhead at 20 and 30 s. Direction of flow is from left to right. Images are representative of three experiments.
TABLE FOUR.
Role of Rac1/Rac2 on platelet adhesion to collagen under flow Murine whole blood specimens were incubated for 10 min at 37 °C prior to being perfused for 4 min over either a collagen-coated or VWF/thrombin-coated surface at 1000 s[H11002]1. Platelet adhesion is expressed as the percentage of surface covered by platelets for five fields of view (1.3 × 104 μm2). Values are reported as mean ± S.E. of three experiments.
| Genotype | Surface | Platelet adhesion to collagen |
|---|---|---|
| % surface coverage | ||
| WT | Collagen | 51.7 ± 2.11 |
| WT | VWF | 35.9 ± 3.07 |
| Rac1-/-Rac2-/- | Collagen | 19.1 ± 1.13a |
| Rac1-/-Rac2-/- | VWF | 9.34 ± 0.80a |
p < 0.01 with respect to no treatment (wild type).
In an attempt to elucidate the mechanisms resulting in the decreased Rac1-/-Rac2-/-thrombus size, we monitored DIOC6-labeled platelet accumulation onto collagen in real time via fluorescent microscopy. As shown in Fig. 9B, a layer of single platelets is initially deposited onto the collagen-coated surface as blood from wild-type mice is perfused. As time increases, these collagen-bound platelets capture free-flowing platelets, resulting in an increase in thrombus size as evidenced by an increase in the fluorescence level of the thrombus because of the accumulation of fluorescently labeled cells. Similarly, when Rac1-/-Rac2-/- murine blood was perfused over collagen, an initial layer of platelets was observed to bind, with no significant difference in the time required for thrombus initiation. However, subsequent Rac1-/-Rac2-/- platelets that were recruited by the collagen-bound cells failed to resist the shear stress and were eventually stripped off and carried away downstream, as exemplified by the arrow at 20 and 30 s, respectively, in Fig. 9B. Thus, Rac1-/-Rac2-/- platelet-embolus formation resulted in a single layer of platelet deposition on the collagen surface, due to the inability of these platelets to support shear-resistant platelet-platelet interactions. A similar process of embolization was observed when Rac1-/-Rac2-/- platelets bound to VWF/thrombin-coated surfaces under flow (not shown). Rac1-/- platelets were observed to form emboli to the same degree as Rac1-/-Rac2-/- platelets, whereas Rac2-/- thrombus formation was similar to that of control platelets (data not shown). Taken together, our data demonstrate that Rac1, but not Rac2, is required for stable thrombus formation under shear flow on both collagen and VWF/thrombin surfaces.
Role of Rac in Thrombus Formation in Vivo—We next wished to investigate the role of Rac in platelet accumulation at sites of injury in a more physiologically relevant environment. We therefore used real time fluorescence and bright field microscopy to examine the dynamic profile of platelet accumulation in arterioles in the mouse cremaster micro-circulation after laser-induced injury (34). In this model of mild injury, platelet accumulation following endothelial damage occurs in a specific temporal pattern (Fig. 10A). A kinetics curve was constructed based on the median value of the integrated fluorescent intensity over a period of time (Fig. 10B). In wild-type mice, during the initial phase of this dynamic process, platelets rapidly accumulated until a maximum thrombus size (741 ± 224; arbitrary units of fluorescence) was obtained (Fig. 10C). Subsequently, a loss of platelets occurred, leading to a diminishing in size over the course of several minutes until the platelet content was stabilized within the thrombi, leaving a flattened mural thrombus at the end of the recording period. This final stage was identified as a plateau in the kinetics curve. In distinct contrast, an atypical pattern of thrombus formation was observed in Rac1-deficient mice (Fig. 10A). More specifically, a decrease in platelet accumulation was observed at all time points for Rac1-/- mice (Fig. 10B), and a significant reduction in the peak thrombus size (173 ± 52) was recorded. Atypical patterns of thrombus formation in Rac1-/- mice included numerous peaks indicative of many emboli detaching from the main thrombus body (data not shown), a “flatter” kinetics profile indicative of poor platelet accumulation, and a lack of a plateau phase at the end suggesting no mural thrombus remained attached to the vessel wall.
FIGURE 10.

Role of Rac in thrombus formation in vivo. A, thrombus formation over time in wild-type (top) and Rac1-/- (bottom) mice. Platelets were labeled in vivo with Alexa 488-conjugated goat anti-rat antibody bound to rat anti-CD41 antibody. The Alexa 488 fluorochrome image, collected digitally and presented as a green pseudocolor, is merged with the bright field image. Blood flow is from right to left in each image, as depicted by an arrow. Lines approximating the walls of the arteriole are shown in the 1st image of the series. B, kinetics of platelet accumulation. Each curve represents the median integrated platelet fluorescence for wild-type (WT) (8 thrombi; n = 2) and Rac1-/-(16 thrombi; n = 3) mice as a function of time after injury. C, corresponding peak thrombus size for wild-type and Rac1-/- mice. Fluorescent intensity of platelets is expressed in arbitrary units (a.u.). *, p = 0.01 with respect to wild-type.
DISCUSSION
Rho GTPases coordinate many cellular responses, often by regulating formation of different actin assemblies. In particular, Rac has been shown to trigger extension of lamellipodia containing arborized actin polymers in a wide variety of cells (4). In the current study, we have shown that Rac1 rather than Rac2 or Rac3 is the predominant isoform in platelets, dissimilar to the trend set by the majority of other cells of hematopoietic origin, where Rac2 is the major isoform. Significantly, we demonstrate that Rac1 and Rac2 are not functionally redundant in platelets, as loss of Rac1 is sufficient to induce identical phenotypes to that observed for Rac1-/-Rac2-/- platelets. Moreover, no phenotype was observed for Rac2-/- platelets in any of the assays performed, consistent with our inability to detect the presence of Rac2 by immunoblotting, although a trace amount of mRNA was found to be present in murine megakaryocytes using SAGE.
Role of Rac in Integrin-mediated Cytoskeletal Changes—Integrin engagement and integrin clustering generates the signals required to mediate certain aspects of cell physiology and morphology. The members of the Rho family of GTPases have been shown to contribute to these integrin-mediated signals in a wide range of cells. More specifically, this work has indicated that Rac is activated downstream of inte-grin-ligand interactions and that it is involved in the formation of lamellipodia (35, 36) and focal adhesions (37, 38). Studies in transfected A5 CHO cell lines, which stably express αIIbβ3, have shown that levels of GTP-bound Rac increase 10-fold upon adhesion to fibrinogen in a manner dependent on Syk (39). This αIIbβ3-regulated signaling pathway generates lamellipodia in the transfected cells (39). A calpain-dependent mechanism of Rac recruitment to sites of occupied integrins leads to Rac activation downstream of αvβ3integrin-ligand adhesion in a bovine aortic endothelial cell line (40). In striking contrast, however, the present study demonstrates that ligand engagement of integrin αIIbβ3 on murine platelets does not induce a detectable increase in Rac activation, instead requiring exogenous stimulation via the G protein-coupled agonists thrombin or ADP (Fig. 2). This finding is consistent with the fact that murine platelet αIIbβ3-mediated spreading on fibrinogen is similar for both wild-type and Rac-deficient platelets under nonstimulated conditions (Fig. 5), revealing that αIIbβ3 integrin-dependent cytoskeletal reorganization is independent of Rac. Moreover, our study demonstrates that Rac is not required for αIIbβ3 integrin-mediated barbed-end exposure in spreading platelets. Therefore, this dynamic remodeling of the murine platelet cytoskeleton downstream of integrin αIIbβ3 engagement of fibrinogen may be attributed to a phospholipase Cλ2-mediated elevation of Ca2+ and activation of protein kinase C (22), leading to actin filament severing by Ca2+-gelsolin (29) and protein kinase C-mediated phosphorylation of α-adducin (41). Alternatively, the initial regulation of the platelet cytoskeleton by integrin αIIbβ3 may be dependent upon the GTPase Cdc42, which has been shown to mediate formation of actin-dependent extensions in NIH 3T3 (37) and Chinese hamster ovary cells (42) downstream of integrin binding. Most interestingly, we were also unable to detect activation of Rac in human platelets on a fibrinogen-coated surface, although these cells do exhibit extensive formation of lamellipodia. However, we do not know whether this is dependent on a low level of activation of Rac that falls below the threshold of detection. This notion will be further discussed in the section below concerning adhesion to collagen and laminin surfaces.
Previous studies examining increases in platelet F-actin in response to stimulation have suggested that the bulk of actin assembly measured derives from the lamellar assembly that fills the spaces between filopodia (29). Most surprisingly, the present study demonstrates that Rac is not essential for thrombin-dependent increases in F-actin levels in murine platelets, although this is the case for the weaker agonist, ADP. Furthermore, the observation that thrombin-stimulated, but not ADP-stimulated, Rac-deficient platelets exhibit the ability to extend elon-gated filopodia, which are significantly longer and thicker compared with nonstimulated conditions, in the absence of lamellipodia formation suggests that the bulk of F-actin assembly detected in platelets in response to thrombin may be due to this enhanced filopodia formation. On the other hand, we show that Rac is essential for ADP-induced increases in F-actin levels, which correlates with the observation that Rac-deficient platelet spreading on fibrinogen is insensitive to ADP stimulation. Taken together, these findings support the notion that G protein-mediated signaling pathways leading to cytoskeletal changes differentially require Rac activation (16), and are consistent with the notion that thrombin-dependent extension of lamellae requires Ca2+-gelsolin-dependent severing of actin filaments in a phosphatidylinositol 3-kinase- and Rac-dependent manner, as suggested previously (5, 17, 29).
It has been proposed that Rac is essential for the uncapping of actin filaments in thrombin receptor-activating peptide-stimulated platelets (5), as well as subsequent actin monomer incorporation into lamellipodia protrusions (24). Recent evidence has demonstrated that Rac localizes to membranes adjacent to actin filaments and at the leading edge of the lamellae of spread platelets (17). This is consistent with the lamellipodial localization of the Arp2/3 complex, which is believed to induce de novo nucleation of actin filaments downstream of Rac (31, 32). Our findings confirm the translocation of Arp2/3 to the lamellae, as well as to an undefined structure in the cell center, upon thrombin stimulation of murine platelets on fibrinogen (Fig. 6). Most strikingly, in thrombin-stimulated Rac-deficient platelets, which only form filopodia, the Arp2/3 complex is associated both at the base and tip as well as within the filopodial protrusions. This might suggest that as filopodia are generated, the Arp2/3 complex is passively transported along with membrane within the filopodia, where it would be poised to generate lamellipodia upon Rac activation. This is supported by previous work demonstrating the presence of the Arp2/3 complex within growing filopodia at early stages of platelet spreading (32).
Role of Rac in Spreading and Aggregation on CollagenπAt sites of vascular injury, platelets come into contact with a number of additional extracellular matrix proteins, including collagen, which powerfully stimulates platelet activation through engagement of GPVI (43). Most interestingly, however, activation of GPVI by specific collagen peptides (18) or by collagen does not induce significant activation of Rac in the presence of inhibitors of the secondary mediators, ADP and thromboxanes. Nevertheless, collagen stimulates lamellipodia formation in platelets through a Rac-dependent pathway, suggesting that Rac is a crucial component of GPVI-mediated spreading. This apparent paradox could be explained by a transient activation of Rac that rapidly returns to basal levels or by the increase in GTP-bound Rac falling below the level of detection. A further possibility is that a basal level of GTP-bound form of Rac is present in platelets and is sufficient to lead to initiation of lamellipodia formation. This has important implications for our inability to detect activation of Rac following adhesion to fibrinogen in human and mouse platelets, although in the latter case formation of lamellipodia was not observed.
It is also noteworthy that a role for Rac in mediating platelet aggregation by the collagen immunoglobulin receptor GPVI was also observed, although it is not required for this response downstream of G protein-coupled receptor agonists. Most intriguingly, this effect of Rac was not associated with a change in tyrosine phosphorylation of Syk and phospholipase Cλ2, two proteins that play a central role in GPVI signaling, or elevation of intracellular calcium. The molecular basis of this effect, and whether it is related to an ability to form lamellipodia-like structures, is presently unclear.
Rac Is Required for Platelet Aggregate Stability under Shear—A major finding from this study is the requirement for Rac signaling for maintaining integrin αIIbβ3 adhesion contacts under shear. In the low levels of shear present in the Born aggregometer, we found that the levels of platelet aggregation induced by either thrombin, ADP, or intermediate to high concentrations of collagen were equivalent for both wild-type and Rac-deficient platelets, although a loss of aggregation to submaximal concentrations of collagen and also the GPVI-specific agonist CRP was observed in the absence of Rac1. More importantly, our data indicate an indispensable role for Rac in thrombus stabilization subjected to increased levels of shear (1000 s-1) in a parallel plate flow chamber. Our data demonstrate that initial recruitment of Rac-deficient platelets to collagen under shear is similar to that observed for wild-type cells. Most strikingly, however, we observed that under flow, Rac-deficient platelet recruitment was counterbalanced by both individual platelets and segments of the thrombus breaking off and flowing downstream in the presence of the torque imposed by shear flow. This dramatic increase in embolization is likely attributed to the decreased cytoskeletal regulation observed for Rac-deficient platelets resulting in a decrease in aggregate stability. Conversely, a second explanation is that this decreased stability observed in Rac-deficient platelet aggregates is because of a defect in GPVI signaling, and therefore platelet activation, following adhesion to collagen. However, our data examining platelet deposition on VWF/ thrombin under flow conclusively demonstrates a role for Rac in supporting thrombus stability independent of GPVI signaling. Furthermore, murine platelets that exhibit a more marked defect in collagen-induced aggregation, such as those that express a reduced level of the ITAM receptor (44) or platelets deficient in the adapter protein LAT,4 do not exhibit a similar degree of thrombus embolization.
The application of intravital microscopy allowed us to examine the role of Rac in thrombus formation at sites of injury in a physiologically relevant context. Most interestingly, we observed that only small, unstable thrombi formed in Rac1-/- mice, as indicated by a dramatic decrease in thrombus peak size and an increase in the number of emboli as compared with wild-type mice. Taken together, these data conclusively demonstrate a critical role for Rac1 in the development of stable thrombi under conditions of shear flow, demonstrating for the first time an essential involvement of a GTPase in the process of thrombus formation in vivo.
In conclusion, our study confirms a pivotal role for Rac1 in regulating lamellipodia formation in platelets following adhesion and activation by G protein-coupled receptors and demonstrates that this response is essential for maintaining integrity of platelet aggregates under physiologically relevant levels of shear.
Supplementary Material
Acknowledgments
We thank Gary Bokoch (Scripps Institute, La Jolla, CA), Ivan de Curtis (San Rafaele Scientific Institute, Milan, Italy), and Michael C. Berndt (Monash University, Clayton, Australia) for the anti-Rac2 and anti-Rac3 antibodies and VWF, respectively. We also thank Mike Tomlinson and John Frampton for the use of and assistance with the SAGE library; Simon Calaminus and Ban Dawood for their help with the F-actin and Ca2+ measurements, respectively; Majd Protty for computational assistance; and Yotis Senis for helpful advice.
Footnotes
This work was supported in part by the Wellcome Trust, British Heart Foundation, and Medical Research Council. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains Video 1, which shows the dynamics of thrombin-stimulated wild-type murine platelets spreading on fibrinogen; Video 2, which shows the dynamics of thrombin-stimulated Rac1-/-Rac2-/ murine platelets spreading on fibrinogen; and supplemental Fig. S1.
The abbreviations used are: BSA, bovine serum albumin; DiOC6, 3,3′-dihexyloxacarbocyanine iodide; VWF, von Willebrand factor; PRP, platelet-rich plasma; Pipes, 1,4-piperazinediethanesulfonic acid; FITC, fluorescein isothiocyanate; DIC, differential interference contrast; SAGE, serial analysis of gene expression.
M. G. Tomlinson, J. Frampton, and S. P. Watson, unpublished observations.
J. M. Auger and S. P. Watson, unpublished observations.
REFERENCES
- 1.Haataja L, Groffen J, Heisterkamp N. J. Biol. Chem. 1997;272:20384–20388. doi: 10.1074/jbc.272.33.20384. [DOI] [PubMed] [Google Scholar]
- 2.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 3.Ridley AJ. Trends Cell Biol. 2001;11:471–477. doi: 10.1016/s0962-8924(01)02153-5. [DOI] [PubMed] [Google Scholar]
- 4.Burridge K, Wennerberg K. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 5.Hartwig JH, Bokoch GM, Carpenter CL, Janmey PA, Taylor LA, Toker A, Stossel TP. Cell. 1995;82:643–653. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- 6.Walmsley MJ, Ooi SK, Reynolds LF, Smith SH, Ruf S, Mathiot A, Vanes L, Williams DA, Cancro MP, Tybulewicz VL. Science. 2003;302:459–462. doi: 10.1126/science.1089709. [DOI] [PubMed] [Google Scholar]
- 7.Gu Y, Filippi MD, Cancelas JA, Siefring JE, Williams EP, Jasti AC, Harris CE, Lee AW, Prabhakar R, Atkinson SJ, Kwiatkowski DJ, Williams DA. Science. 2003;302:445–449. doi: 10.1126/science.1088485. [DOI] [PubMed] [Google Scholar]
- 8.Miki H, Yamaguchi H, Suetsugu S, Takenawa T. Nature. 2000;408:732–735. doi: 10.1038/35047107. [DOI] [PubMed] [Google Scholar]
- 9.Machesky LM, Insall RH. Curr. Biol. 1998;8:1347–1356. doi: 10.1016/s0960-9822(98)00015-3. [DOI] [PubMed] [Google Scholar]
- 10.Biyasheva A, Svitkina T, Kunda P, Baum B, Borisy G. J. Cell Sci. 2004;117:837–848. doi: 10.1242/jcs.00921. [DOI] [PubMed] [Google Scholar]
- 11.Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM. Science. 2000;290:333–337. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- 12.Gregg D, Rauscher FM, Goldschmidt-Clermont PJ. Am. J. Physiol. 2003;285:C723–C734. doi: 10.1152/ajpcell.00230.2003. [DOI] [PubMed] [Google Scholar]
- 13.Barkalow K, Witke W, Kwiatkowski DJ, Hartwig JH. J. Cell Biol. 1996;134:389–399. doi: 10.1083/jcb.134.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falet H, Hoffmeister KM, Neujahr R, Italiano JE, Jr., Stossel TP, Southwick FS, Hartwig JH. Proc. Natl. Acad. Sci. U. S. A. 2002;99:16782–16787. doi: 10.1073/pnas.222652499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oda A, Miki H, Wada I, Yamaguchi H, Yamazaki D, Suetsugu S, Nakajima M, Nakayama A, Okawa K, Miyazaki H, Matsuno K, Ochs HD, Machesky LM, Fujita H, Takenawa T. Blood. 2005;105:3141–3148. doi: 10.1182/blood-2003-04-1319. [DOI] [PubMed] [Google Scholar]
- 16.Gratacap MP, Payrastre B, Nieswandt B, Offermanns S. J. Biol. Chem. 2001;276:47906–47913. doi: 10.1074/jbc.M104442200. [DOI] [PubMed] [Google Scholar]
- 17.Azim AC, Barkalow K, Chou J, Hartwig JH. Blood. 2000;95:959–964. [PubMed] [Google Scholar]
- 18.Pearce AC, Wilde JI, Doody GM, Best D, Inoue O, Vigorito E, Tybulewicz VL, Turner M, Watson SP. Blood. 2002;100:3561–3569. doi: 10.1182/blood.V100.10.3561. [DOI] [PubMed] [Google Scholar]
- 19.Soulet C, Gendreau S, Missy K, Benard V, Plantavid M, Payrastre B. FEBS Lett. 2001;507:253–258. doi: 10.1016/s0014-5793(01)02984-2. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki-Inoue K, Yatomi Y, Asazuma N, Kainoh M, Tanaka T, Satoh K, Ozaki Y. Blood. 2001;98:3708–3716. doi: 10.1182/blood.v98.13.3708. [DOI] [PubMed] [Google Scholar]
- 21.Benard V, Bohl BP, Bokoch GM. J. Biol. Chem. 1999;274:13198–13204. doi: 10.1074/jbc.274.19.13198. [DOI] [PubMed] [Google Scholar]
- 22.Wonerow P, Pearce AC, Vaux DJ, Watson SP. J. Biol. Chem. 2003;278:37520–37529. doi: 10.1074/jbc.M305077200. [DOI] [PubMed] [Google Scholar]
- 23.McCarty OJ, Zhao Y, Andrew N, Machesky LM, Staunton D, Frampton J, Watson SP. J. Thromb. Haemostasis. 2004;2:1823–1833. doi: 10.1111/j.1538-7836.2004.00925.x. [DOI] [PubMed] [Google Scholar]
- 24.Machesky LM, Hall A. J. Cell Biol. 1997;138:913–926. doi: 10.1083/jcb.138.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Nat. Med. 2002;8:1175–1181. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 26.Didsbury J, Weber RF, Bokoch GM, Evans T, Snyderman R. J. Biol. Chem. 1989;264:16378–16382. [PubMed] [Google Scholar]
- 27.Bolis A, Corbetta S, Cioce A, de Curtis I. Eur. J. Neurosci. 2003;18:2417–2424. doi: 10.1046/j.1460-9568.2003.02938.x. [DOI] [PubMed] [Google Scholar]
- 28.Hartwig JH. J. Cell Biol. 1992;118:1421–1442. doi: 10.1083/jcb.118.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Falet H, Barkalow KL, Pivniouk VI, Barnes MJ, Geha RS, Hartwig JH. Blood. 2000;96:3786–3792. [PubMed] [Google Scholar]
- 30.Fox JE, Shattil SJ, Kinlough-Rathbone RL, Richardson M, Packham MA, Sanan DA. J. Biol. Chem. 1996;271:7004–7011. doi: 10.1074/jbc.271.12.7004. [DOI] [PubMed] [Google Scholar]
- 31.Falet H, Hoffmeister KM, Neujahr R, Hartwig JH. Blood. 2002;100:2113–2122. [PubMed] [Google Scholar]
- 32.Li Z, Kim ES, Bearer EL. Blood. 2002;99:4466–4474. doi: 10.1182/blood.v99.12.4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abulencia JP, Tien N, McCarty OJ, Plymire D, Mousa SA, Konstantopoulos K. Arterioscler. Thromb. Vasc. Biol. 2001;21:149–156. doi: 10.1161/01.atv.21.1.149. [DOI] [PubMed] [Google Scholar]
- 34.Celi A, Merrill-Skoloff G, Gross P, Falati S, Sim DS, Flaumenhaft R, Furie BC, Furie B. J. Thromb. Haemostasis. 2003;1:60–68. doi: 10.1046/j.1538-7836.2003.t01-1-00033.x. [DOI] [PubMed] [Google Scholar]
- 35.Small JV, Stradal T, Vignal E, Rottner K. Trends Cell Biol. 2002;12:112–120. doi: 10.1016/s0962-8924(01)02237-1. [DOI] [PubMed] [Google Scholar]
- 36.Hall A. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 37.Price LS, Leng J, Schwartz MA, Bokoch GM. Mol. Biol. Cell. 1998;9:1863–1871. doi: 10.1091/mbc.9.7.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clark EA, King WG, Brugge JS, Symons M, Hynes RO. J. Cell Biol. 1998;142:573–586. doi: 10.1083/jcb.142.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Obergfell A, Judd BA, del Pozo MA, Schwartz MA, Koretzky GA, Shattil SJ. J. Biol. Chem. 2001;276:5916–5923. doi: 10.1074/jbc.M010639200. [DOI] [PubMed] [Google Scholar]
- 40.Bialkowska K, Kulkarni S, Du X, Goll DE, Saido TC, Fox JE. J. Cell Biol. 2000;151:685–696. doi: 10.1083/jcb.151.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barkalow KL, Italiano JE, Jr., Chou DE, Matsuoka Y, Bennett V, Hartwig JH. J. Cell Biol. 2003;161:557–570. doi: 10.1083/jcb.200211122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bialkowska K, Zaffran Y, Meyer SC, Fox JE. J. Biol. Chem. 2003;278:33342–33350. doi: 10.1074/jbc.M301217200. [DOI] [PubMed] [Google Scholar]
- 43.Nieswandt B, Watson SP. Blood. 2003;102:449–461. doi: 10.1182/blood-2002-12-3882. [DOI] [PubMed] [Google Scholar]
- 44.Best D, Senis YA, Jarvis GE, Eagleton HJ, Roberts DJ, Saito T, Jung SM, Moroi M, Harrison P, Green FR, Watson SP. Blood. 2003;102:2811–2818. doi: 10.1182/blood-2003-01-0231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.